
Abstract
Background
The objective of this study was to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of PF‑06835375, a potent selective afucosyl immunoglobulin G1 antibody targeting C-X-C chemokine receptor type 5 (CXCR5) that potentially depletes B cells, follicular T helper (Tfh) cells, and circulating Tfh-like (cTfh) cells, in patients with systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA).
Methods
This first-in-human, multicenter, double-blind, sponsor-open, placebo-controlled Phase 1 study recruited patients aged 18–70 years with SLE or RA. In Part A, patients received single doses of intravenous PF-06835375 (dose range: 0.03–6 mg) or placebo in six sequential single ascending dose (SAD) cohorts. In Part B, patients received repeat doses of subcutaneous PF-06835375 (dose range: 0.3–10 mg) or placebo on Days 1 and 29 in five multiple ascending dose (MAD) cohorts. Tetanus/Diphtheria (Td) and Meningococcal B (MenB/Trumenba™) vaccines were administered at Day 4 (Td and MenB) and Week 8 (MenB only) to assess PF-06835375 functional effects. Endpoints included treatment-emergent adverse events (TEAEs), pharmacokinetic parameters, pharmacodynamic effects on B and cTfh cells, and biomarker counts, vaccine response, and exploratory differential gene expression analysis. Safety, pharmacokinetic, and pharmacodynamic endpoints are summarized descriptively. The change from baseline of B and Tfh cell-specific genes over time was calculated using a prespecified mixed-effects model, with a false discovery rate < 0.05 considered statistically significant.
Results
In total, 73 patients were treated (SAD cohorts: SLE, n = 17; RA, n = 14; MAD cohorts: SLE, n = 22; RA, n = 20). Mean age was 53.3 years. Sixty-two (84.9%) patients experienced TEAEs (placebo n = 17; PF-06835375 n = 45); most were mild or moderate. Three (9.7%) patients experienced serious adverse events. Mean t1/2 ranged from 3.4–121.4 h (SAD cohorts) and 162.0–234.0 h (MAD cohorts, Day 29). B and cTfh cell counts generally showed dose-dependent reductions across cohorts (range of mean maximum depletion: 67.3–99.3%/62.4–98.7% [SAD] and 91.1–99.6%/89.5–98.1% [MAD], respectively). B cell-related genes and pathways were significantly downregulated in patients treated with PF-06835375.
Conclusions
These data support further development of PF-06835375 to assess the clinical potential for B and Tfh cell depletion as a treatment for autoimmune diseases.
Trial registration
ClinicalTrials.gov identifier: NCT03334851.
Similar content being viewed by others


Introduction
Systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) are systemic autoimmune diseases with substantial morbidity and mortality [1,2,3,4]. The global prevalence of SLE and RA has been estimated at 43.7 per 100,000 and 246.6 per 100,000 individuals, respectively [2, 5]. The prevalence of both SLE and RA varies by global region, although a trend of increasing prevalence has been reported over the last few decades [2, 5].
Treatment options for patients with SLE and RA have improved over the last two decades, with a better understanding of the pathobiology of the diseases, coupled with the development of targeted therapies, such as cytokine inhibitors and Janus kinase inhibitors for RA, and B cell and Type 1 interferon-directed therapy for SLE [6,7,8,9]. Even with improved treatments, 37% of patients with RA continue to experience active disease despite therapy [10], and a large proportion of patients with SLE still require disease-modifying antirheumatic drugs [11], despite their toxicity [11], and corticosteroids [11]. Given the risks associated with the long-term use of many existing treatments for SLE and RA, and that they are frequently insufficient to adequately control active disease [11, 12], there remains a need for safer and more effective therapies that can induce long-lasting remission.
B cell and T cell dysregulation is involved in the pathology of SLE and RA [13,14,15]. In SLE, T cells amplify inflammation by secreting pro-inflammatory cytokines, thus mediating autoantibody production by B cells, which in turn maintain disease through the accumulation of autoreactive memory T cells [16]. Meanwhile, in RA, B cells secrete proteins implicated in disease pathogenesis, such as rheumatoid factors, anti-citrullinated protein antibodies, and pro-inflammatory cytokines [17]. A major role of T cells in RA is to activate macrophages and fibroblasts, leading to the production of various cytokines and chemokines which exacerbate joint inflammation [17]. Follicular T helper (Tfh) cells are also implicated in the pathogenesis of autoimmune diseases, due to their role in the germinal center reaction, affinity maturation, and autoantibody generation [18, 19], and are elevated in both SLE and RA [20, 21]. The dysregulation of these immune cell types, and their involvement in the pathogenesis of SLE and RA, suggests therapeutic potential for targeting B and Tfh cells together in autoimmune diseases in order to inhibit the production of autoantibodies targeting self-antigens.
PF-06835375 is a humanized, afucosyl immunoglobulin G1 antibody selective against C-X-C chemokine receptor type 5 (CXCR5) expressed on B cells, Tfh cells, and circulating Tfh-like (cTfh) cells. PF-06835375 is in development for the treatment of autoimmune diseases through depletion of CXCR5-positive B and Tfh cells and antagonism of C-X-C motif chemokine ligand 13-dependent signaling, thereby representing a new strategy for treating SLE and RA.
This first-in-human study (NCT03334851) evaluated the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of PF‑06835375 in patients with seropositive SLE and RA, and assessed the functional effect of PF-06835375 on vaccine responses in this pool of autoimmune patients.
Methods
Patients
Patients were eligible for inclusion if they were aged 18–70 years, with a body mass index of 17.5–40 kg/m2, and a total body weight > 45 kg. In this Phase 1 study, patients were required to have either seropositive SLE or RA but without minimum disease activity requirement for inclusion.
SLE diagnosis was confirmed using the Systemic Lupus International Collaborating Clinics classification criteria and a positive anti-nuclear antibodies titer ≥ 1:80, and/or anti-dsDNA, and/or anti-Smith antibodies at screening. Patients with a clinical Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) score > 8 were excluded from the three lowest single dose cohorts in Part A of the study but were permitted to enroll in all other cohorts in Parts A and B, following consultation with the medical monitor.
RA diagnosis was confirmed using the 2010 American College for Rheumatology/European Alliance of Associations for Rheumatology criteria and positive rheumatoid factor and/or anti-citrullinated peptide antibody. Patients with a Disease Activity Score 28 score > 5.1 were excluded from the three lowest single dose cohorts in Part A of the study but were permitted to enroll in all other cohorts in Parts A and B, following consultation with the medical monitor.
The use of certain non-prescription concomitant treatments was permissible during the study, although patients were asked to abstain from initiating new treatments. All concomitant treatments taken during the study were recorded with indication, daily dose, and start and stop dates of administration. Concomitant treatments were recorded at each clinic visit.
Study design
The study was a multicenter, randomized, double-blind, sponsor-open, placebo-controlled Phase 1 single and multiple-dose escalation study consisting of two parts. The study design is illustrated in Fig. S1.
In Part A, patients with SLE or RA were randomized 3:3:3:3:6:5 to intravenous (IV) PF-06835375 (0.03, 0.1, 0.3, 1, 3, or 6 mg) in six sequential single ascending dose (SAD) cohorts. Nine patients were randomized to placebo across the six SAD cohorts. Patients randomized in Part A were not eligible to enroll in Part B. In Part B, patients with SLE or RA were randomized 6:6:7:6:6 to subcutaneous (SC) PF-06835375 (0.3, 1, 3, 6, or 10 mg) administered on Days 1 and 29 in five multiple ascending dose (MAD) cohorts. Eleven patients were randomized to placebo across the five MAD cohorts. Additional pre-dose medications, including corticosteroids, were administered based on emerging safety and tolerability data, and post-dose treatments were allowed at the investigator’s discretion.
In Part A, 100 mg IV methylprednisolone was administered pre-dose to all patients in the cohort receiving PF-06835375 6 mg IV to mitigate the potential for infusion-related reactions. In Part B, oral prednisone 40 mg pre-dose and 20 mg post-dose were administered to the remaining patients to be enrolled into the cohort receiving PF-06835375 3 mg SC, as the first patients who received this dose experienced infusion-related reactions. Oral prednisone 40 mg pre-dose and 20 mg post-dose were also administered to all patients in the cohorts receiving PF-06835375 6 mg and 10 mg SC.
The selected starting doses (SAD and MAD) were planned to provide predicted exposure margins of multiple thousands (> 10,000) compared to the exposure-stopping limit at no observed adverse effect level (NOAEL) while having minimal B-cell depletion. Maximum doses were planned such that at least a tenfold exposure margin compared to NOAEL exists with a maximal predicted B-cell depletion. MAD doses are higher compared to SAD doses, as the route of administration is different. SC SAD dosing has reduced absorption compared to IV MAD dosing, which reduces the amount of drug that reaches the systemic circulation.
To mitigate unanticipated safety risks, the study utilized sentinel dosing for cohorts of more than four patients. This involved dosing of additional patients following communication between the sponsor's medical monitor and investigators from sites at which sentinel patients were first enrolled.
In order to provide an understanding of PD properties related to the degree of B and cTfh cell depletion, the functional effects of PF-06835375 were assessed with two vaccine challenges. On Day 4, the tetanus/diphtheria (Td) vaccine was administered. On Day 4 and Week 8, the Meningococcal B (MenB/Trumenba™) vaccine was administered. The Td vaccine was used as a recall challenge as it induces clear secondary antibody and immune cell responses. The MenB vaccine was used as a neoantigen when administered at Day 4 and as a recall challenge when administered at Week 8.
All patients were followed for a minimum of 16 weeks after the last dose of PF-06835375 was administered; study completion criteria were based on B cell counts meeting ≥ 1 of the following criteria: (1) ≥ 50% of baseline value and stable or increasing between Weeks 12–16; (2) ≥ lower limits of normal (80 cells/µL) and stable or increasing between Weeks 12–16. Patients not meeting either criterion at Week 16, continued in the study until B cell counts met ≥ 1 of the criteria, or until B cell counts reached a new stable level and the patient was clinically stable for ≥ 3 consecutive visits, each at least two weeks apart, based on assessment by both the sponsor and the investigator.
The first visit of the first patient took place in November 2017 and the last visit of the last patient took place in February 2022, which encompassed the period during the COVID-19 pandemic.
This study was approved by the Schulman Associates Institutional Review Board, Inc. (reference number IORG000063) and was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and all International Council for Harmonisation Good Clinical Practice guidelines and International Ethical Guidelines for Biomedical Research Involving Human Subjects (Council for International Organizations of Medical Sciences 2022. Written informed consent was required from each patient before any study-specific activity.
Endpoints
Primary endpoints
The primary objectives of the study were to examine the safety and tolerability of PF-06835375. Primary endpoints were the incidence of dose-limiting or treatment-related treatment-emergent adverse events (TEAEs); incidence, severity, and causal relationship of TEAEs and withdrawals due to TEAEs; incidence of chemistry, hematology, and urinalysis laboratory findings through the end of the study; abnormal and clinically relevant changes in vital signs and electrocardiogram (ECG) parameters; and incidence of infections.
Secondary pharmacokinetic and pharmacodynamic endpoints
Serum PF-06835375 concentrations after single (Day 1 IV SAD cohorts) and multiple (Day 1 and 29 SC MAD cohorts) PF‑06835375 doses were determined. PK parameters were generated by noncompartmental methods.
For SAD cohorts (Part A), the following PK parameters were analyzed: area under the concentration–time profile from time 0 extrapolated to infinite time (AUCinf); area under the concentration–time profile from time 0 to the time of the last quantifiable concentration (AUClast), maximum serum concentration (Cmax); time at which Cmax occurred (Tmax), terminal half-life (t½), and clearance (CL).
For MAD cohorts (Part B), the following PK parameters were analyzed: area under the concentration–time profile from Day 1 to Day 29 (AUCtau), AUClast, Cmax, Tmax, t½, and apparent clearance (CL/F).
The absolute count of circulating CXCR5-positive B cells and cTfh cells over time was assessed for the SAD and MAD cohorts following administration of PF-06835375. T cell and B cell absolute counts were determined with flow cytometry using BD Multitest™ 6-color TBNK with BD Trucount™ tubes according to the manufacturer’s instructions (BD Biosciences, San Jose, CA). Data were acquired and analyzed with a FACSCantoII cytometer using BD FACSCanto Clinical Software. In addition, a flow cytometry panel that monitored T helper cell subsets, including CD3+CD4+CD45RO+CXCR5+ cTfh, was also performed. Whole blood was stained with an antibody cocktail containing anti-human monoclonal antibodies against CD45-AF700 and CCR6-BV421 (Biolegend, San Diego, CA), CD3-APC-H7, CD4 PerCP-Cy5.5, CD45RO-BV510, PD1-PE, ICOS-AF647, CD183-PE-Cy7 (BD Biosciences), and a non-drug competitive CXCR5-AF488 antibody (Creative Diagnostics, Shirley, NY), for 20 min at room temperature. Red blood cells were lyzed using FACSLyse (BD Biosciences), washed with staining buffer, and data were immediately acquired on a BD FACSCanto II flow cytometer. The absolute count of CD4+ cTfh cells was calculated by multiplying the frequency of Tfh cells of total CD4 T cells by the absolute count of CD4+ T cells derived from the BD Multitest™ Trucount™ assay. Depletion was defined as cell counts below 10 cells/μL. The incidence of the development of anti-drug antibodies (ADAs) and neutralizing antibodies (NAbs) were also examined.
Tertiary and exploratory pharmacodynamic and biomarker endpoints
The geometric means of B cell activating factor (BAFF) at baseline, as well as the geometric means of antibody response to the Td and MenB vaccines were assessed over time.
Whole blood collected in PAXgene tubes (BD Biosciences) was used to generate RNA sequencing (RNA-seq) datasets from Day 1 (pre-dose), 29 (pre-dose), 57, and 113. Samples collected from patients in all treatment groups in the SC MAD cohort and from patients in the 6 mg IV SAD cohort were used for data generation and analysis. Briefly, globin-depleted RNA was used for library preparation using the TruSeq Stranded mRNA Library Prep Kit (Illumina) and sequenced via next-generation sequencing on the Illumina NovaSeq 6000 platform. All samples were sequenced using paired-end 100 bp reads and mapped to the human genome assembly GRCh38. Samples were analyzed for the following genes: CD19, CXCR5, interleukin (IL)-6, and tumor necrosis factor (TNF) receptor superfamily members (TNFRSF)13B (TNFRSF13B), 13C (TNFRSF13C), and 17 (TNFRSF17).
Statistical analyses
Safety data and PK parameters were presented using descriptive summary statistics.
R programming language (www.r-projects.com) [22] was used to perform all transcriptomic analyses. The transcriptomic data were modeled longitudinally using a mixed-effects model with random subject effect, fixed treatment, and visit effects, using log2 counts per million (CPM) values following trimmed mean of M-values normalization [23] using the R package limma [24]. Due to minimal variance observed in a principal component analysis, samples from the SAD and MAD placebo cohorts were pooled into a single placebo group for statistical analysis. The Benjamini–Hochberg procedure [25] was used to adjust p-values for multiple hypotheses by controlling the false discovery rate (FDR).
Gene set analysis was performed using Fast Gene Set Enrichment Analysis (FGSEA) method [26] implemented within the Gene Set Enrichment Analysis package and the Molecular Signatures Database (mSigDB). A pre-ranked list of genes based on estimates of the summary statistics from a mixed-effects model was used as the input for pathway analysis with a significance of p < 0.05. Sample level log2 CPM was calculated for the selected significant gene sets. Modeling was performed using the same approach described for single genes [27]. The change from baseline of B and Tfh cell-specific genes over time was calculated using the mixed-effects model described previously. Differentially expressed genes for any post- versus pre-treatment comparison with FDR < 0.05 were considered statistically significant.
A total of four different analysis populations were employed in this study. The PK analysis population included all patients who received at least one dose of the study treatment and had evaluable PK data. The PD analysis population included all patients who received at least one dose of the study treatment and had at least one PD measurement. The safety analysis population included all patients who received at least one dose of the study treatment. The pooled-placebo analysis population included all patients enrolled in the placebo groups, regardless of SAD or MAD cohorts.
Results
Patients
Patient disposition is illustrated in Fig. 1. In total, 74 patients were randomized and 73 patients were treated (SAD cohorts: SLE, n = 17; RA, n = 14; MAD cohorts: SLE, n = 22; RA, n = 20). Of patients receiving placebo in the MAD cohort, one discontinued due to a protocol deviation, and two patients discontinued during the follow-up phase; of patients receiving PF-06835375 3 mg SC, three discontinued during follow-up; of patients receiving PF-06835375 6 mg and 10 mg, one in each group discontinued during follow-up. Pre- and post-dose corticosteroids were administered in the PF-06835375 3 and 6 mg IV cohorts and 3, 6, and 10 mg SC cohorts. Patient demographics and baseline characteristics are presented in Table 1. Mean (standard deviation) age was 53.3 (10.7) years. Most patients were female (n = 65, 89.0%) and White (n = 54, 74.0%). At baseline, the mean SLEDAI-2K and Disease Activity Score 28 C-reactive protein values were 0.58 and 0.46, in patients with SLE and RA, respectively.

Patient disposition in single ascending dose and multiple ascending dose cohorts. AE adverse event, IV intravenous, MAD multiple ascending dose, RA rheumatoid arthritis, SAD single ascending dose, SC subcutaneous, SLE systemic lupus erythematous
Primary safety endpoints
A total of 62 (84.9%) patients across the SAD and MAD cohorts experienced TEAEs (Table 2), most of which were mild or moderate in severity. A total of three (9.7%) patients (all in the SAD cohort) experienced serious adverse events (SAEs) unrelated to the study treatment, and one (9.1%) patient discontinued due to a TEAE of disease progression unrelated to the study treatment in the placebo SC cohort.
In the SAD cohort, a total of 15 (48.4%) patients experienced treatment-related TEAEs: three (37.5%) patients receiving placebo IV, one (33.3%) patient receiving PF-06835375 0.1 mg IV, two (66.7%) patients receiving PF-06835375 1 mg IV, six (100.0%) patients receiving PF-06835375 3 mg IV, and three (60.0%) patients receiving PF-06835375 6 mg IV. In the MAD cohort, 16 (38.1%) patients experienced treatment-related TEAEs: three (27.3%) patients receiving placebo SC, three (50.0%) patients receiving PF-06835375 0.3 mg SC, one (16.7%) patient receiving PF-06835375 1 mg SC, five (71.4%) patients receiving PF-06835375 3 mg SC, two (33.3%) patients receiving PF-06835375 6 mg SC, and two (33.3%) patients receiving PF-06835375 10 mg SC.
The most common TEAEs in the SAD and MAD cohorts combined during the active collection period (from informed consent through at least 90 days after the last administration of PF-06835375) were headache (n = 18, 24.7%), pyrexia (n = 11, 15.1%), and urinary tract infection (n = 9, 12.3%). The most common infections after urinary tract infection were upper respiratory tract infection (n = 6, 8.2%) and viral upper respiratory tract infection (n = 3, 4.1%). All infections were mild or moderate in severity.
Two patients who both received PF-06835375 3 mg IV were reported to have mild infusion-related reaction, with onset at Day 1 and resolved at Day 14 and Day 15, and one patient who received PF-06835375 3 mg SC was reported to have mild allergic reaction, with onset at Day 1 and resolved at Day 2. All were deemed related to the study treatment. None of these events required treatment discontinuation.
Laboratory and ECG abnormalities (n = 3, each) considered clinically significant are shown in Table 2. All ECG abnormalities occurred in the SAD cohorts (PF-06835375 0.1, 1, and 6 mg). Of these ECG abnormalities, two patients experienced increased heart rate, whereas one patient experienced decreased ECG T wave amplitude. Each of these events were mild or moderate in severity and were resolved within the same day of dosing. No clinically significant changes in vital signs or deaths occurred.
Secondary pharmacokinetic and pharmacodynamic endpoints.
Pharmacokinetic endpoints
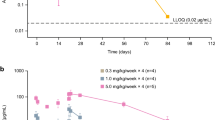
PK parameters are shown in Tables 3 and 4; concentration–time profiles for SAD and MAD cohorts are shown in Fig. 2A and B, respectively. Among the SAD cohorts (Part A), median Tmax ranged from 2–4 h and mean CL from 0.021–0.313 L/h. Mean t1/2 ranged from 3.40–121.4 h. Exposure (AUC and Cmax) generally appeared to increase dose-proportionally for doses ≤ 1 mg and more than dose-proportionally for doses > 1 mg. Among MAD cohorts, Day 1 median Tmax ranged from 144–170 h; the increase in exposure (AUCtau and Cmax) generally appeared to be greater than dose-proportional. Due to high affinity of the antibody to CXCR5 on the B-cell surface, significant target-mediated drug disposition at lower concentrations was observed. A reduction in clearance was also observed with higher doses and hence higher concentrations, which is indicative of target saturation and target-mediated drug disposition behavior. Day 29 median Tmax ranged from 121–171 h and mean CL/F from 0.0785–0.117 L/h. Day 29 mean t1/2 ranged from 162.0–234.0 h. Exposure (AUCtau and Cmax) generally appeared to be dose-proportional.

Serum PF-06835375 concentration–time profiles in the A single ascending dose and B multiple ascending dose cohorts (pharmacokinetic analysis population). The PK analysis population included all patients who received at least one dose of the study treatment and had evaluable PK data. The plots presented are semi-logarithmic. IV intravenous, MAD multiple ascending dose, PK pharmacokinetic, SAD single ascending dose, SC subcutaneous
Pharmacodynamic endpoints
B and cTfh cell counts generally showed dose-dependent reductions across cohorts (range of mean maximum depletion: 67.3–99.3% and 62.4–98.7%, respectively, in SAD cohorts, and 91.1–99.6% and 89.5–98.1%, respectively, in MAD cohorts; Fig. 3). The mean duration of B and cTfh depletion extended up to 71.6 and 62.0 days, respectively, in SAD, and 78.5 and 109.5 days, respectively, in MAD cohorts.

Mean A, B CXCR5-positive B and C, D cTfh cell counts in the single ascending dose and multiple ascending dose cohorts (PD analysis population). The PD analysis population included all patients who received at least one dose of the study treatment and had at least one PD measurement. A single IV dose was administered in SAD cohorts (designated Day 1), and multiple SC doses were administered in MAD cohorts (Day 1 and 29); the study duration including follow-up was 4–10 months from the screening. The plots presented are semi-logarithmic. cTfh circulating follicular T helper-like, CXCR5 C-X-C chemokine receptor type 5, IV intravenous, MAD multiple ascending dose, PD pharmacodynamic, SAD single ascending dose, SC subcutaneous
A total of 438 samples were analyzed for ADA, and 77 (17.6%) samples tested positive; these ADA-positive samples were further tested for NAb. Of these 77 samples, 60 (77.9%) samples tested NAb positive. In the IV SAD cohorts, a total of 23 ADA/NAb evaluable patients were analyzed, of these patients, seven (30.4%) tested positive for ADA and seven (30.4%) tested positive for NAb. In the SC MAD cohorts, a total of 31 ADA/NAb evaluable patients were analyzed; of these patients, 10 (32.3%) tested positive for ADA and seven (22.6%) tested positive for NAb.
No notable changes were seen in PK parameters, B and cTfh cell counts, and safety based on the ADA and NAb status of patients.
Exploratory (tertiary) pharmacodynamic and biomarker endpoints
To assess the mechanism of PF-06835375 and the functional effects of combination B and Tfh cell depletion, both primary neoantigen and recall vaccine responses were assessed. There were no changes in post- versus pre-vaccination B cell counts in participants who received placebo. In participants who received placebo, cTfh cell counts at Day 8 and Day 15 fluctuated around the baseline level (cTfh cell ratio range from 0.1 to 3) (Fig. S2 and S3). All study participants who received PF-06835375 had lower B cell counts compared to baseline after Day 1, with decreases to < 100 cells/µL in the lower doses and to < 10 cells/µL with higher doses, to Day 15. Almost all study participants who received PF-06835375 had lower cTfh cell ratios, which were ≤ 1 at Day 4 and remained low at Day 8 and Day 15. This observation confirms that the cTfh-inhibitory aspect of PF-06835375’s mechanism of action includes inhibition of cTfh cells in addition to its B cell-depleting properties, and indicates that PF-06835375 has the potential to reduce the number of newly formed autoreactive B cells and consequently autoantibody levels.
Regarding recall vaccine responses, the geometric means of diphtheria and tetanus antibody concentrations for the placebo group in both the SAD and MAD cohorts increased from Day 1 to Day 29, and were relatively stable from Day 29 to Day 85 (Fig. 4A–D). At Day 57, the geometric means of the MenB antibody concentrations showed increases in the recall responses in most treatment groups until Day 85 in both cohorts (Fig. 4E–F). Regarding neoantigen responses, the means of the MenB antibody concentrations in the placebo groups also increased from Day 1 to Day 29, and remained stable (SAD) or decreased (MAD) to Day 57 (Fig. 4E–F).

Geometric means of antibody responses to the A, B, C, D Tetanus/Diphtheria vaccine in the single ascending dose and multiple ascending dose cohorts and the E, F Meningococcal B vaccine in the single ascending dose and multiple ascending dose cohorts (PD analysis population). The PD analysis population included all patients who received at least one dose of the study treatment and had at least one PD measurement. Data were excluded from the plot if patients did not receive the Td or MenB vaccine at Day 4. If patients did not receive the MenB vaccine at Week 8, their Week 12 data were excluded from the plot. The lower limit of detection for anti-MenB was 4, a titer of 2 was reported in case of non-detectable MenB antibody. The plots presented are semi-logarithmic. IV intravenous, MAD multiple ascending dose, MenB Meningococcal B, PD pharmacodynamic, SAD single ascending dose, SC subcutaneous, Td tetanus/diphtheria
The geometric means of diphtheria and tetanus antibody concentrations for all the active treatment groups in both cohorts followed a similar trend to those of the placebo groups, increasing from Day 1 to Day 29 and remaining relatively stable from Day 29 to Day 85 (Fig. 4A–D). At Day 57, the geometric means of the MenB antibody concentrations showed increases overall until Day 85 for most active treatment groups in both cohorts (Fig. 4E–F). The geometric means of MenB antibody titers were seen to increase from Day 1 to Day 29 across all doses in both cohorts, and remained elevated until Day 57 (Fig. 4E and F). A trend in dose–response to the Td and the MenB vaccinations was not observed in patients in either of the SAD or MAD cohorts (Fig. 4A–F).
Regarding serum BAFF, the geometric means at baseline were 1196.23 pg/mL and 1052.83 pg/mL in the SAD and MAD cohorts, respectively. The geometric means of BAFF over time are presented in Fig. 5A and B. Overall, BAFF levels increased over time, but there was some variation across doses. Increases were visible especially in the 0.03 mg SAD and placebo SAD cohort between Days 113 and 141, and in the MAD active treatment cohort across time points.

Geometric means of BAFF over time in the A single ascending dose and B multiple ascending dose cohorts (PD analysis population). The PD analysis population included all patients who received at least one dose of the study treatment and had at least one PD measurement. The plots presented are semi-logarithmic. BAFF B-cell activating factor, IV intravenous, MAD multiple ascending dose, PD pharmacodynamic, SAD single ascending dose, SC subcutaneous
Exploratory transcriptomic data generated using RNA-seq were analyzed for gene expression changes following B and cTfh cell depletion. The pathways that were most significantly affected at Days 29, 57, and 113 were B cell-related pathways/gene sets, which were downregulated in patients who received active treatment (Fig. 6; FDR < 0.05). Representative genes from pathways related to B-cell activation, differentiation, and receptor signaling showed a change in expression in samples from both SAD and MAD cohorts, whereas no significant changes were observed from baseline in the placebo group. Although changes in Tfh-specific gene signature remain to be determined, the expression of CXCR5 (B and Tfh cell surface marker) and CD19 (B cell surface marker) genes decreased significantly at all three time points assessed from Day 1 (Fig. 7). A significant decrease from baseline in the expression of the inflammatory cytokine gene IL-6 was seen across all doses and time points, with the exception of PF-06835375 6 mg IV in the SAD cohort at Day 113 (Fig. 7). Significant decreases in the expression of TNFRSF genes expressed on early-to-late B cell lineages TNFRSF17, TNFRSF13B, and TNFRSF13C were observed at most time points in patients treated with PF-06835375 (Fig. 7).

B cell-related pathways in patients in the multiple ascending and single ascending dose cohorts and the pooled placebo group. aPatients receiving placebo in the MAD and SAD cohort were pooled into a single placebo group for analysis. Heatmap represents longitudinal modulation of gene sets upon treatment in 1, 3, 6, and 10 mg MAD and 6 mg SAD cohorts. The color scale depicts the range of change from baseline in normalized expression level for each gene set (a gene set is a group of genes represented by a gene ontology term shown to the right of the heatmap, in rows). “Day” and “Dose” legends provide column annotations for the heatmap. *FDR < 0.05. FDR, false discovery rate; MAD multiple ascending dose, PBO placebo, SAD single ascending dose

Mean (95% CI) change from baseline in the expression of B and Tfh cell-specific markers over time analyzed by RNA sequencing: A CD19, B CXCR5, C IL-6, D TNFRSF17, E TNFRSF13B, and F TNFRSF13C. aPatients receiving placebo in the MAD and SAD cohort were pooled into a single placebo group for analysis. TNFRSF13B, TNFRSF13C, and TNFRSF17 encode transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI), B-cell activating factor receptor (BAFF-R), and B cell maturation antigen (BCMA) proteins, respectively. CI, confidence interval; CXCR5 C-X-C chemokine receptor type 5, IL interleukin, IV intravenous, MAD multiple ascending dose, SAD single ascending dose, TNFRSF tumor necrosis factor receptor superfamily member. *p ≤ 0.05; **p ≤ 0.01; ***p < 0.001
Discussion
This first-in-human Phase 1 study evaluated the safety, tolerability, PK, and PD of PF‑06835375, an antibody against CXCR5, and its functional effect on vaccine responses in patients with seropositive SLE and RA. PF-06835375 was generally well tolerated in patients with seropositive SLE and RA; most TEAEs were mild or moderate in severity and only one patient discontinued due to a TEAE of disease progression in the placebo group. PD findings indicated potent and prolonged depletion of both CXCR5-positive populations of B and cTfh cells, but with intact vaccination responses across treatment groups and consistent with the mechanism of action of PF-0683575.
The TEAEs, including infections, and SAEs noted in this study were consistent with those observed with other B cell-depleting autoimmune therapies, such as constitutional symptoms [28] and infusion-induced side effects [29, 30]. In the current study, two patients receiving 3 mg IV had mild infusion-related reaction and one patient receiving 3 mg SC had mild allergic reaction, with the addition of steroids for the 3 mg and subsequent SC MAD cohorts. Notably, infections, overall, were the second most common TEAEs reported in the current study, including urinary tract infection, upper respiratory tract infection, and viral upper respiratory tract infection; all were mild or moderate in severity. The small number of serious infections reported in this Phase 1 study is comparable with that reported in a Phase 3 study of patients with RA receiving the T cell-targeted costimulatory modulator abatacept, among whom 1.9% developed serious infections (compared with 8.5% of patients receiving TNF inhibitor therapy with infliximab) [31], and a Phase 2b study in patients with RA receiving B-cell depleting therapy with rituximab, among whom 2.0% developed serious infections [32]. There were no reported events of herpes zoster or SARS-CoV-2 infection during the course of the study. The overall impact of the COVID-19 pandemic on this study was minimal, by extending the overall time span of the study. Mild or moderate abnormalities in ECGs were reported in three individuals, each resolving within the same day of dosing. Collectively, these findings did not identify safety concerns related to the B and Tfh cell depletion with PF-06835375.
In both the SAD and MAD cohorts of the current study, robust B- and cTfh-cell depletion were observed post-dose in all PF-06835375 groups, compared with patients receiving placebo. B cell counts in these patients markedly decreased starting at 8 h post-dose on Day 1. Additionally, B and Tfh cell depletion was similar in participants with RA or SLE. Sufficient and timely depletion of B cells may be important for the overall effectiveness of immunosuppressant agents given that, among patients with SLE or RA, complete B-cell depletion following rituximab infusion was associated with improved clinical responses over time compared with those whose B-cell depletion was incomplete at the same time point [33, 34]. We also observed that the cTfh cell absolute counts in all patients receiving higher doses of PF-06835375 (1, 3, and 6 mg IV in SAD cohorts; 3, 6, and 10 mg in MAD cohorts) markedly decreased at 8 h post-dose on Day 1; in MAD cohorts, low cTfh cell counts persisted then decreased again after the second study dosing on Day 29. Given that cTfh cells are reported to be elevated in both SLE and RA [20, 21], and promote the germinal center reaction and generation of autoantibody secreting plasma cells [19, 35], the reduction of cTfh cells in patients receiving higher doses of PF-06835375 has the potential to reduce disease activity.
All cohorts, except the lowest IV SAD doses of PF-06835375 (0.03 mg and 0.1 mg), had at least one ADA-positive patient; the median time of onset for treatment-induced ADA and NAb was 31.5 and 36.0 days, respectively. The immunogenicity data did not suggest any clinically relevant impact of PF-06835375 on PK, B and cTfh cell depletion, or safety.
The recall immune response of patients receiving PF-06835375 were assessed by Td vaccination, while MenB vaccination was used to assess neoantigen immune responses. These data demonstrate that patients receiving PF-0683575 exhibited intact humoral immune responses to vaccinations, despite potent B and cTfh cell depletion, and low post- versus pre-vaccination cTfh cell ratios, demonstrating patients receiving standard of care immunosuppressive agents were still able to mount a protective antibody response. Our data, regarding functional vaccine recall responses, are consistent with previous findings in belimumab-treated patients with SLE, among whom antibody responses to pneumococcal, tetanus, and influenza antigens were not reduced, indicating preservation of the memory B cell compartment [36]. Markedly, our data contrast with data from a Phase 2 study of patients with active RA and background methotrexate treated with rituximab, in which B-cell depletion reduced responses to neoantigen (keyhole limpet hemocyanin), but did not reduce recall responses to tetanus and diphtheria vaccines [37]. Our findings suggest that the ability of PF-06835375, at the exposures studied in this trial, to deplete B and cTfh cells did not appear to have a major impact on patients’ humoral immune responses to vaccines. However, it should be noted that the sample size was small, resulting in variability in the estimates of vaccine responses. Differences between our results and those reported previously may be attributed to differences in the study populations and background medications, and may be pertinent, given that treatment with immunosuppressants has been identified as a risk factor for inadequate responses to vaccines, including SARS-CoV-2 vaccination in patients with both SLE and RA [38, 39].
Regarding BAFF levels, observed increases over time have also been reported in studies using rituximab in patients with active RA and non-responders to anti-TNF-α, and in patients with active refractory SLE [40, 41].
RNA-seq data indicated that blood transcriptomic changes were dominated by significant decreases in the expression of B cell-related genes and pathways. While only a representative set of genes was depicted, B and Tfh cell depletion, and potential changes in inflammation-associated genes (as shown by IL-6), have been included in this manuscript. Decreased expression of the TNFRSF17 gene, which encodes B cell maturation antigen protein, primarily expressed on late-stage B cells, plasmablasts, and long-lived plasma cells [42], could be indicative of the effect of PF-06835375 in depleting late-stage, antibody-producing circulating B cells. Likewise, lower expression of TNFRSF13B and 13C genes, which respectively encode transmembrane activator and calcium modulator and cyclophilin ligand interactor and BAFF receptor proteins [43], could potentially dampen signaling-mediated B cell autoreactivity. A deeper investigation of the transcriptomic data could lead to a refined transcriptomic signature for PF-06835375 treatment, provide further insight into its mechanism of action, and allow for an assessment of disease-associated molecular changes. However, changes in protein levels remain to be addressed.
There have been many recent advances in therapies for autoimmunity, including B cell-targeted agents that build on approved approaches (i.e., CD20 depletion and BAFF inhibition). B-cell depletion with potent next-generation antibodies directed against CD20 along with both B- and CAR-T-cell therapies that specifically target CD19-expressing cells have emerged as novel approaches expanding into refractory patient populations [44, 45]. However, PF-06835375 is the first agent with dual function to target B cells and Tfh cells. Tfh cells play a critical role in immune responses by promoting B-cell development, germinal center formation, and antibody production. Aberrant proliferation and function of Tfh cells can lead to autoantibody production and development of autoimmune diseases [46, 47]. Although usually located in secondary lymphoid organs, Tfh cells can also be identified in human blood, and their frequency and phenotype are often altered in patients with autoimmune diseases including SLE and RA [46, 47]. Although the data in this manuscript show lower cTfh cell ratios post-dosing, there were still sufficient neoantigen and functional recall vaccination antibody responses, indicating that humoral immunity functionality remains preserved in participants receiving PF-06835375.
The strengths of this Phase 1 study include that it was conducted in patients with seropositive SLE and RA, and not in healthy volunteers, and that the majority of patients recruited to this study were female, which mirrors the global populations of patients with these conditions [2, 48, 49]. This study investigated a range of PF-06835375 doses and different administration routes, meaning that a variety of potential impacts on drug characteristics and PK were explored. A vaccination challenge was included for assessing the mechanism of action and safety, with respect to demonstrating the ability to produce neoantigen and functional recall responses to Td and MenB vaccinations over a range of B and Tfh cell depletion levels.
Limitations of this study include the small sample size and the lack of entry criteria requiring minimum disease activity, which may not be representative of the wider patient population seeking treatment. Also, the majority of patients were White, with a mean age of 53.3 years; prevalence rates for SLE are higher in non-White populations and RA has a bimodal age distribution with incidence peaks in the 25–45 and > 65 age groups [48, 50]. Furthermore, since the main aims of this Phase 1 study were to assess safety, tolerability, PK, and PD to evaluate the mechanistic aspects of PF-06835375. Efficacy was not evaluated as there was no minimum disease activity requirement for patients to enroll in the study. The lack of disease activity requirement for inclusion in the study limited the ability to interpret the effects of PF-06835375 on disease-related parameters. A Phase 2 study in primary immune thrombocytopenia (NCT05070845) is ongoing to explore clinical efficacy related to combination B- and Tfh-cell depletion.
Conclusions
These first-in-human data support further development of PF-06835375 to assess the clinical potential for B and Tfh cell depletion as a treatment for autoimmune diseases.
Availability of data and materials
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Abbreviations
- ACR:
-
American college of rheumatology
- ADA:
-
Anti-drug antibodies
- AE:
-
Adverse event
- ANA:
-
Anti-nuclear antibodies
- Anti-dsDNA:
-
Anti-double-stranded deoxyribonucleic acid
- AUCinf:
-
Area under the concentration–time profile from time 0 extrapolated to infinite time
- AUClast:
-
Area under the concentration–time profile from time 0 to the time of the last quantifiable concentration
- AUCtau :
-
Area under the serum concentration–time profile from time 0 to time tau, the dosing interval, where tau = 28 days
- BAFF:
-
B-cell activating factor
- BAFF-R:
-
B-cell activating factor receptor
- BMI:
-
Body mass index
- CL:
-
Clearance
- CL/F:
-
Apparent clearance
- Cmax :
-
Maximum serum concentration during the dosing interval
- CPM:
-
Counts per million
- cTfh:
-
Circulating follicular T helper cell
- CXCR5:
-
C-X-C chemokine receptor type 5
- hsCRP:
-
High-sensitive C-reactive protein
- ECG:
-
Electrocardiogram
- EULAR:
-
European alliance of associations for rheumatology
- FDR:
-
False discovery rate
- FGSEA:
-
Fast gene set enrichment analysis
- Ig:
-
Immunoglobulin
- IL:
-
Interleukin
- IFN-γ:
-
Interferon-gamma
- IV:
-
Intravenous
- MAD:
-
Multiple ascending dose
- MenB:
-
Meningococcal B
- NAb:
-
Neutralizing antibodies
- NOAEL:
-
No observed adverse effect level
- PCA:
-
Principal component analysis
- PD:
-
Pharmacodynamics
- PK:
-
Pharmacokinetics
- RA:
-
Rheumatoid arthritis
- RNA-seq:
-
RNA sequencing
- SAD:
-
Single ascending dose
- SAE:
-
Serious adverse event
- SC:
-
Subcutaneous
- SLE:
-
Systemic lupus erythematous
- SLEDAI-2K:
-
Systemic Lupus Erythematosus Disease Activity Index 2000
- SLICC:
-
Systemic Lupus International Collaborating Clinics
- t1/2 :
-
Terminal half-life
- Td::
-
Tetanus/Diphtheria
- TEAE:
-
Treatment-emergent adverse event
- Tfh:
-
Follicular T helper
- Tmax :
-
Time for Cmax
- TNF:
-
Tumor necrosis factor
- TNFRSF:
-
Tumor necrosis factor receptor superfamily
References
Jorge AM, Lu N, Zhang Y, Rai SK, Choi HK. Unchanging premature mortality trends in systemic lupus erythematosus: a general population-based study (1999–2014). Rheumatology (Oxford). 2018;57:337–44.
Safiri S, Kolahi AA, Hoy D, Smith E, Bettampadi D, Mansournia MA, et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: a systematic analysis of the Global Burden of Disease study 2017. Ann Rheum Dis. 2019;78:1463–71.
Heckert SL, Maassen JM, le Cessie S, Goekoop-Ruiterman YPM, Güler-Yüksel M, Lems W, et al. Long-term mortality in treated-to-target RA and UA: results of the BeSt and IMPROVED cohort. Ann Rheum Dis. 2024;83:161–8.
Figueroa-Parra G, Meade-Aguilar JA, Hulshizer CA, Gunderson TM, Chamberlain AM, Thanarajasingam U, et al. Multimorbidity in systemic lupus erythematosus in a population-based cohort: the lupus Midwest network. Rheumatology (Oxford). 2023:kead617.
Tian J, Zhang D, Yao X, Huang Y, Lu Q. Global epidemiology of systemic lupus erythematosus: a comprehensive systematic analysis and modelling study. Ann Rheum Dis. 2022;82:351–6.
Sim TM, Ong SJ, Mak A, Tay SH. Type I interferons in systemic lupus erythematosus: a journey from bench to bedside. Int J Mol Sci. 2022;23:2505.
Bruera S, Chavula T, Madan R, Agarwal SK. Targeting type I interferons in systemic lupus erythematous. Front Pharmacol. 2022;13:1046687.
Parodis I, Stockfelt M, Sjöwall C. B cell therapy in systemic lupus erythematosus: from rationale to clinical practice. Front Med (Lausanne). 2020;7:316.
Gadina M, Le MT, Schwartz DM, Silvennoinen O, Nakayamada S, Yamaoka K, et al. Janus kinases to jakinibs: from basic insights to clinical practice. Rheumatology (Oxford). 2019;58:i4–16.
Svensson B, Andersson M, Forslind K, Ajeganova S, Hafström I. Persistently active disease is common in patients with rheumatoid arthritis, particularly in women: a long-term inception cohort study. Scand J Rheumatol. 2016;45:448–55.
Fanouriakis A, Kostopoulou M, Alunno A, Aringer M, Bajema I, Boletis JN, et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. 2019;78:736–45.
Fraenkel L, Bathon JM, England BR, St Clair EW, Arayssi T, Carandang K, et al. 2021 American college of rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken). 2021;73:924–39.
Becker AM, Dao KH, Han BK, Kornu R, Lakhanpal S, Mobley AB, et al. SLE peripheral blood B cell, T cell and myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature. PLoS ONE. 2013;8:e67003.
Gunawan M, Her Z, Liu M, Tan SY, Chan XY, Tan WWS, et al. A novel human systemic lupus erythematosus model in humanised mice. Sci Rep. 2017;7:16642.
Takemura S, Klimiuk PA, Braun A, Goronzy JJ, Weyand CM. T cell activation in rheumatoid synovium is B cell dependent. J Immunol. 2001;167:4710–8.
Suarez-Fueyo A, Bradley SJ, Tsokos GC. T cells in Systemic Lupus Erythematosus. Curr Opin Immunol. 2016;43:32–8.
Yap HY, Tee SZ, Wong MM, Chow SK, Peh SC, Teow SY. Pathogenic role of immune cells in rheumatoid arthritis: implications in clinical treatment and biomarker development. Cells. 2018;7:161.
Ding T, Su R, Wu R, Xue H, Wang Y, Su R, et al. Frontiers of autoantibodies in autoimmune disorders: crosstalk between Tfh/Tfr and regulatory B cells. Front Immunol. 2021;12:641013.
Gatto D, Brink R. The germinal center reaction. J Allergy Clin Immunol. 2010;126:898–907;quiz 8–9.
Arroyo-Villa I, Bautista-Caro MB, Balsa A, Aguado-Acin P, Bonilla-Hernan MG, Plasencia C, et al. Constitutively altered frequencies of circulating follicullar helper T cell counterparts and their subsets in rheumatoid arthritis. Arthritis Res Ther. 2014;16:500.
Choi JY, Ho JH, Pasoto SG, Bunin V, Kim ST, Carrasco S, et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol. 2015;67:988–99.
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing. 2021. https://www.R-project.org/. Accessed 05 Apr 2024.
Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc: Ser B (Methodol). 2018;57:289–300.
Korotkevich G, Sukhov V, Sergushichev AA. Fast gene set enrichment analysis. bioRxiv. 2019. https://doi.org/10.1101/060012.
Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7.
Cohen S, Clowse M, Pardo P, Bhattacharya I, Menon S, Gourley I, et al. Safety, tolerability, pharmacokinetic and pharmacodynamic properties of SBI-087, a CD20-Directed B-cell depleting agent: Phase 1 dose escalating studies in patients with either mild rheumatoid arthritis or systemic lupus. Clin Ther. 2016;38:1417–34.e2.
Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222–33.
Rubbert-Roth A, Tak PP, Zerbini C, Tremblay JL, Carreño L, Armstrong G, et al. Efficacy and safety of various repeat treatment dosing regimens of rituximab in patients with active rheumatoid arthritis: results of a Phase III randomized study (MIRROR). Rheumatology (Oxford). 2010;49:1683–93.
Schiff M, Keiserman M, Codding C, Songcharoen S, Berman A, Nayiager S, et al. Efficacy and safety of abatacept or infliximab vs placebo in ATTEST: a phase III, multi-centre, randomised, double-blind, placebo-controlled study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Ann Rheum Dis. 2008;67:1096–103.
Emery P, Fleischmann R, Filipowicz-Sosnowska A, Schechtman J, Szczepanski L, Kavanaugh A, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54:1390–400.
Dass S, Rawstron AC, Vital EM, Henshaw K, McGonagle D, Emery P. Highly sensitive B cell analysis predicts response to rituximab therapy in rheumatoid arthritis. Arthritis Rheum. 2008;58:2993–9.
Vital EM, Dass S, Buch MH, Henshaw K, Pease CT, Martin MF, et al. B cell biomarkers of rituximab responses in systemic lupus erythematosus. Arthritis Rheum. 2011;63:3038–47.
Mesquita D, Cruvinel WdM, Resende LdS, Mesquita FV, Silva NPd, Câmara NOS, et al. Follicular helper T cell in immunity and autoimmunity. Braz J Med Biol Res. 2016;49:e5209.
Chatham WW, Wallace DJ, Stohl W, Latinis KM, Manzi S, McCune WJ, et al. Effect of belimumab on vaccine antigen antibodies to influenza, pneumococcal, and tetanus vaccines in patients with systemic lupus erythematosus in the BLISS-76 trial. J Rheumatol. 2012;39:1632–40.
Bingham CO 3rd, Looney RJ, Deodhar A, Halsey N, Greenwald M, Codding C, et al. Immunization responses in rheumatoid arthritis patients treated with rituximab: results from a controlled clinical trial. Arthritis Rheum. 2010;62:64–74.
Izmirly PM, Kim MY, Samanovic M, Fernandez-Ruiz R, Ohana S, Deonaraine KK, et al. Evaluation of immune response and disease status in systemic lupus erythematosus patients following SARS-CoV-2 Vaccination. Arthritis Rheumatol. 2022;74:284–94.
Medeiros-Ribeiro AC, Bonfiglioli KR, Domiciano DS, Shimabuco AY, da Silva HC, Saad CGS, et al. Distinct impact of DMARD combination and monotherapy in immunogenicity of an inactivated SARS-CoV-2 vaccine in rheumatoid arthritis. Ann Rheum Dis. 2022;81:710–9.
Vallerskog T, Heimbürger M, Gunnarsson I, Zhou W, Wahren-Herlenius M, Trollmo C, et al. Differential effects on BAFF and APRIL levels in rituximab-treated patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res Ther. 2006;8:R167.
Lavie F, Miceli-Richard C, Ittah M, Sellam J, Gottenberg JE, Mariette X. Increase of B cell-activating factor of the TNF family (BAFF) after rituximab treatment: insights into a new regulating system of BAFF production. Ann Rheum Dis. 2007;66:700–3.
O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. 2004;199:91–8.
Rawlings DJ, Metzler G, Wray-Dutra M, Jackson SW. Altered B cell signalling in autoimmunity. Nat Rev Immunol. 2017;17:421–36.
Frampton JE. Inebilizumab: first approval. Drugs. 2020;80:1259–64.
Muller F, Taubmann J, Bucci L, Wilhelm A, Bergmann C, Volkl S, et al. CD19 CAR T-cell therapy in autoimmune disease - a case series with follow-up. N Engl J Med. 2024;390:687–700.
Wei X, Niu X. T follicular helper cells in autoimmune diseases. J Autoimmun. 2023;134:102976.
Walker LSK. The link between circulating follicular helper T cells and autoimmunity. Nat Rev Immunol. 2022;22:567–75.
Barber MRW, Drenkard C, Falasinnu T, Hoi A, Mak A, Kow NY, et al. Global epidemiology of systemic lupus erythematosus. Nat Rev Rheumatol. 2021;17:515–32.
Rees F, Doherty M, Grainge MJ, Lanyon P, Zhang W. The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies. Rheumatology. 2017;56:1945–61.
Kobak S, Bes C. An autumn tale: geriatric rheumatoid arthritis. Ther Adv Musculoskelet Dis. 2018;10:3–11.
Acknowledgements
This study was sponsored by Pfizer Inc. Medical writing support, under the guidance of the authors, was provided by Karen Thompson, PhD, CMC Connect, a division of IPG Health Medical Communications, and was funded by Pfizer Inc, New York, NY, USA, in accordance with Good Publication Practice (GPP 2022) guidelines (Ann Intern Med. 2022;175(9):1298-1304. doi:10.7326/M22-1460).
Funding
This study was sponsored by Pfizer Inc.
Author information
Authors and Affiliations
Contributions
SK, DAM, and EP contributed to the study conception and design. VC, SK, AS, MC, and ES contributed to data acquisition, analysis, and interpretation. All authors have edited and substantively revised all drafts and have approved the submitted version. All authors have agreed both to be personally accountable for the author's own contributions and to ensure that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and the resolution documented in the literature.
Authors’ information
Pfizer Inc, Cambridge, MA, USA: Jean S. Beebe, Mina Hassan-Zahraee, and Sarita Koride affiliation at the time of the study.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Schulman Associates Institutional Review Board, Inc. (reference number IORG000063) and patients provided written informed consent before performing any study-specific activity.
Consent for publication
Not applicable.
Competing interests
SC is a consultant and investigator for Pfizer Inc. JSB, SK, and MHZ are former employees of Pfizer Inc. VC is an investigator for Pfizer Inc. RL is a consultant for AbbVie, AstraZeneca, and Janssen; investigator for AbbVie, Amgen, AstraZeneca, BMS, Dr. Reddy’s Laboratories, Equillium, GlaxoSmithKline, Idorsia, Janssen, Kangpu, Lilly, Novartis, Pfizer, RemeGen, SunPharm, and Viela Bio; and speaker for AbbVie and GlaxoSmithKline. MV is an employee of and shareholder in Pfizer Inc. SG, LX, MS, CH, SL, MS, AS, ES, EP, DAM, and MC are employees of Pfizer Inc.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
13075_2024_3337_MOESM1_ESM.docx
Additional file1: Fig. S1 Study design, Fig. S2 cTfh cell ratios by participant type in the single ascending dose cohort (PD analysis population), Fig. S3 cTfh cell ratios by participant type in the multiple ascending dose cohort (PD analysis population). Overview of study design for Part 1 and Part 2, cTfh cell ratios by participant type in the single and multiple ascending dose cohorts (PD analysis population).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cohen, S., Beebe, J.S., Chindalore, V. et al. A Phase 1, randomized, double-blind, placebo-controlled, single- and multiple-dose escalation study to evaluate the safety and pharmacokinetics/pharmacodynamics of PF-06835375, a C-X-C chemokine receptor type 5 directed antibody, in patients with systemic lupus erythematosus or rheumatoid arthritis. Arthritis Res Ther 26, 117 (2024). https://doi.org/10.1186/s13075-024-03337-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-024-03337-2