
Abstract
Background
Rituximab is used for the treatment of active rheumatoid arthritis. In the present study, we examined the long-term flare risk and safety of reduced doses of rituximab.
Patients-methods
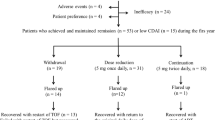
This was a prospective, observational, single-center study of patients starting rituximab on standard dose (SD). Patients were switched to low dose (LD) (1 g every 6 months), based on the treating rheumatologist’s decision after having achieved sustained clinical responses, while the rest of the patients continued on standard dose (SD). During a 60-month period, we assessed (Kaplan–Meier survival analysis) the relapse rate (increase ≥ 1.2 in DAS28-ESR for ≥ 6 months) and discontinuations due to treatment failure in the low dose group, and we compared the incidence of serious adverse events (SAEs) between LD and SD groups.
Results
Out of 361 patients [females 83.4%, mean age 61.9 (10.6) years, seropositive 50.3%, median total comorbidities count 4], 81 patients (22.4%) entered LD in a median time of 24 months (95% CI 18–30 months). Seropositivity (OR 1.823), more than 2 previous bDMARDs failures (OR 0.428), and DAS28 < 4.88 at 6 months (OR 2.329) predicted the odds of entering LD (p < 0.05 for all). During 60 months of follow-up, only 7.5% of patients on LD relapsed. Patients on LD had significantly less SAEs and all-cause hospitalizations as compared to the SD group (p < 0.05 for all). Linear regression analysis showed that previous hospitalization while on bDMARDs (p < 0.0001), use of prednisolone > 5 mg/day while on rituximab (p < 0.0001), and a history of ≥ 2 previous csDMARDs (p = 0.041) predicted the risk of SAEs.
Conclusion
In a cohort of patients with established RA and significant comorbidities who taper rituximab after substantial initial disease activity improvement, a low rate of relapses and lower risk of SAEs compared to SD were recorded. Seropositivity, a lower number of previous bDMARDs use, and lower DAS28 at 6 months predicted the probability of entering the LD regimen.
Similar content being viewed by others


Background
Rituximab is among the biologic agents approved for the treatment of active rheumatoid arthritis (RA) not responding or intolerant to conventional synthetic disease-modifying drugs (csDMARDs) or biologic DMARDs (bDMARDs) [1]. The approved dosing regimen is 1000 mg for two consecutive infusions 2 weeks apart. The need for further courses is recommended to be evaluated 24 weeks following the initial course. Randomized controlled trials (RCTs) of rituximab in methotrexate (MTX)-inadequate responders and MTX-naive patients, showed comparable efficacy of reduced doses of RTX (two infusions of 500 mg) to that of standard doses in terms of reduction of disease activity [2,3,4]. Moreover, a meta-analysis of RCTs and cohort studies comparing low-dose (1000 mg) to “standard dose” of RTX (2000 mg) found similar effectiveness and advocated the use of the low-dose regimen, considering cost benefits [5]. The practice of tapering bDMARDs in RA patients on sustained remission or low disease activity, has been proved in several clinical trials mostly for tumor necrosis factor (TNF) inhibitors and it is proposed by current treatment guidelines [1]. Lowering the cost of RA treatment and reducing long-term cumulative bDMARDs doses is the main reasons supporting this practice.
In the present observational study, we evaluated the long-term effectiveness and safety of reduced doses of rituximab in a large cohort of RA patients of clinical practice, who achieved clinical responses while on initial standard doses of rituximab.
Methods
We analyzed data from the University of Crete Rheumatology Clinic Registry (UCRCR), a single-center prospective cohort study initiated in 2004. According to the protocol—and after their informed consent—patients with inflammatory arthritis are included in the registry at the time they start their first bDMARD. Demographics, comorbidities assessed both as comorbidities count (CC) and based on the “Rheumatic Disease Comorbidities Index” (RCDI), disease characteristics, and extra-articular manifestations, as well as disease activity and function indices, are recorded at baseline. Patients are followed-up every 3–6 months with disease activity [disease activity score using 28 joints (DAS28), 28 swollen joint count (28SJC), 28 tender joint count (28TJC), 44SJC, 44TJC, visual analog score (VAS) global, VAS pain, VAS physician, erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP)], function [modified Health Assessment Quaestionnaire (mHAQ)], and quality of life (EQ-5D) indices in every follow-up visit. Detailed data regarding rheumatologic drugs and their dosages, as well as treatment discontinuations and all adverse events (based on MedDRA coding) during follow-up, are also recorded.
Since 2015, patients who achieve remission or low disease activity for ≥ 2 consecutive 3-month visits or who have a sustained significant improvement (DAS28 decrease > 1.2) from baseline and, based on the clinical judgment of the treating rheumatologist would be benefited from rituximab tapering, enter the low dose (LD; 1 g/6 months) scheme. All other patients continue standard treatment and follow-up on standard dose (SD; 2 g). For the present study we included all patients in UCRCR who received rituximab and entered LD or continued in SD scheme. In patients who entered the LD regimen, flare was defined as the increase in DAS28 from LD start of ≥ 1.2 for 2 consecutive 3-month visits.
All demographic and baseline characteristics of patients were presented using descriptive statistics. Between-group comparisons were performed using Pearson’s chi-square tests or Fisher’s exact test in cases of categorical variables. Between-group comparisons were also performed using independent samples T-test or Mann–Whitney U test in cases of normally or non-normally distributed numeric variables respectively. A Kaplan–Meier survival analysis was performed using flare (yes/no) as the event variable and time of flare as the time-to-event variable. Non-parametric pairwise comparisons were performed using Wilcoxon signed-ranked test. A repeated-measures linear mixed model was performed with the dependent variable as the DAS28-ESR score and the dosage group (low dose vs high dose) as the independent variable. A multi-variable linear regression model was performed using the number of serious adverse events as an independent variable and gender (male/female), age, RCDI, the number of concomitant non-biologic medications, the number of previous biologics (≥ 2 biologics versus < 2 biologics), the concomitant prednisolone > 5 mg/day (yes/no), the past hospitalization for serious infection (yes/no) and the study group (low dose vs high dose) was performed. A multi-variable logistic regression model was performed using as dependent variable the dose scheme (low dose vs high dose) and independent variables gender (male vs female), age, disease duration (in years), the RCDI, patient being seropositive (yes/no), the number of previous biologics (> 2 biologics vs ≤ 2 biologics), the DAS28-ESR scores at baseline, and the DAS28-ESR category levels at 6 months which resulted upon median split (≥ 4.88 versus < 4.88). An intention-to-treat analysis was carried throughout. The level of statistical significance was set to α = 0.05 and the statistical software that was used was IBM SPSS version 25 and STATA SE 11.
Results
Patients’ characteristics
We studied 361 patients who started rituximab according to the treating rheumatologist’s decision following the national and EULAR guidelines for the treatment of RA. Patients have mostly established RA [median disease duration 5 years (min 0.5, max 51; IQR 9 years)], half of them were seropositive for rheumatoid factor (RF) or anti-CCP antibodies (ACPA) and received rituximab as the third (median) bDMARD treatment (min = 1, max = 7) (Table 1). After a median time of 24 months (95% CI for the median 18–30 months) therapy with the standard dose of rituximab, 81 patients (22.4%) entered the LD scheme. Forty-five out of the 81 (55.5%) patients satisfied the “criterion” of sustained remission or low DAS28 score (DAS28 < 3.2). An additional 28.4% (n = 23) of the patients had tender or swollen joint count \(\le\) 3 while having high patient’s VAS global (> 60), and were considered as having “well-controlled disease” according to their rheumatologist’s opinion. The remaining 14% switched to the LD scheme due to older age/frailty and patient’s choice. Patients on LD were more often seropositive for RF or ACPAs (p = 0.023 and p = 0.016, respectively) and received rituximab earlier as a bDMARD compared to patients remaining on SD (RTX as ≤ 2nd treatment bDMARD line vs ≥ 3rd line, p = 0.023). All patients have had a prior exposure to a high number of csDMARDs, (methotrexate: 90.9%, leflunomide: 64.8%), while csDMARDs history as well as prior steroids’ exposure (63.4% on prednisolone ≤ 10 mg/day) were comparable among SD and LD groups (Supplementary Table 1). Furthermore, 77.0% were exposed to anti-TNFα agents and 33.8% to non-TNFα inhibitors prior to starting rituximab.
A significant burden of comorbidities was recorded at the treatment baseline [median total comorbidities count (CC): 4; median RDCI: 2], comparable between the 2 groups (Table 1). As expected, hypertension and dyslipidemia were the most commonly reported comorbidities (46% and 44% of patients respectively). Interestingly, 23.3% and 8% of the patients had a history of hospitalization or recurrent hospitalizations for serious infection respectively while on previous bDMARDs, comparable between the two groups. Ten patients had “past HBV”, 2 HCV infections, and one patient concomitant HCV and past HBV [anti-HBc ( +), anti-HBs ( −)], while 8 had a history of past reactivation of varicella-zoster virus during treatment with a previous biologic agent. During follow-up, we observed no new cases of hepatitis or HBV reactivation in patients with past infection in either group. Finally, 13 (3.6%) and 23 (6.4%) patients had a history of hematologic or solid tumor malignancy respectively.
Disease characteristics at rituximab initiation and predictors of entering the LD group
At rituximab initiation, patients had highly active RA [median DAS28 5.73 (1.74, 8.69; 1.43)] and compromised functionality [median mHAQ 1.0 (0, 2.8; 0.8)], comparable between LD and SD groups (Table 2). Patients eventually entering the LD regimen had higher baseline inflammatory markers, both ESR and CRP (p = 0.003 and p = 0.006, respectively), while patients on SD had more swollen joints (p = 0.015). Rituximab was given as monotherapy in 15.2% of the patients, while 61.5% were on combination with MTX and 45.1% on prednisolone (Table 2). Concomitant RA-related medications were comparable between the two groups. Logistic regression analysis revealed that seropositivity (for RF or ACPAs) [OR 1.823 (1.009–3.292), p = 0.046], past use of > 2 bDMARDs [OR 0.428 (0.204–0.898, p = 0.025] and a DAS28 lower than 4.88 at 6 months [OR 2.329 (1.254–4.325), p = 0.007] predicted entering the LD group (Table 3).
Flare risk during follow-up in the low dose group
After a median time of treatment with SD of 24 months (95% CI 18–30 months), 81 (22.4%) patients tapered rituximab to 1 g/6 months. Disease activity in both SD and LD groups improved significantly during the first 24 months of treatment from 5.77 (1.93, 8.69; 1.42) and 5.66 (1.74, 8.66; 1.59) to 4.56 (1.81,7.38;1.96), and 3.76 (1.54, 7.80;1.94), respectively, (p < 0.0001 for both). As expected, those who tapered had a significantly higher improvement at 6 months compared to baseline [median DAS28 difference 0.65 (− 3.00, 4.20; 1.64) for the SD and 1.11 (− 2.20, 4.54; 1.78) for the LD (p = 0.017)].
After rituximab tapering, patients were prospectively followed for a median of 56 (1, 177; 59) months. For the LD group, DAS28 ESR at last visit [median 3.33 (1.05, 7.82; 2.12)] was comparable to DAS28 at rituximab taper baseline visit [median 3.28 (1.05, 6.58;1.88)] (p = 0.639) (Fig. 1). We further assessed for disease flares in both groups. During 60 months of follow-up, only 5.9% and 7.5% of the patients in SD and LD, respectively, experienced a flare of RA (p = 0.6) (Table 4). The K-M survival analysis (having as event a flare of the disease), is given in Fig. 2. Moreover, and in order to assess the clinical utility of the tapering strategy, we assessed for discontinuations due to treatment failure. We found that patients on LD had significantly less discontinuations due to failure as compared to those on SD (37.9% vs 63.6%, respectively, p < 0.0001) (Table 4). A total of 18 of 177 (10.2%) discontinuations were due to infections. Interestingly, 3 out of 18 (16.7%) patients who discontinued due to infections had low IgG levels (two in the standard dose group, one in the low dose group).

Linear mixed model predictions of DAS28-ESR score according to rituximab dosing group

Survival plot of flare probability within the low dose group
Safety and events
We followed LD patients for a median of 56 (1, 177; 59) months, total period of 7287 person-months, while patients on SD were followed up for 14.5 (1, 127; 27) months or 4824 person-months (Table 4). Lower follow-up period was due to a higher discontinuation rate of patients on SD (52.9%) as compared to that of patients on LD (35.8%). Patients on LD were cumulatively exposed to 90 g (2, 246; 78) of rituximab as compared to 28 g (2, 254; 52) in SD groups. During this period, a total of 45 and 137 serious adverse events (SAEs) in LD and SD respectively accounting for an incidence rate of 0.77 and 1.57 events per 1000 person-years, were documented (p < 0.0001) (Table 5). Incidence rates for serious infections (0.49 vs 0.88, p = 0.0026) and all-cause hospitalizations (0.53 vs 1.08, p = 0.0002), were lower in LD than in SD group. As a sensitivity analysis, given the heterogeneity and the differences of rituximab exposure between SD and LD groups, we also compared adverse events between more “homogenous” subgroups of SD and LD. Thus, we compared the incidence rates of adverse events (events per 1000 person-years) for those patients with a total follow-up time of ≥ 24 months in both groups. In accordance to the findings of the main analysis, a more favorable safety profile of LD as compared to the SD group was found for moderate/serious AE, serious infections, and hospitalizations (p < 0.001 for all, Supplementary Table 2).
We finally assessed for predictors of serious adverse events. Linear regression analysis showed that past hospitalization for serious infection while one a previous bDMARD (beta 0.974, p < 0.0001), sustained use for at least 6 months of prednisolone > 5 mg/day while on rituximab (beta 0.689, p < 0.0001) and > 2 previous csDMARDs use (beta 0.320, p = 0.041) were all independent predictors of a higher number of serious adverse events in the cohort (Table 6).
Discussion
In this observational study we found that in RA patients with initial sustained clinical response to standard doses of rituximab, reduced doses during a 5-year period have a low risk of disease’s flare and discontinuations due to treatment failure, while patients experienced significantly less serious adverse events as compared to patients who continue with standard doses. In our group of patients with established disease and a high comorbidities burden, predictors of entering the LD scheme as well as factors predicting serious events while on LD were identified.
Tapering bDMARDs has been a strategy suggested for RA patients with persistent clinical responses (mostly in remission) upon initially standard doses of bDMARDs [1]. Most of the data supporting this approach are based on trials of TNF inhibitors [6]. Controlled clinical trials of rituximab in standard and reduced doses have been shown to have comparable both biological effects (CD19 + ve B cells depletion/repopulation) and clinical responses [7]. Reduced doses of rituximab have been assessed either from the first dose or after initial standard doses [2,3,4, 7]. The above mentioned are randomized studies, with mostly a short follow-up period, except the study of Mariette et al. which has a follow-up of 104 weeks. Concerning the effect of reduced rituximab doses on joint damage protection, data from RCTs are not consistent, and one could argue that the protective effect on joint damage of standard compared to lower doses could be different. In MTX-naïve patients, a differential protective effect between standard and a low dose of rituximab after 1 year has been found [4]. Interestingly enough, the 2 years analysis of the IMAGE study showed a favorable protective effect of rituximab low dose [8]. Nevertheless, this study was not powered to compare rituximab doses and patients were MTX-naïve, a population for whom rituximab is not yet approved.
Of note, in clinical practice, the common scheme is that of the “standard dose” (1000 mg 2 times 15 days apart). Interestingly, a more recent observational, prospective study [Autoimmunity and Rituximab (AIR)] showed that after a first course of rituximab at standard dose patients that have been re-treated with low dose of RTX remained on therapy at 5 years and received reduced cumulatively doses of the monoclonal [9]. Our study and that of Henry et al., have the longest follow-up and comparable results. Both these observational studies showed that in the long term, low doses of rituximab have a high drug retention rate (53.8% in the French study), while in our study we documented only 7.5% relapse rate. In both studies, drug retention or frequency of relapses was comparable between low and standard doses of rituximab. Further supporting the value of the concept to taper rituximab in patients with the initial response, we found that only 37.9% vs 63.6% of patients discontinued therapy due to treatment failure on LD and SD respectively (p < 0.0001).
In our cohort of 361 RA patients, we aimed to identify factors at rituximab initiation which predict who will enter the LD group. Logistic regression analysis revealed that seropositivity (OR 1.823), past use of > 2 bDMARDs (OR 0.428), and a DAS28 lower than 4.88 at 6 months (OR 2.329) predicted entering the low-dose group (Table 3). In our study, patients who entered the low dose were responders to initial standard doses of rituximab, and thus the above predictors can be considered as factors predicting long-term clinical responses to rituximab. Seropositivity and a lower number of previous bDMARDs failures have been shown as predictors for response to rituximab in several studies [10,11,12,13]. We and others have shown that an early (during the first year of treatment) clinical response to TNFα inhibitors may predict anti-TNFα survival [14, 15]. Herein and comparable to the above anti-TNFα related data, we showed that an improvement in DAS28 during the first 6 months of treatment may predict long-term rituximab drug survival. All the above factors were predictors of long-term rituximab efficacy and drug survival, and we consider this as a clinically important finding of our analysis.
The second main finding of our study was that the LD group experienced a significantly lower number of all adverse events, SAE, and hospitalizations as compared to SD group (Table 5). Our findings are in line with the results reported by Henry et al., showing a lower rate of serious infections in the reduced dose group [9]. Both studies, as mentioned above, are the observational studies with the longest follow-up of patients in everyday clinical practice. Interestingly, a numerically lower risk of serious infections in patients on reduced doses of RTX has been reported in a meta-analysis of randomized controlled trials (RCTs) comparing standard and reduced doses (0.73 (0.37–1.47), p = 0.38) [5]. Patient selection and the short-term follow-up of the patients in RCTs may explain differences between observational studies and the aforementioned meta-analysis. We further explored for predictors of serious adverse events. Interestingly, we found that past hospitalization for serious infection while one a previous bDMARD as the most important risk factor for SAE. This is a novel finding not reported in the literature before. Since our registry is focused in all patients starting the first bDMARD, we were able to analyze prior history (while on bDMARDs) of AE and medications as predisposing factors for SAE while on rituximab. As expected, sustained use for at least 6 months of prednisolone > 5 mg/day while on rituximab was also an independent risk factor for SAE, underling the importance of tapering/stopping steroids in order to minimize the risk for SAE in our patients. The association of low doses of steroids with adverse events (mostly infections) has been reported multiple times in different RA cohorts under multiple therapies [14, 16, 17].
This was an observational, single-center study, of patients followed by a standard protocol. Patients’ selection for tapering rituximab was allowed to be the treating physician’s decision, based on clinical responses and patient profile and not based on a standard protocol. Thus, 44 out of the 81 (55.5%) patients satisfied the “criterion” of sustained remission or low DAS28 score (DAS28 < 3.2), while an additional 28.4% (n = 23) of the patients had tender or swollen joint count \(\le\) 3 while having high patient’s VAS global (> 60), and were considered as having “well-controlled disease” according to their rheumatologist’s opinion. Moreover, although disease activity in both SD and LD groups improved significantly during the first 24 months of treatment, those who tapered had a significantly higher improvement at as early as 6 months [median DAS28 difference 0.65 (− 3.00, 4.20; 1.64) for the SD and 1.11 (− 2.20, 4.54; 1.78) for the LD (p = 0.017)]. Thus, we consider that although we do not follow a strict protocol for tapering rituximab, patients who entered the LD scheme were those with significant improvement in disease activity after initial standard doses of rituximab.
One could argue that the practice of continuing rituximab for those patients not achieving “treat-to-target” goals is not acceptable in clinical practice. Nevertheless, SD patients had clinically and statistically important improvement in disease activity during the first 24 months of treatment from 5.77 (1.93, 8.69; 1.42) to 4.56 (1.81,7.38;1.96) (p < 0.0001). Moreover, as pointed out in cohort characteristics, patients on SD had established, active RA after a failure of a median of 2 (up to 6) bDMARDs and 2 (up to 7) of csDMARDs with a high burden of comorbidities 4 (up to 15). We consider that treatment switches in a cohort like this, are not strictly based on the “treat-to-target” approach; both patients and rheumatologists are considering several other factors for treatment decisions.
The percentage of rheumatoid factor or anti-CCP antibodies positivity was relatively low in our cohort as compared to other observational studies [9]. Given the data from RCTs and observational studies supporting a higher response rate of rituximab in seropositive patients, many clinicians apply seropositivity as a “biomarker” favoring rituximab among other bDMARDs. Nevertheless, a metanalysis of RCTs showed a modest differential effect in seropositive patients [18] and in observational studies, despite the higher responses in seropositive patients, clinically and statistically important improvements were found also in seronegative patients [10]. Moreover, the seronegative RA population is increased during the last decade compared to earlier studies [19]. Finally, data supporting that seropositive respond better than seronegative patients have been also published for TNFα inhibitors, abatacept, and tocilizumab [11, 20]. The above could explain the lower percentage of seropositive patients in our group as compared to other cohorts.
The two groups were heterogenous regarding their baseline characteristics, time of follow-up, and exposure to rituximab. Even though this is common in observational studies, it raises concerns for the interpretation of the findings. Nevertheless, we have to emphasize that SD group was a “control” group to LD only in terms of comparing the safety of the two groups and not for long-term clinical efficacy. Moreover, the sensitivity analysis of the rate of adverse events restricted to patients with a total follow-up time of ≥ 24 months, revealed a better safety profile of LD, similarly to the whole group analysis (Supplementary Table 2). Finally, although patients on LD had a higher exposure to rituximab compared to SD (90 vs 28 g) and longer time on rituximab (56 vs 14.5 months), yet they had a lower incidence of serious/moderate AE, lower incidence of serious infections and hospitalizations (Table 5). Thus, although and as expected due to the observational nature of the study, there were differences between the two groups, we consider the results of the comparisons clinically important, with a special focus on the safety of LD.
Conclusion
Our prospective 5-year analysis of established RA patients with multiple bDMARDs failures and high comorbidities burden confirms that reduced doses of rituximab is a valid approach for patients with initial sustained clinical response. Patients starting rituximab after a failure to less than 2 bDMARDs, who are seropositive and improve disease activity at 6 months while on rituximab are those who have a higher probability to enter the LD. Rheumatologists should be aware of the increased risk of SAE in patients with a prior history of hospitalization and sustained use of steroids.
Availability of data and materials
The data and analytic methods that support the findings of this study are available to qualified investigators by request to the corresponding author.
Abbreviations
- SD:
-
Standard dose
- LD:
-
Low dose
- ESR:
-
Erythrocyte sedimentation rate
- SAEs:
-
Serious adverse events
- bDMARDs:
-
Biologic disease-modifying anti-rheumatic drugs
- csDMARDs:
-
Conventional synthetic disease-modifying anti-rheumatic drugs
- RCTs:
-
Randomized controlled trials
- MTX:
-
Methotrexate
- RTX:
-
Rituximab
- TNF:
-
Tumor necrosis factor
- RA:
-
Rheumatoid arthritis
- UCRCR:
-
University of Crete Rheumatology Clinic Registry
- RCDI:
-
Rheumatic Disease Comorbidities Index
- DAS28:
-
Disease activity score using 28 joints
- 28SJC:
-
28 Swollen joint count
- 28TJC:
-
28 Tender joint count
- 44SJC:
-
44 Swollen joint count
- 44TJC:
-
44 Tender joint count
- VAS:
-
Visual analog score
- CRP:
-
C-reactive protein
- mHAQ:
-
Modified Health Assessment Questionnaire
- EQ-5D:
-
Quality of life
- MedDRA:
-
Medical Dictionary for Regulatory Activities
- EULAR:
-
European League Against Rheumatism
- RF:
-
Rheumatoid factor
- ACPA:
-
Anti-CCP antibodies
- TNF:
-
Tumor necrosis factor
- CC:
-
Comorbidities count
- AIR:
-
Autoimmunity and rituximab
- TIA:
-
Transient ischemic attack
- COPD:
-
Chronic obstructive pulmonary disease
- TB:
-
Tuberculosis
- IQR:
-
Inter quartile range
References
Smolen JS, Landewe RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. 2020;79(6):685–99.
Emery P, Deodhar A, Rigby WF, Isaacs JD, Combe B, Racewicz AJ, et al. Efficacy and safety of different doses and retreatment of rituximab: a randomised, placebo-controlled trial in patients who are biological naive with active rheumatoid arthritis and an inadequate response to methotrexate (Study Evaluating Rituximab’s Efficacy in MTX iNadequate rEsponders (SERENE)). Ann Rheum Dis. 2010;69(9):1629–35.
Rubbert-Roth A, Tak PP, Zerbini C, Tremblay JL, Carreno L, Armstrong G, et al. Efficacy and safety of various repeat treatment dosing regimens of rituximab in patients with active rheumatoid arthritis: results of a Phase III randomized study (MIRROR). Rheumatology (Oxford). 2010;49(9):1683–93.
Tak PP, Rigby WF, Rubbert-Roth A, Peterfy CG, van Vollenhoven RF, Stohl W, et al. Inhibition of joint damage and improved clinical outcomes with rituximab plus methotrexate in early active rheumatoid arthritis: the IMAGE trial. Ann Rheum Dis. 2011;70(1):39–46.
Bredemeier M, de Oliveira FK, Rocha CM. Low- versus high-dose rituximab for rheumatoid arthritis: a systematic review and meta-analysis. Arthritis Care Res (Hoboken). 2014;66(2):228–35.
Kerschbaumer A, Sepriano A, Smolen JS, van der Heijde D, Dougados M, van Vollenhoven R, et al. Efficacy of pharmacological treatment in rheumatoid arthritis: a systematic literature research informing the 2019 update of the EULAR recommendations for management of rheumatoid arthritis. Ann Rheum Dis. 2020;79(6):744–59.
Emery P, Fleischmann R, Filipowicz-Sosnowska A, Schechtman J, Szczepanski L, Kavanaugh A, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54(5):1390–400.
Tak PP, Rigby W, Rubbert-Roth A, Peterfy C, van Vollenhoven RF, Stohl W, et al. Sustained inhibition of progressive joint damage with rituximab plus methotrexate in early active rheumatoid arthritis: 2-year results from the randomised controlled trial IMAGE. Ann Rheum Dis. 2012;71(3):351–7.
Henry J, Gottenberg JE, Rouanet S, Pavy S, Sellam J, Tubach F, et al. Doses of rituximab for retreatment in rheumatoid arthritis: influence on maintenance and risk of serious infection. Rheumatology (Oxford). 2018;57(3):538–47.
Chatzidionysiou K, Lie E, Nasonov E, Lukina G, Hetland ML, Tarp U, et al. Highest clinical effectiveness of rituximab in autoantibody-positive patients with rheumatoid arthritis and in those for whom no more than one previous TNF antagonist has failed: pooled data from 10 European registries. Ann Rheum Dis. 2011;70(9):1575–80.
Maneiro RJ, Salgado E, Carmona L, Gomez-Reino JJ. Rheumatoid factor as predictor of response to abatacept, rituximab and tocilizumab in rheumatoid arthritis: Systematic review and meta-analysis. Semin Arthritis Rheum. 2013;43(1):9–17.
Couderc M, Mathieu S, Pereira B, Glace B, Soubrier M. Predictive factors of rituximab response in rheumatoid arthritis: results from a French university hospital. Arthritis Care Res (Hoboken). 2013;65(4):648–52.
Gardette A, Ottaviani S, Tubach F, Roy C, Nicaise-Roland P, Palazzo E, et al. High anti-CCP antibody titres predict good response to rituximab in patients with active rheumatoid arthritis. Joint Bone Spine. 2014;81(5):416–20.
Flouri I, Markatseli TE, Voulgari PV, Boki KA, Papadopoulos I, Settas L, et al. Comparative effectiveness and survival of infliximab, adalimumab, and etanercept for rheumatoid arthritis patients in the Hellenic Registry of Biologics: Low rates of remission and 5-year drug survival. Semin Arthritis Rheum. 2014;43(4):447–57.
Gulfe A, Kristensen LE, Geborek P. Six and 12 weeks treatment response predicts continuation of tumor necrosis factor blockade in rheumatoid arthritis: an observational cohort study from southern Sweden. J Rheumatol. 2009;36(3):517–21.
Dixon WG, Abrahamowicz M, Beauchamp ME, Ray DW, Bernatsky S, Suissa S, et al. Immediate and delayed impact of oral glucocorticoid therapy on risk of serious infection in older patients with rheumatoid arthritis: a nested case-control analysis. Ann Rheum Dis. 2012;71(7):1128–33.
George MD, Baker JF, Winthrop KL, Goldstein SD, Alemao E, Chen L, et al. Immunosuppression and the risk of readmission and mortality in patients with rheumatoid arthritis undergoing hip fracture, abdominopelvic and cardiac surgery. Ann Rheum Dis. 2020;79(5):573–80.
Isaacs JD, Cohen SB, Emery P, Tak PP, Wang J, Lei G, et al. Effect of baseline rheumatoid factor and anticitrullinated peptide antibody serotype on rituximab clinical response: a meta-analysis. Ann Rheum Dis. 2013;72(3):329–36.
Myasoedova E, Davis J, Matteson EL, Crowson CS. Is the epidemiology of rheumatoid arthritis changing? Results from a population-based incidence study, 1985–2014. Ann Rheum Dis. 2020;79(4):440–4.
Gottenberg JE, Courvoisier DS, Hernandez MV, Iannone F, Lie E, Canhao H, et al. Brief Report: Association of Rheumatoid Factor and Anti-Citrullinated Protein Antibody Positivity With Better Effectiveness of Abatacept: Results From the Pan-European Registry Analysis. Arthritis Rheumatol. 2016;68(6):1346–52.
Acknowledgements
The authors are grateful to Mrs Eleni Kampouraki and Styliani Polia (rheumatology nurses) for patients’ care and data entry.
Funding
This work was supported in part by the “Pancretan Health Association”.
Author information
Authors and Affiliations
Contributions
PS, IF, and GB were involved in the design and statistical analysis of the study as well as manuscript drafting and approval of the final version. AB and NA were involved in the statistical analysis and drafting of the manuscript. The rest of the authors contributed equally in consulting, manuscript proofreading, and approval.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was obtained from the Institutional Review-Board of the University Hospital of Heraklion, Crete (decision-number:1476/20–03-2012). Participants provided informed consents.
Consent of publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Previous anti-rheumatic medications, and uni-variate comparisons between previous anti-rheumatic medication of patients under SD or LD RTX. Table S2. Comparison of incidence rates of adverse events (events per 1000 person-years) for all patients and by dose group for patients with a total follow-up time ≥24 months.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bertsias, A., Avgoustidis, N., Papalopoulos, I. et al. Rheumatoid arthritis patients initiating rituximab with low number of previous bDMARDs failures may effectively reduce rituximab dose and experience fewer serious adverse events than patients on full dose: a 5-year cohort study. Arthritis Res Ther 24, 132 (2022). https://doi.org/10.1186/s13075-022-02826-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-022-02826-6