
Abstract
Objectives
The aim of this study was to compare the clinical effectiveness of tofacitinib and abatacept and clarify the impact of the HLA-DRB1 shared epitope (SE) on responses to these treatments in patients with rheumatoid arthritis (RA).
Methods
After adjustments by propensity score matching, 70 out of 161 patients receiving tofacitinib and 70 out of 131 receiving abatacept were extracted. The clinical effectiveness of both drugs over 24 weeks and the impact of the copy numbers of SE on effectiveness outcomes were investigated.
Results
The percentage of patients in remission in the 28-joint count disease activity score using the erythrocyte sedimentation rate (DAS28-ESR) did not significantly differ between patients receiving tofacitinib and abatacept at week 24 (32% vs 37%, p = 0.359). The mean change at week 4 in DAS28-ESR from baseline was significantly greater in patients receiving tofacitinib than in those receiving abatacept (− 1.516 vs − 0.827, p = 0.0003). The percentage of patients in remission at week 4 was 30% with tofacitinib and 15% with abatacept (p = 0.016). When patients were stratified by the copy numbers of SE alleles, differences in these numbers did not affect DAS28-ESR scores of patients receiving tofacitinib. However, among patients receiving abatacept, DAS28-ESR scores were significantly lower in patients carrying 2 copies of SE alleles than in those carrying 0 copies at each time point throughout the 24-week period. Furthermore, the percentage of patients in remission with DAS28-ESR at week 24 was not affected by the copy numbers of SE alleles in patients receiving tofacitinib (p = 0.947), whereas it significantly increased as the copy numbers became higher in patients receiving abatacept (p = 0.00309). Multivariable logistic regression analyses showed a correlation between the presence of SE and DAS28-ESR remission in patients receiving abatacept (OR = 25.881, 95% CI = 3.140–213.351, p = 0.0025), but not in those receiving tofacitinib (OR = 1.473, 95% CI = 0.291–7.446, p = 0.639).
Conclusions
Although the clinical effectiveness of tofacitinib and abatacept was similar at week 24, tofacitinib was superior to abatacept for changes from baseline in DAS28-ESR and the achievement of remission at week 4. SE positivity was associated with the achievement of DAS28-ESR remission by week 24 in patients receiving abatacept, but not in those receiving tofacitinib.
Similar content being viewed by others


Background
Rheumatoid arthritis (RA) is a complex autoimmune disease that develops from the combined effects of genetic and environmental factors. Genetic factors make a significant contribution to the development of RA in population, accounting for approximately 60% of population susceptibility to the disease [1]. Among the susceptibility genes to RA, the strongest relationship was reported with the HLA region, particularly HLA-DRB1 alleles that share a similar amino acid sequence, called the shared epitope (SE) [2]. Although the SE hypothesis was initially proposed to explain genetic susceptibility to RA, subsequent investigations suggested that the primary role of SE may be in the development of more severe disease manifestations [3, 4]. The well-known SE-coding alleles include members of the HLA-DRB1 *04 allele group, *0101, *1402, and *1001 [5]. SE may modulate the severity of RA in affected patients [3, 4, 6]. Autoantibodies, such as the anti-citrullinated peptide antibody (ACPA), are more likely to occur in patients with RA who are positive for SE [7,8,9]. SE has also been linked to progressive joint damage [10]. A recent study revealed that the presence of Val and Leu at HLA-DRB1 position 11, other than SE, were associated with more radiographic progression [11]. Furthermore, the presence of the SE may affect responses to treatment [12, 13]. However, the role of SE alleles in therapeutic responses remains unclear.
The introduction of multiple biologic disease modifying antirheumatic drugs (bDMARDs) and targeted synthetic DMARDs (tsDMARDs) has significantly improved the treatment of RA. According to the European League Against Rheumatism (EULAR) 2019 update recommendations, if the treatment target is not achieved with the first conventional synthetic DMARD (csDMARD) strategy and poor prognostic factors are present, bDMARD or tsDMARD is recommended in the next step [14]. Among candidate drugs, tofacitinib is an oral Janus kinase (JAK) inhibitor that preferentially reduces signaling from type I and II cytokine receptors by inhibiting JAK3 and/or JAK1 [15, 16], while abatacept is a genetically engineered fusion protein that selectively inhibits T cell activation by binding to CD 80/86 and modulating their interaction with CD28 [17]. Both tofacitinib and abatacept have been shown to be similarly efficacious based on clinical outcomes in randomized controlled trials (RCTs) compared to adalimumab [18, 19]. In Japan, abatacept use comprised 17% of overall bDMARDs use in fiscal year 2017 and the proportion was higher especially in elderly populations [20], indicating that a comparative effectiveness study of abatacept with targeted therapies other than TNF inhibitors is necessary for clinical decision making. However, to date, there are no clinical studies that directly compare the efficacies of both drugs. Therefore, this study was undertaken as the first comparison of tofacitinib and abatacept for the treatment of RA. RCTs are regarded as a reliable means of obtaining evidence on the efficacy and safety of drugs. However, there are limitations to RCTs. The most important limitation is that the study participants are selected using inclusion and exclusion criteria. On the other hand, observational studies involve patients who are commonly encountered in daily clinical practice. But the study participants are subjected to selection bias due to uncontrolled differences between the treatment and control groups. Therefore, a precise comparison of the effectiveness of drugs is difficult in observational studies. In recent years, propensity score (PS) matching has been shown to reduce limitations, such as selection bias arising from observational studies, by adjusting for potential confounders and producing similar data to RCTs [21, 22].
In the present study, we used the PS matching method to compare clinical outcomes for 24 weeks between patients receiving tofacitinib and abatacept and attempted to clarify whether the copy numbers of SE alleles affect responses to treatment with tofacitinib or abatacept during 24 weeks in patients with RA.
Methods
Patients and study design
This was a multicenter, retrospective, longitudinal observational study conducted at 12 hospitals and clinics for rheumatology in Japan. We enrolled patients aged ≥ 20 years who fulfilled the 2010 American College of Rheumatology (ACR)/EULAR criteria [23] for RA and started treatment with tofacitinib or abatacept between January 2015 and September 2019. The prior use of bDMARDs or JAK inhibitors did not limit patient enrollment in the present study. Data in this study were collected prospectively from January 2018 as well as retrospectively for patients who had been treated with tofacitinib or abatacept until December 2017. The clinical effectiveness of the tofacitinib and abatacept treatments was evaluated over 24 weeks. An HLA-DRB1 allele analysis was performed at enrollment. Written informed consent was obtained according to the Declaration of Helsinki. This study design was initially approved by the Ethics Committee of Toho University School of Medicine in January 2018 (approved number, A17085) and then by each participating center or institution. This study was registered with the University Hospital Medical Information Network Clinical Trial Registry (UMIN000037418).
Tofacitinib and abatacept treatments
Tofacitinib and abatacept were prescribed to patients with RA at the discretion of the treating physician. The dosage of tofacitinib was adjusted by renal function. Patients with an estimated glomerular filtration rate (eGFR) ≥ 60 ml/min/1.73m2 received 5 mg of tofacitinib orally twice daily, while those with eGFR< 60 ml/min/1.73m2 received 5 mg of tofacitinib orally once daily. Abatacept was administered as an intravenous infusion (500 mg for patients of < 60 kg, 750 mg for 60–100 kg and 1000 mg for > 100 kg) on weeks 0, 2, and 4, and then every 4 weeks thereafter. Alternatively, patients received 125 mg by a subcutaneous injection once weekly [24].
Clinical effectiveness
Disease activity was assessed by the 28-joint count disease activity score using the erythrocyte sedimentation rate (DAS28-ESR) [25], the Simplified Disease Activity Index (SDAI) [26], and the Clinical Disease Activity Index (CDAI) [27] at baseline, 4, 12, and 24 weeks. The EULAR response was evaluated at 4, 12, and 24 weeks [28].
HLA-DRB1 genotyping and autoantibody detection
The HLA-DRB1 allele was genotyped by the SeCore DRB1 Locus Exon 2 & 3 Sequencing kit (One Lambda) with the polymerase chain reaction-sequencing based typing method. HLA-DRB1 *01:01, *04:01, *04:04, *04:05, *04:10, *10:01, *14:02, and *14:06 were defined as SE [29]. ACPA was detected using a second-generation anti-CCP CLIA kit (Abbott Japan Laboratories, Tokyo, Japan). A cutoff value of 4.5 U/ml was used for anti-CCP antibody positivity.
Statistical analysis
We compared the baseline characteristics of patients treated with tofacitinib and abatacept. To simultaneously control for potential confounders, we generated PS to predict the probability of a patient initiating tofacitinib by a multiple logistic regression model using the following key variables at baseline: age, sex, disease duration, body mass index, number of SE, biologic-naïve, tender joint count (TJC), swollen joint count (SJC), patient’s global assessment (PGA), physician’s global assessment (PGA), DAS28-ESR, the rheumatoid factor (RF) titer, ACPA titer, ESR, C-reactive protein (CRP), number of lymphocytes, hemoglobin, and Health Assessment Questionnaires Disability Index (HAQ-DI). We then performed 1:1 nearest neighbor matching using a caliper of 1 of the standard deviation of the logit of the PS scale for tofacitinib [30, 31]. After confirming the sufficient accuracy of this method (the area under the receiver operating characteristic curve was 0.86 when all samples before PS matching was used.), we matched 70 patients to each group. Differences between groups of normally distributed continuous data were examined using the Student’s t test. Differences between groups of non-normally distributed continuous data were tested for significance as follows: non-parametric Mann-Whitney U test with Bonferroni corrections to compare two groups and the Kruskal-Wallis test to compare three groups. Pearson’s χ2 test was used for categorized variables. The Kaplan-Meier method was used to assess retention rates and differences were analyzed by the Log-rank test. The effects of the number of SE copies on the proportion of patients who achieved a good EULAR response or DAS28-ESR remission at 24 weeks in each treatment group were assessed using the Cochran-Armitage test. The impact of SE on DAS28-ESR at week 24 was examined by a multivariable conditional logistic regression model, which was adjusted for age, sex, disease duration, biologic-naïve, the ACPA titer, HAQ-DI at baseline, and ΔDAS28, which indicates the magnitude of changes from baseline to week 4 in DAS28-ESR after PS matching. P values less than 0.05 were considered to be significant. The last observation carried forward method was used for patients who discontinued treatment before week 24 to include all patients in the analysis. All statistical analyses were performed with R version 3.6.1 (R Core Team, 2019, Vienna, Austria).
Results
Enrollment of study participants and baseline characteristics
One hundred and sixty-four patients treated with tofacitinib and 131 patients treated with abatacept were enrolled (see Supplementary Table S1). All patients provided written informed consent for the present study. To avoid treatment-selection bias, PS matching was performed, resulting in 70 matched pairs of patients treated with tofacitinib or abatacept. No significant differences were observed in the baseline characteristics of the two groups (Table 1). Unless otherwise stated, data after the PS matching was used for subsequent analyses.
Comparison of clinical efficacies between tofacitinib and abatacept
Retention rates are shown in Supplementary Figure S1. At week 24, 92.9% of patients in both groups were still receiving tofacitinib or abatacept. Retention rates were not significantly different between the two groups (p = 0.606 by Log-rank test for evaluating time to discontinuation). The disease activity scores of DAS28-ESR, CDAI, and SDAI recorded over 24 weeks are shown in Fig. 1. No significant differences were observed in improved DAS28-ESR, CDAI, or SDAI scores at week 24 between the two treatment groups. However, tofacitinib was superior to abatacept for the mean disease activity scores of DAS28-ESR (tofacitinib vs abatacept; 3.3 vs 3.9, p = 0.011), CDAI (tofacitinib vs abatacept; 8.5 vs 12.2, p = 0.0043), and SDAI (tofacitinib vs abatacept; 9.1 vs 12.9, p = 0.0037) at week 4 as well as for the mean changes from baseline at week 4 in DAS28-ESR (tofacitinib vs abatacept; − 1.52 vs − 0.83, p = 0.0003), CDAI (tofacitinib vs abatacept; − 11.86 vs − 6.73, p = 0.00087), and SDAI (tofacitinib vs abatacept; − 12.86 vs − 7.46, p = 0.0018) scores. Changes in disease activity categories according to DAS28-ESR are shown in Fig. 2 A. Tofacitinib was superior to abatacept for the percentage of patients in DAS28-ESR remission at week 4 (tofacitinib vs abatacept; 30% vs 15%, p = 0.016), while no significant difference was observed at week 24 (tofacitinib vs abatacept; 32% vs 37%, p = 0.359). Changes in disease activity categories according to CDAI and SDAI were also examined as shown in Supplementary Figure S2. Tofacitinib was superior to abatacept for the percentage of patients in remission at week 4 (tofacitinib vs abatacept; 20% vs 7%, p = 0.044) and week 12 (tofacitinib vs abatacept; 34% vs 17%, p = 0.016) in CDAI and at week 12 (tofacitinib vs abatacept; 34% vs 16%, p = 0.022) in SDAI, while no significant differences were observed in the percentage of patients in remission in CDAI and SDAI at week 24 between the two groups.

Disease activity scores in DAS28-ESR, CDAI, and SDAI. Mean disease activity scores and mean changes from baseline scores in DAS28-ESR (A), CDAI (B), and SDAI (C) recorded over 24 weeks are shown after the initiation of treatment with tofacitinib or abatacept. RA disease activity scores between the two treatment groups were compared at each time point. Error bars indicate 95% confidence intervals. *p < 0.05 by the Student’s t test. CDAI, Clinical Disease Activity Index; DAS28-ESR, Disease Activity Score in 28 joints using the erythrocyte sedimentation rate: SDAI, Simplified Disease Activity Index

Time course of DAS28-ESR and EULAR responses. A Changes in disease activity categories according to DAS28-ESR are shown at baseline, 4, 12, and 24 weeks after the treatment with tofacitinib or abatacept. B The proportion of patients meeting the EULAR response criteria are shown 4, 12, and 24 weeks after the start of treatment with tofacitinib or abatacept. The percentage of patients who achieved good or moderate EULAR responses or DAS28-ESR remission were compared between the two treatment groups at each time point. DAS28-ESR, Disease Activity Score in 28 joints using the erythrocyte sedimentation rate; EULAR, European League Against Rheumatism
The time courses of EULAR responses are shown in Fig. 2 B. At week 24, the percentages of patients who achieved good or moderate EULAR responses in the tofacitinib and abatacept groups were 78 and 81%, respectively. No significant differences were observed between the two treatment groups (p = 0.72). However, at week 4, the percentage of good or moderate EULAR responses was significantly higher in the tofacitinib group than in the abatacept group (tofacitinib vs abatacept; 73% vs 50%, p = 0.0026). The proportion of patients with a good EULAR response reached a plateau at week 12 in the tofacitinib group, while it continuously increased for 24 weeks in the abatacept group.
Influence of HLA-DRB1 SE on responses to treatment
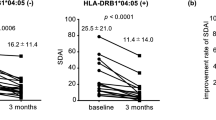
The time courses of scores in DAS28-ESR, CDAI, and SDAI stratified according to the copy numbers of SE alleles are shown in Fig. 3. RA disease activity scores in each treatment group at each time point were compared between 0 SE alleles and 1 SE allele, 0 SE alleles and 2 SE alleles, and 1 SE allele and 2 SE alleles by the Mann-Whitney U test with Bonferroni corrections which adjust p values less than 0.0167 to be significant. In patients receiving tofacitinib, no significant differences were observed in RA disease activity scores regardless of the copy numbers of SE alleles and the composite measures for disease activities used in the analyses. In contrast, in patients receiving abatacept, significant differences were observed in RA disease activity scores that depended on the copy numbers of SE alleles. The following analyses showed significant differences in DAS28-ESR: 1 SE allele vs 2 SE alleles (4.0 vs 2.9, p = 0.0127) and 0 SE alleles vs 2 SE alleles (4.1 vs 2.9, p = 0.00986) at week 4, 0 SE alleles vs 2 SE alleles (3.6 vs 2.6, p = 0.0106) at week 12, and 0 SE alleles vs 2 SE alleles (3.5 vs 2.3, p = 0.000959) at week 24 (Fig. 3 A). The following significant differences were observed in CDAI: 0 SE alleles vs 2 SE alleles (14.1 vs 7.2, p = 0.00162) at week 4, 0 SE alleles vs 2 SE alleles (9.4 vs 5.5, p = 0.00167) at week 12, and 0 SE alleles vs 2 SE alleles (8.4 vs 4.2, p = 0.00456) at week 24 (Fig. 3 B). The following significant differences were observed in SDAI: 0 SE alleles vs 2 SE alleles (15.1 vs 7.7, p = 0.00240) at week 4, 0 SE alleles vs 2 SE alleles (9.8 vs 6.0, p = 0.0150) at week 12, and 0 SE alleles vs 2 SE alleles (8.8 vs 4.7, p = 0.00173) at week 24 (Fig. 3 C).

Copy numbers of SE alleles and RA disease activity scores. The mean values of scores in DAS28-ESR (A), CDAI (B), and SDAI (C) stratified according to the copy number of SE alleles are shown at baseline, 4, 12, and 24 weeks after the start of treatment with tofacitinib or abatacept. RA disease activity scores in DAS28-ESR, CDAI and SDAI were compared between 0 SE alleles and 1 SE allele, 0 SE alleles and 2 SE alleles, and 1 SE allele and 2 SE alleles at each time point in each treatment by the Mann-Whitney U test with Bonferroni corrections. Error bars indicate 95% confidence intervals. P values less than 0.0167 were considered to be significant. †p < 0.0167, 1 SE allele and 2 SE alleles were compared; **p < 0.0167, 0 SE alleles and 2 SE alleles were compared; ***p < 0.0033, 0 SE alleles and 2 SE alleles were compared. CDAI, Clinical Disease Activity Index; DAS28-ESR, Disease Activity Score in 28 joints using the erythrocyte sedimentation rate; SDAI, Simplified Disease Activity Index; RA, rheumatoid arthritis; SE 0, 0 copies of shared epitope alleles; SE 1, 1 copy of shared epitope alleles; SE 2, 2 copies of shared epitope alleles
Figure 4 shows the percentage of patients in DAS28-ESR remission with tofacitinib and abatacept stratified according to the copy numbers of SE alleles. The effects of the copy numbers of SE alleles on DAS28-ESR remission at week 24 were examined using the Cochran-Armitage test. The percentage of patients in DAS28-ESR remission at week 24 was not affected by the copy numbers of SE alleles in the tofacitinib group (32% for 0 SE alleles, 26% for 1 SE allele, and 40% for 2 SE alleles, p = 0.947), whereas it significantly increased as the copy numbers of SE alleles became higher in the abatacept group (10% for 0 SE alleles, 45% for 1 SE allele, and 58% for 2 SE alleles, p = 0.00309). These results were confirmed in an analysis using the EULAR response criteria shown in Fig. 5. The effects of the copy numbers of SE alleles on a good response versus a moderate or no response at week 24 were examined using the Cochran-Armitage test. The percentage of patients who achieved a good EULAR response at week 24 was not affected by the copy numbers of SE alleles in patients receiving tofacitinib (50% for 0 SE alleles, 42% for 1 SE allele, and 60% for 2 SE alleles, p = 0.924), but significantly increased as the copy numbers of SE alleles became higher in patients receiving abatacept (20% for 0 SE alleles, 45% for 1 SE allele, and 75% for 2 SE alleles, p = 0.0182).

Copy numbers of SE alleles and remission in DAS28-ESR. The percentage of patients in DAS28-ESR remission stratified according to the copy numbers of SE alleles are shown 4, 12, and 24 weeks after the start of treatment with tofacitinib (A) or abatacept (B). The effects of the copy numbers of SE alleles on the percentage of patients achieving remission in DAS28-ESR at 24 weeks in each treatment group were assessed by the Cochran-Armitage test; not significant for tofacitinib, p = 0.00309 for abatacept. DAS28-ESR, Disease Activity Score in 28 joints using the erythrocyte sedimentation rate; SE 0, 0 copies of shared epitope alleles; SE 1, 1 copy of shared epitope alleles; SE 2, 2 copies of shared epitope alleles

Copy numbers of SE alleles and EULAR response criteria. The percentage of patients meeting EULAR responses criteria stratified according to the copy numbers of SE alleles are shown 4, 12, and 24 weeks after the start of treatment with tofacitinib (A) or abatacept (B). The effects of the copy numbers of SE alleles on the percentage of patients who achieved a good EULAR response at 24 weeks in each treatment group was assessed by the Cochran-Armitage test; not significant for tofacitinib, p = 0.0182 for abatacept. EULAR, European League Against Rheumatism; SE 0, 0 copies of shared epitope alleles; SE 1, 1 copy of shared epitope alleles; SE 2, 2 copies of shared epitope alleles
Impact of SE on DAS28-ESR remission
The impact of the presence of SE alleles on DAS28-ESR remission was assessed using a multivariable conditional logistic regression analysis adjusted for age, sex, RA disease duration, biologic-naïve, the ACPA titer, ΔDAS28-ESR, HAQ-DI, and PS. The presence of SE alleles (vs the absence of SE) correlated with achieving DAS28-ESR remission with abatacept at week 24 (OR = 25.881, 95% CI = 3.140–213.351, p = 0.0025), whereas the presence of SE did not affect responses to treatment with tofacitinib (OR = 1.473, 95% CI = 0.291–7.446, p = 0.639) (Table 2). Similarly, in the analysis using data before PS matching, the presence of SE alleles correlated with achieving DAS28-ESR remission with abatacept at week 24 (OR = 3.845, 95% CI = 1.386–10.669, p = 0.0097). In contrast, SE positivity did not affect responses to treatment with tofacitinib (OR = 1.910, 95% CI = 0.796–4.583, p = 0.1471) (see Supplementary Table S2).
Discussion
To the best of our knowledge, this is the first study to compare the clinical effectiveness of tofacitinib and abatacept. The clinically important results obtained demonstrated that tofacitinib and abatacept had similar clinical effectiveness at week 24, whereas tofacitinib exerted therapeutic effects earlier than abatacept. The significant clinical effects of tofacitinib assessed by ACR20 responses were previously detected as early as 2 weeks after the initiation of treatment [32]. Furthermore, a RCT comparing abatacept or infliximab with placebo reported that the onset of responses assessed by ACR20 initially appeared more rapidly with infliximab, whereas similar response rates were noted with abatacept and infliximab by day 85 [33]. Our results are consistent with these findings.
We also observed that the percentage of patients with EULAR good responses or DAS28-ESR remission significantly increased as the copy numbers of SE alleles became higher in patients receiving abatacept. In contrast, SE positivity had no effect on responses to treatment with tofacitinib. The different mechanistic roles of SE in response to these treatments may be attributed to differences in the mode of actions of both drugs. Previous studies reported that T cells were more strongly activated in ACPA-positive patients than in ACPA-negative patients [34], and SE alleles are more frequently detected in ACPA-positive patients (82–89.6%) than in ACPA-negative patients (53–70%) [35]. These findings suggest that T cells are activated more in SE-positive patients than in SE-negative patients. Therefore, abatacept, which selectively inhibits T cell activation, may be more effective in SE-positive patients. In contrast, tofacitinib has been shown to affect not only CD4+T cells, but also dendritic cells and B cells. Tofacitinib suppressed the production and stimulation of loop of a type-I interferon through JAK1/JAK3, decreased CD80/CD86 expression, and suppressed the T cell stimulatory capacity of dendritic cells. Furthermore, tofacitinib inhibited IgG production and IL-6 gene expression in the activated B cells [36]. Therefore, tofacitinib may exhibit its clinical effectiveness in SE-negative and -positive patients through pathways other than T cells.
Following the inclusion of SE positivity in a multivariable logistic analysis, the ACPA titter was not a significant predictor of DAS28-ESR remission at week 24 in patients receiving abatacept (see Supplementary Table S3). Since the presence of SE was significantly associated with ACPA titer (see Supplementary Table S6), we considered possible multicollinearity between ACPA and SE positivity and analyzed their impacts separately on achieving remission or low disease activity (LDA) at week 24 in patients receiving abatacept. We confirmed that the presence of SE significantly associated with the achievement of remission or LDA in DAS28-ESR, but ACPA positivity did not in conditional multivariable logistic analyses (see Supplementary Table S4 and S5). Previous studies reported a relationship between the increased efficacy of abatacept and ACPA positivity [37, 38]. Our results suggest that the better clinical response to abatacept in patients positive for ACPA observed in the previous studies may have been attributed to the presence of SE alleles, because the presence of SE were previously shown to be associated with the high titers of ACPA [9], which was also confirmed in the present study (see Supplementary Table S6). Since ACPA titers are also affected by non-SE alleles, such as HLA-DRB1 *0901 and *15 [39, 40], SE positivity may be a more accurate predictor of the efficacy of abatacept. In the present study, 30.0 and 25.7% of patients in the abatacept group had HLA-DR*0901 and *15, respectively.
Relationships have been reported between HLA-DRB1 alleles and clinical manifestations, including mortality risk [41], risk of severe joint destruction [10], and extra-articular manifestations, such as rheumatoid vasculitis [42]. Some amino acid haplotypes in HLA-DRB1 may be useful for the stratification of patients in terms of long-term outcomes, i.e., all-cause mortality, the risk of radiographic damage and laboratory measures of disease activity. Therefore, the prior identification of HLA-DRB1 alleles may be useful not only for selecting an appropriate molecular targeted therapy, but also for predicting disease progression in patients with RA in daily clinical practice. The present study has several limitations. First, we were unable to exclude all selection bias, even after PS matching. However, no significant differences were observed between the two treatment groups after PS matching. Furthermore, a certain number of cases were excluded after PS matching. We examined the relationship between SE and the effectiveness of these two drugs with and without PS matching and obtained similar results. In addition, due to the small sample size of this study, the caliper for PS matching was loosely set at width equal to 1 of the SD. However, a caliper width equal to 0.2–0.25 of the SD has generally been recommended [43, 44], and we performed PS matching again with a caliper width equal to 0.25 of the SD. After the PS matching, it was found that the impact of SE positivity on DAS28-ESR remission at week 24 in each treatment group was not different between patients selected with a caliper width equal to 0.25 of the SD and those selected with a caliper width equal to 1 of the SD. Second, all subjects in our study were Japanese descent and showed the similar proportion of patients with positive SE to the studies conducted in Korea [45], UK [46], and Sweden [47] except in a study conducted in Malaysia [48]. Proportion of RA patients with SE positivity may affect generalizability of our results in other regions or ethnicities. Third, the evaluation was set at week 24; however, longer observations may be required.
Conclusions
The present study demonstrated that tofacitinib and abatacept had similar clinical effectiveness at week 24, whereas tofacitinib was superior to abatacept for changes from baseline and achieving DAS28-ESR remission at week 4. Furthermore, the presence of SE correlated with the achievement of DAS28-ESR remission at week 24 in patients receiving abatacept, but not in those receiving tofacitinib. Collectively, genetic information on HLA-DRB1 alleles and the present results are expected to be important factors facilitating shared decision making by rheumatologists discussing the advantages and disadvantages of different therapeutic options with their patients.
Availability of data and materials
The datasets generated and/or analyzed in the present study are available from the corresponding author upon reasonable request.
Abbreviations
- ACPA:
-
Anticitrullinated peptide antibody
- ACR:
-
American College of Rheumatology
- CCP:
-
Cyclic citrullinated peptide
- CD:
-
Cluster of differentiation
- CDAI:
-
Clinical Disease Activity Index
- CRP:
-
C-reactive protein
- DAS28-ESR:
-
Disease Activity Score using the erythrocyte sedimentation rate
- DMARDs:
-
Disease-modifying antirheumatic drugs
- ESR:
-
Erythrocyte sedimentation rate
- EULAR:
-
European League Against Rheumatism
- HAQ-DI:
-
Health Assessment Questionnaires Disability Index
- HLA:
-
Human leukocyte antigen
- JAK:
-
Janus kinase
- RA:
-
Rheumatoid arthritis
- RF:
-
Rheumatoid factor
- SDAI:
-
Simplified Disease Activity Index
- SE:
-
Shared epitope
References
MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000;43(1):30–7. https://doi.org/10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B.
Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understand the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30(11):1205–13. https://doi.org/10.1002/art.1780301102.
Gonzalez-Gay MA, Garcia-Porrua C, Hajeer AH. Influence of human leukocyte antigen-DRB1 on the susceptibility and severity of rheumatoid arthritis. Semin Arthritis Rheum. 2002;31:335–60.
Gorman JD, Lum RF, Chen JJ, Suarez-Almazor ME, Thomson G, Criswell LA. Impact of shared epitope genotype and ethnicity on erosive disease. A metanalysis of 3,240 rheumatoid arthritis patients. Arthritis Rheum. 2004;50(2):400–12. https://doi.org/10.1002/art.20006.
Holoshitz J. The rheumatoid arthritis HLA-DR shared epitope. Curr Opin Rheumatol. 2010;22(3):293–8. https://doi.org/10.1097/BOR.0b013e328336ba63.
Turesson C, Schaid DJ, Weyand CM, Jacobsson LTH, Goronzy JJ, Petersson IF, et al. The impact of HLA-DRB1 genes on extra-articular disease manifestations in rheumatoid arthritis. Arthritis Res Ther. 2005;7:R1386–93.
Huizinga TMJ, Amos CI, van der Helm-van Mil AHM, Chen W, van Gaalen FA, Jawaheer D, et al. Refining the complex rheumatoid arthritis phenotype based on specificity of the HLA-DRB1 shared epitope for antibodies to citrullinated protein. Arthritis Rheum. 2005;52(11):3433–8. https://doi.org/10.1002/art.21385.
McInnes SG. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–19. https://doi.org/10.1056/NEJMra1004965.
van der Helm-van Mil AHM, Verpoort KN, Breedveld FC, Huizinga TWJ, Toes REM, Vires RRP. The HLA-DRB1 shared epitope alleles are primarily a risk factor for anti-cyclic citrullinated peptide antibodies and are not an independent risk factor for development of rheumatoid arthritis. Arthritis Rheum. 2006;54:1117–21.
Wagner U, Kaltenhauser S, Pierer M, Seidel W, Trolzsch M, Hantzschel H, et al. Prospective analysis of the impact of HLA-DR and -DQ on joint destruction in recent-onset rheumatoid arthritis. Rheumatology. 2003;42(4):553–62. https://doi.org/10.1093/rheumatology/keg190.
van Steenbergen HW, Raychaughuri S, Rodríguez-Rodríguez L, Rantapää-Dahlqvist S, Berglin E, Toes REM, et al. Association of valine and leucine at HLA-DRB1 position 11 with radiographic progression in rheumatoid arthritis, independent of the shared epitope alleles but not independent of anti-citrullinated protein antibodies. Arthritis Rheumatol. 2015;67(4):877–86. https://doi.org/10.1002/art.39018.
Oryoji K, Yoshida K, Kashiwado Y, Tanaka K, Mizuki S, Tsukamoto H, et al. Shared epitope positivity is related to efficacy of abatacept in rheumatoid arthritis. Ann Rheum Dis. 2018;77(8):1234–6. https://doi.org/10.1136/annrheumdis-2017-211430.
Criswell LA, Lum RF, Turner KN, Woehl B, Zhu Y, Wang J, et al. The influence of genetic variation in the HLA-DRB1 and LTA-TNF regions on the response to treatment of early rheumatoid arthritis with methotrexate or etanercept. Arthritis Rheum. 2004;50(9):2750–6. https://doi.org/10.1002/art.20469.
Smolen JS, Landewé RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. 2020;79(6):685–99. https://doi.org/10.1136/annrheumdis-2019-216655.
Tanaka Y, Yamaoka K. JAK inhibitor tofacitinib for treating rheumatoid arthritis: from basic to clinical. Mod Rheumatol. 2012;23:415–24.
Rochman Y, Spolski R, Leonard WJ. New insight into the regulation of T cells by γc family cytokines. Nat Immunol Rev. 2009;9(7):480–90. https://doi.org/10.1038/nri2580.
Genovese MC, Becker JC, Schiff M, Luggen M, Sherrer Y, Kremer J, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Eng J Med. 2005;353(11):1114–23. https://doi.org/10.1056/NEJMoa050524.
van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367(6):508–19. https://doi.org/10.1056/NEJMoa1112072.
Schiff M, Weinblatt ME, Valente R, van der Heijde D, Citera G, Elegbe A, et al. Head-to-head comparison of subcutaneous abatacept versus adalimumab for rheumatoid arthritis: two-year efficacy and safety findings from AMPLE trial. Ann Rheum Dis. 2014;73(1):86–94. https://doi.org/10.1136/annrheumdis-2013-203843.
Nakajima A, Sakai R, Inoue E, Harigai M. Prevalence of patients with rheumatoid arthritis and age-stratified trends in clinical characteristics and treatment, based on the National Database of Health Insurance Claims and Specific Health Checkups of Japan. Int J Rheum Dis. 2020;23(12):1676–84. https://doi.org/10.1111/1756-185X.13974.
Conner AF, Speroff T, Dawson NV, Thomas C, Harrell FE Jr, Wagner D, et al. The effectiveness of right heart catheterization in the initial care of critically III patients. JAMA. 1996;276:889–97.
Kubo S, Nakayamada S, Nakano K, Hirata S, Fukuyo S, Miyagawa I, et al. Comparison of the efficacies of abatacept and tocilizumab in patients with rheumatoid arthritis by propensity score matching. Ann Rhuem Dis. 2016;75(7):1321–7. https://doi.org/10.1136/annrheumdis-2015-207784.
Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, et al. Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League against rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569–81. https://doi.org/10.1002/art.27584.
Genevese MC, Covarrubias A, Leon G, Mysler E, Keiserman M, Valente R, et al. Subcutaneous abatacept versus intravenous abatacept. A phase IIIb noninferiority study in patients with an inadequate response to methotrexate. Arthritis Rheum. 2011;63(10):2854–64. https://doi.org/10.1002/art.30463.
Prevoo MLL. vanʻT Hof MA, Kuper HH, van Leeuwen MA, van de Putte LBA, van Riel PLCM. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995;38(1):44–8. https://doi.org/10.1002/art.1780380107.
Smolen JS, Breedveld FC, Schiff MH, Kalden JR, Emery P, Eberl G, et al. A simplified disease activity index for rheumatoid arthritis for use in clinical practice. Rheumatology (Oxford). 2003;42(2):244–57. https://doi.org/10.1093/rheumatology/keg072.
Aletaha D, Nell VP, Stamm T, Uffman M, Pflugbeil S, Machold K, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther. 2005;7(4):R796–806. https://doi.org/10.1186/ar1740.
van Gestel AM, Prevoo MLL, MA VH, Van Rijswijk MH, de Putte LBA V, PLCM VR. Development and validation of the European league against rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism Criteria. Arthritis Rheum. 1996;39(1):34–40. https://doi.org/10.1002/art.1780390105.
Ishikawa Y, Ikari K, Hashimoto M, Ohmura K, Tanaka M, Ito H, et al. Shared epitope defines distinct associations of cigarette smoking with levels of anticitrullinated protein antibody and rheumatoid factor. Ann Rheum Dis. 2019;78(11):1480–7. https://doi.org/10.1136/annrheumdis-2019-215463.
Lunt M. Selecting an appropriate caliper can be essential for achieving good balance with propensity score matching. Am J Epidemiol. 2014;179(2):226–35. https://doi.org/10.1093/aje/kwt212.
Toyoshima O, Nishizawa T, Yoshida S, Sekiba K, Kataoka Y, Hata K, et al. Expert endoscopists with high adenoma detection rates frequently detect diminutive adenoma in proximal colon. Endosc Int Open. 2020;08:E775–82.
Burmester GR, Blanco R, Charles-Schoeman C, Wollenhaupt J, Zerbini C, Benda B, et al. Tofacitinib (CP-690, 550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumor necrosis factor inhibitors: a randomized phase 3 trial. Lancet. 2013;381(9865):451–60. https://doi.org/10.1016/S0140-6736(12)61424-X.
Schiff M, Keiserman M, Codding C, Songcharoen S, Berman A, Nayiager S, et al. Efficacy and safety of abatacept or infliximab vs placebo in ATTEST: a phase III, multi-centre, randomized, double-blind, placebo-controlled study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Ann Rheum Dis. 2008;67(8):1096–103. https://doi.org/10.1136/ard.2007.080002.
Pieper J, Herrath J, Raghavan S, Muhammad K, van Vollenhoven R, Malmström V. CTLA4-Ig (abatacept) therapy modulates T cell effector functions in autoantibody-positive rheumatoid arthritis patients. BMC Immunol. 2013;14:R34.
Kallberg H, Padyukov L, Plenge RM, Ronnelid J, Gregersen PK, van der Helm-van Mil AH, et al. Gene-gene and gene-environment interactions involving HLA-DRB1, PTPN22, and smoking in two subsets of rheumatoid arthritis. Am J Hum Genet. 2007;80(5):867–75. https://doi.org/10.1086/516736.
Tanaka Y. Recent progress and perspective in JAK inhibitors for rheumatoid arthritis: from bench to bedside. J Biochem. 2015;158(3):173–9. https://doi.org/10.1093/jb/mvv069.
Sokolove J, Schiff M, Fleischmann R, Weinblatt ME, Connolly SE, Johnsen A, et al. Impact of baseline anti-cyclic citrullinated peptide-2 antibody concentration on efficacy outcomes following treatment with subcutaneous abatacept or adalimumab: 2-year results from AMPLE trial. Ann Rheum Dis. 2016;75(4):709–14. https://doi.org/10.1136/annrheumdis-2015-207942.
Harrold LR, Litman HI, Connolly SE, Kelly S, Hua W, Alemao E, et al. Effect of anticitrullinated protein antibody status on response to abatacept or antitumor necrosis factor-ɑ therapy in patients with rheumatoid arthritis: A US national observational study. J Rheumatol. 2018;45:35–9.
Laki J, Lundström E, Snir O, Rönnelid J, Ganji I, Catrina AI, et al. Very high levels of anti-citrullinated protein antibodies are associated with HLA-DRB1*15 non-shared epitope allele in patients with rheumatoid arthritis. Arthritis Rheum. 2012;64(7):2078–84. https://doi.org/10.1002/art.34421.
Terao C, Ikari K, Ohmura K, Suzuki T, Iwamoto T, Takasugi K, et al. Quantitative effect of HLA-DRB1 alleles to ACPA levels in Japanese rheumatoid arthritis: no strong genetic impact of shared epitope to ACPA levels after stratification of HLA-DRB1*0901. Ann Rheum Dis. 2012;71(6):1095–7. https://doi.org/10.1136/annrheumdis-2011-200907.
Viatte S, Plant D, Han B, Fu B, Yarwood A, Thomson W, et al. Association of HLA-DRB1 haplotypes with rheumatoid arthritis severity, mortality and treatment response. JAMA. 2015;131:1645–56.
Gorman JD, David-Vaudey E, Pai M, Lum RF, Criswell LA. Particular HLA-DRB1 shared epitope genotypes are strongly associated with rheumatoid vasculitis. Arthritis Rheum. 2004;50(11):3476–84. https://doi.org/10.1002/art.20588.
Rosenbaum PR, Rubin DB. Constructing a control group using multivariate matched sampling methods that incorporate the propensity score. Am Stat. 1985;39:33–8.
Austin PC. Optimal caliper widths for propensity-score matching when estimating differences in means and differences in proportions in observational studies. Pharm Stat. 2011;10(2):150–61. https://doi.org/10.1002/pst.433.
Bang SY, Lee KH, Cho SK, Lee SK, Lee KW, Bae SC. Smoking increases rheumatoid arthritis susceptibility in individuals carrying HLA-DRB1 shared epitope, regardless of rheumatoid factor or anti-cyclic citrullinated peptide antibody status. Arthritis Rheumatology. 2010;62:369–77.
Murphy D, Mattey D, Hutchinson D. Anti-citrullinated protein antibody-positive rheumatoid arthritis is primarily determined by rheumatoid factor titre and the shared epitope rather than smoking per se. Plos One. 2017;12(7):e0180655. https://doi.org/10.1371/journal.pone.0180655.
Hedstrӧm AK, Rӧnnelid J, Klareskog L, Alfredsson L. Complex relationship of smoking, HLA-DRB1 genes, and serologic profiles in patients with early rheumatoid arthritis: update from a Swedish population-based case-control study. Arthritis Rheumatology. 2019;71(9):1504–11. https://doi.org/10.1002/art.40852.
Too CL, Yahya A, Murad S, Dhaliwal JS, Larsson PT, Muhamad NA, et al. Smoking interacts with HLA-DRB1 shared epitope in the development of anti-citrullinated protein antibody-positive rheumatoid arthritis: results from Malaysian Epidemiological Investigation of rheumatoid arthritis (MyEIRA). Arthritis Res Ther. 2012;14(2):R89. https://doi.org/10.1186/ar3813.
Acknowledgements
The authors would like to thank the study patients and investigators. The TOF-ABT Study Group: Yuji Akiyama (Ogawa Red Cross Hospital), Souichirou Ando (Ando Clinic of Rheumatology), Yayoi Hashiba (Zenjinkai Shimin no mori Hospital), Motohide Kaneko (Kaneko Clinic of Rheumatology), Mitsuhiro Kawagoe (Aoki Clinic of Rheumatology), Tsuneo Kondo (Saitama Medical Center, Saitama Medical University), Kazuyoshi Kubo (Zenjinkai Shimin no mori Hospital), Ikuko Masuda (Jujo Takeda Rehabilitation Hospital), Mitsuyo Matsumoto (Hirose Clinic of Rheumatology), Yusuke Okada (Saitama Medical Center, Saitama Medical University), Akiko Shibata (Saitama Medical Center, Saitama Medical University), Kimihiro Suzuki (Suzuki Clinic of Rheumatology), Ko Takamatsu (National Defense Medical College), and Hirofumi Takei (Kanoiwa Hospital).
Disclosure statement
MH has received research grants from AbbVie Japan GK, Asahi Kasei Corp., Astellas Pharma Inc., Ayumi Pharmaceutical Co., Bristol Myers Squibb Co., Ltd., Chugai Pharmaceutical Co., Ltd., Daiichi-Sankyo, Inc., Eisai Co., Ltd., Kissei Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corp., Nippon Kayaku Co., Ltd., Sekiui Medical, Shionogi & Co., Ltd., Taisho Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., and Teijin Pharma Ltd. MH has received speaker’s fee from AbbVie Japan GK, Ayumi Pharmaceutical Co., Boehringer Ingelheim Japan, Inc., Bristol Myers Squibb Co., Ltd., Chugai Pharmaceutical Co., Ltd., Eisai Co., Ltd., Eli Lilly Japan K.K., GlaxoSmithKline K.K., Kissei Pharmaceutical Co., Ltd., Pfizer Japan Inc., Takeda Pharmaceutical Co., Ltd., and Teijin Pharma Ltd. MH is a consultant for AbbVie, Boehringer-Ingelheim, Bristol Myers Squibb Co., Kissei Pharmaceutical Co., Ltd., and Teijin Pharma.; KA has received consulting fees from Mitsubishi-Tanabe Pharma Corp., AbbVie GK., Astellas Pharma Inc., Glaxo Smith Klein plc, Gilead Sciences Inc., Pfizer Japan Inc. and Eli Lilly Japan K.K., and has received research grants from Chugai Pharmaceutical Co., Ltd. and Asahi Kasei Corp.; TH has received consulting fees from Takeda Pharmaceutical Co., Ltd., Mitsubishi-Tanabe Pharma Corp., AbbVie Japan GK, Eisai Co., Ltd., Janssen Inc., and Asahi Kasei Corp.; TN has received research grants from Bristol-Myers Squibb Co., Ltd., and Ono Pharmaceutical Co., Ltd.
Funding
No financial support has been received for this work.
Author information
Authors and Affiliations
Consortia
Contributions
WH, MH, and TN participated in the study design, data interpretation, and manuscript preparation. WH, MH, KI, and TN critically reviewed the manuscript. WH, KA, TH, KI, MN, KA, HN, YK, and TN recruited patients and collected data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The present study was performed in accordance with the Helsinki Declaration and was approved by the Ethics Committee of Toho University School of Medicine (approval number A17085). All participants gave their written informed consent.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hirose, W., Harigai, M., Amano, K. et al. Impact of the HLA-DRB1 shared epitope on responses to treatment with tofacitinib or abatacept in patients with rheumatoid arthritis. Arthritis Res Ther 23, 228 (2021). https://doi.org/10.1186/s13075-021-02612-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-021-02612-w