
Abstract
Background
Despite the success of TNF-inhibitor therapy in rheumatoid arthritis treatment, up to 40% of patients fail to respond adequately. This study aimed to identify transcriptome-based biomarkers of adalimumab response in rheumatoid arthritis (RA) to aid timely switching in non-responder patients and provide a better mechanistic understanding of the pathways involved in response/non-response.
Methods
The Affymetrix Human Transcriptome Array 2.0 (HTA) was used to measure the transcriptome in whole blood at pre-treatment and at 3 months in EULAR good- and non-responders to adalimumab therapy. Differential expression of transcripts was analysed at the transcript level using multiple linear regression. Differentially expressed genes were validated in independent samples using OpenArray™ RT-qPCR.
Results
In total, 813 transcripts were differentially expressed between pre-treatment and 3 months in adalimumab good-responders. No significant differential expression was observed between good- and non-responders at either time-point and no significant changes were observed in non-responders between time-points. OpenArray™ RT-qPCR was performed for 104 differentially expressed transcripts in good-responders, selected based on magnitude of effect or p value or based on prior association with RA or the immune system, validating differential expression for 17 transcripts.
Conclusions
An early transcriptome signature of DAS28 response to adalimumab has been identified and replicated in independent datasets. Whilst treat-to-target approaches encourage early switching in non-responsive patients, registry evidence suggests that this does not always occur. The results herein could guide the development of a blood test to distinguish responders from non-responders at 3 months and support clinical decisions to switch non-responsive patients to an alternative therapy.
Similar content being viewed by others


Background
TNF-inhibitor (TNFi) therapies have revolutionised the treatment of rheumatoid arthritis (RA) for many patients, reducing synovial inflammation and long-term disability attributed to cartilage and bone destruction [1,2,3]. Despite their success, up to 40% of patients fail to respond adequately leaving them vulnerable to further disease progression and potential adverse effects of treatment [4, 5]. In addition, non-response is economically inefficient; the cost of TNFi therapy is estimated to be £3000–10,000 per patient per year. Thus, early identification of non-responders for switching to an alternative therapy is a research priority for improved long-term outcomes and the responsible use of limited healthcare resources.
Currently, non-responder patients can be identified and switched to an alternative therapy at 3 months, but many remain on an ineffective therapy for much longer periods [6]. Whilst clinical markers explain some of the variability in response, alone they offer insufficient predictive capability [7]. Ideally, a reliable biological biomarker or panel of biomarkers would be measured in newly diagnosed patients and throughout the treatment time-course to predict and monitor response to therapy. This would aid timely therapeutic switching in patients in whom the treatment is unlikely to be effective but requires the identification of reliable biomarkers and development of a statistical classifier of treatment response.
Prediction of TNFi response in RA has so far been disappointing with little evidence of replication between studies [8]. Progress has been hampered by small sample sizes, lack of replication, and a paucity of reliable biomarkers of TNFi response that are needed to drive advanced statistical approaches to develop robust classifiers [9,10,11].
The aim of this study was to discover and validate biomarkers that are associated with DAS28 response to TNFi by comparing changes in the transcriptome of peripheral blood from RA patients who were good- and non-responders to adalimumab therapy. Whilst previous studies have utilised microarrays to investigate TNF treatment response in RA [12], to our knowledge, this is the first to utilise the human transcriptome array (HTA), which facilitates study of both gene and exon-level data.
Methods
Patient selection
Seventy patients were selected from the Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate (BRAGGSS), previously described [7], from contributing UK centres. Inclusion criteria specified that participants provided informed written consent, were Caucasian, were over 18 years of age, and fulfilled the 2010 American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) criteria for RA [13].
All patients were biologic naïve, had received previous treatment with DMARDs, and were selected if they were treated with adalimumab and could be categorised as good or non-responders to treatment at 3 months. Stringent inclusion criteria were applied to select responder groups. Good-responders were defined by a 28-joint count disease activity score (DAS28) of < 2.6 at follow-up (i.e. clinical remission) and an improvement of > 1.2. Non-responders were included if their improvement in DAS28 was < 0.6 with an endpoint DAS28 of > 5.1 (i.e. high disease activity). Non-responder patients were excluded if anti-drug antibodies, measured by radioimmunoassay at 3-month follow-up, were detected in serum samples and/or if they self-reported non-adherence [14, 15].
Blood collection
Pre-treatment and 3-month post-treatment blood samples were collected into Tempus™ Blood RNA Tubes (3 ml) (Applied Biosystems, Foster City, CA, USA). Once collected, samples were shipped to the Versus Arthritis Centre for Genetics and Genomics laboratory for central processing. Samples were logged onto the laboratory information management system (LIMS) and were stored at − 80 °C until RNA isolation.
RNA isolation
Total RNA was isolated from Tempus™ whole blood using the MagMAX™ for Stabilised Blood Tubes RNA Isolation Kit, compatible with Tempus™ Blood RNA Tubes (Life Technologies, Carlsbad, CA, USA), according to manufacturer’s instructions. Extraction batches were mixed for responders and non-responders in order to reduce technical bias. Extracted RNA samples were treated with DNase to eliminate any potential genomic DNA contamination from downstream transcriptome measurement. RNA was quantified using the Nanodrop ND-1000 (Thermo Scientific, Waltham, MA, USA) and quality-assessed using the 2100 Bioanalyzer to generate an RNA integrity number (RIN; Agilent, Santa Clara, CA, USA).
Transcriptome measurement
Total RNA (100 ng) was amplified and converted into biotinylated sense-strand cDNA targets using the Affymetrix WT PLUS kit according to the manufacturer’s instructions (Affymetrix, Santa Clara, CA, USA). All samples were collected using the same study protocol, irrespective of clinical features. We adhered to the manufacturer’s recommendations regarding RNA quality control (260/280 ratio between 1.7 and 2.1). The Nanodrop ND-1000 was used to monitor and normalise cDNA concentration across samples throughout the target preparation. Samples of different response status and time-point were arranged in the 96-well plate at random to avoid cDNA conversion bias. For each sample, 5-μg of fragmented, end-labelled sense-strand target cDNA was hybridised to a GeneChip Human Transcriptome Array (HTA) 2.0 before incubation for 16 h at 45 °C in the GeneChip® Hybridization Oven 645. Following hybridisation, arrays were washed and stained using the GeneChip® Fluidics Station 450 and scanned using the GeneChip® Scanner 3000 7G with Autoloader to generate a raw CEL data file for each sample.
Statistical analysis of transcriptome
Raw CEL files were quality control assessed using the Affymetrix Expression Console software (version 1.1). All array files were then processed in the programming language ‘R’ using Bioconductor packages: The pd.hta.2.0 package was used for platform design information annotation and the affy package was used to summarise probe-level data into a single expression measure for each individual transcript and pre-process the data. The affy package was used to perform normalisation and probe specific background correction before summarising the probe set values into a single expression measure according to default settings. Highly variable probes were used to cluster samples and produce a dissimilarity matrix. The highly variable probes were used to create a cluster dendrogram of samples based on both transcript- and probe-level differences and illustrate how samples cluster to identify any major outliers of which there were none. The PCAmethods library was used to conduct principal component analysis (PCA) to test for run order effects and limma was used for differential expression analysis. The arrayWeights function in limma was used to assess array quality using default parameters. Differential expression analysis was adjusted for baseline DAS28, age, gender, concurrent DMARD use, and array weights. Pathway analysis was performed using the Ingenuity Pathway Analysis (IPA) tool (version 33559992) according to default settings.
RT-qPCR validation
Results from the discovery analyses were ranked according to p value and also fold change. The top 10 hits according to most significant p value and the top 10 according to largest fold change were selected for validation. A further 84 transcripts that satisfied a fold change > 1.2 and a false discovery rate (FDR) < 0.05 were selected according to a known biological association with RA or the immune system. TaqMan™ assays were selected for a total of 104 transcripts from the initial adalimumab transcriptome study for custom design of a gene expression OpenArray™ Plate, 112 assay format. Housekeeping genes were used to normalise targets and calculate relative expression values: ACTB (Hs99999903_m1), B2M (Hs00187842_m1), GAPDH (Hs99999905_m1), and HPRT1 (Hs02800695_m1). Responder groups were less stringently defined than in the discovery study. That is to say good-response to adalimumab were not necessarily in clinical remission at follow-up but did show large DAS28 improvements and were in low disease activity states (change in DAS28 > 2.78; mean = 3.08; n = 11) versus poor-responders (change in DAS28 < 1.04; mean = 0.83; n = 11). This enabled us to test whether the changes identified in the discovery cohort could be identified in patients with a less extreme response. Total RNA was reverse transcribed into single-stranded cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to manufacturer’s instructions. Sample cDNA and TaqMan™ OpenArray™ Gene Expression Master Mix were loaded on OpenArray™ plates using the OpenArray™ AccuFill® software and OpenArray™ AccuFill® system (Applied Biosystems). RT-qPCR was performed on the QuantStudio™ 12 K Flex real-time qPCR system (Applied Biosystems) and analysed in the Relative Quantification Application on the Thermo Fisher Cloud (Thermo Scientific).
Results
Cohort characteristics
Seventy patients receiving adalimumab therapy were included in the initial study. Baseline characteristics for the patients are presented in Table 1.
Transcriptome measurement
PCA of the dataset revealed one principal component contributing significantly to sample variance (> 10%) which was adjusted for during downstream differential expression analysis. There was no correlation between this principal component and age, gender, DMARD use, baseline DAS28, DAS28 components, RIN, or RNA extraction batch. Differentially expressed transcripts between response groups and time-points were defined by a fold change > 1.2 and a false discovery rate (FDR) < 0.05. No significant differential expression was observed between good- and non-responders at either pre-treatment or 3 months and no significant changes were observed in non-responders between pre-treatment and 3 months. However, 813 transcripts were differentially expressed between time-points in good-responders, mapping to 491 unique genes. This comprised 202 transcripts that were more- and 611 transcripts that were less-abundant at 3 months, compared with the pre-treatment sample (Additional file 1: Table S1). Testing was performed to identify whether adjusting for RIN in the analysis was important, but this adjustment did not qualitatively change the results. RIN was not included in the final analysis because it was not available for all samples (n = 20).
Ingenuity pathway analysis was performed to identify enrichment of relevant pathways involving differentially expressed transcripts, which are either up- or downregulated (Table 2 or Additional file 2: Table S2). The most significantly enriched pathway within the differential expression dataset for adalimumab good-responders was in ‘B and T cell signalling in rheumatoid arthritis’ (p = 1.4E− 10) with TNF identified as one of the top upstream regulators (p = 1.53E− 06).
RT-qPCR validation
A total of 11 good- and 11 non-responders were available to validate the top discovery results. Cohort characteristics were broadly similar to the discovery cohort with a minor difference in the mean baseline DAS28 (good-responders, mean = 6.27; non-responders, mean = 5.33; p = 0.0005). Relative quantification was used to compare expression between baseline and 3 months of adalimumab treatment for both response groups (Fig. 1). In good-responders differential expression was validated for 17 transcripts in the same direction of change as the initial discovery cohort (fold change > 1.2, FDR p < 0.05; Table 3). In the equivalent analysis of non-responders, no transcripts were differentially expressed at a significant level.

Heatmap of adalimumab good-responders and non-responders following real-time quantitative polymerase chain reaction (RT-qPCR) analysis of gene expression. Heatmap and hierarchal clustering is based on pairwise similarity in gene expression for the 104 transcripts measured in the RT-qPCR validation study. Good-responders and non-responders are represented in the upper and lower heatmaps, respectively. Heatmap rows and columns represent baseline and 3 months samples for 11 rheumatoid arthritis patients on adalimumab TNF-inhibitor therapy. BL = baseline and 3M = 3-months
Discussion
This study sought to identify biomarkers of adalimumab TNFi therapy. We observed a disease-relevant panel of transcriptomic changes in good-responders. In particular, significant differential expression was observed in good-responders between pre-treatment and 3 months, attributed to genes in RA-associated pathways that are responsive to relevant upstream regulators including TNF and CSF2 (otherwise known as Granulocyte-macrophage colony-stimulating factor (GM-CSF)), a promising target for therapy in RA [16].
Differential expression of seventeen transcripts observed in good-responders were validated in a small independent sample (n = 22) using OpenArray qPCR analysis. The validated transcripts were differentially expressed in the same direction as the initial discovery cohort and included ENTPD1 (otherwise known as CD39), which is primarily expressed on activated lymphoid cells. A previous study reported an expansion of CD39 positive regulatory T cells following successful treatment with methotrexate [17], whilst a more recent study reported that a higher genetic score for CD39 expression on T cells at the ENTPD1 locus was associated with a poor response to TNFi [18]. Here, we observed relatively higher levels of ENTPD1 at pre-treatment in good-responders compared with the 3-month sample. However, in the current study, we cannot say whether the observed change in ENTPD1 expression is reflected at the protein level, if a particular cell type is prominently affected or if expression is modified by genotype. Further studies of ENTPD1 locus in relevant cell populations are now required in RA to resolve these interesting observations. In addition, this study identified increased expression of CD40LG in good-responders. The CD40LG gene encodes the complementary ligand for the CD40 transmembrane protein which is expressed on both B cells and antigen-presenting cells (APCs) and has been associated with increased TNF expression. Furthermore, transcription of both genes is reportedly elevated in the synovial tissue of RA patients [19]. More recently, increased CD40 transcription and lower CD40 methylation in whole blood were associated with improved TNFi response [18].
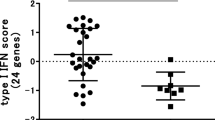
High expression of type I interferon genes has previously been associated with improved response to TNFi therapy [20] and, more recently, non-response to methotrexate therapy [21]. However, whilst expression of a number of interferon genes was initially associated with good-response in this study (IFNG, IFNG-AS1, LY6E, MX2, SERPING1, OAS2), only an association with IFNG-AS1 was retained following adjustment for baseline DAS28 score and none were validated by RT-qPCR. Overall, this study identified many immune-related genes with increased expression in the whole blood of good-responders, whilst seemingly paradoxical, this may represent migration of RA-associated inflammatory factors out of the affected joint and into the peripheral blood in a positive response to TNFi therapy.
No differential expression was observed at pre-treatment when good- and non-responders were analysed, in keeping with previous results [8, 22]. One possible reason could be that immune pathways are saturated at baseline since patients included the study had very high disease activity at baseline. Only after treatment is administered do the different gene expression groups emerge. We found that good-responders showed enrichment in pathways linked to TNF, supporting the hypothesis that there are different key drivers in different subgroups of patients. In future studies, it will be important to test the differentially expressed transcripts identified herein against different drugs in order to elucidate whether they are general markers of response or specific to this class of drug. The inability to identify baseline markers suggests that further discovery studies should redirect efforts to other data types or include on-treatment sampling as part of the study design. It is possible that the high levels of disease activity seen at pre-treatment in all study participants may be obscuring detection of pre-treatment gene expression signatures that are relevant to future treatment response.
This study benefited from the use of a stringent sample selection process including good-responders in clinical remission with absence of anti-drug antibody or inadequate adherence explaining non-response. In addition, the use of a longitudinal approach has allowed the identification of transcripts and pathways that change in response to successful treatment. The signatures were observed in whole blood which requires fewer sample processing steps. However, this may dilute subtle changes that occur in cell types of low abundance that would otherwise be apparent in cell-specific studies. This is most likely pertinent to B and T cells, which this study has highlighted as important contributors to the transcriptomic response in good-responders. Whilst it would be interesting to identify the cell types in which these gene expression changes occur, to our knowledge, the reference datasets required to resolve cell subtypes from whole blood are not currently available for the array type used in this investigation.
We recognise that the initial discovery cohort contained more responders than non-responders, which may contribute towards a lack of power for finding significant changes between baseline and 3 months in non-responders. However, in order to maximise power and mimic a case-control approach, the replication phase examined an equal number of responders and non-responders. Furthermore, significant changes were identified only in the good-responder group, in keeping with findings in the discovery cohort. Whilst there was no statistically significant difference in concurrent DMARD use at baseline, we recognise that confounding could still occur and so adjusted for this during differential expression analysis.
Whilst 813 differentially expressed transcripts were found in responders between baseline and 3 months, no significant differences in gene expression were identified between responders and non-responders at 3 months. This is likely due to power and inter-patient differences in baseline gene expression levels. We also tested for correlation between change in transcript abundance and change in DAS28 and sub-components in the good-responder subgroup (Additional file 3: Figure S1–S4). In this unadjusted analysis, no significant p values were observed; however, the sample size was small. These analyses are based on good- and poor-EULAR response. Future studies should extend this analysis to include patients across the full spectrum of response, including intermediate responders, and analyse continuous measures of response, including the DAS28 composite score and its sub-components, as secondary outcomes.
We cannot exclude the possibility that other true associations were not detected in the replication phase of the study as the sample size was small, limiting power. Nonetheless, the findings support further testing of transcripts that have been independently replicated to develop a statistical model to stratify patients according to likely treatment response. Whilst the transcriptomic response was detected at 3 months, it is possible that changes in transcript levels could manifest much earlier. Furthermore, it will be important to test if these findings are specific to adalimumab response or generalise to other therapies licenced for use in RA. Future similar studies should therefore seek to collect samples at earlier time-points and from patients treated with other classes of therapy to address these important questions. Whilst the inclusion of the extremes of response phenotype in the current study maximised the power to detect associations, the inclusion of intermediate DAS28 responders in future studies will be necessary to allow an estimation of the predictive ability of the biomarker panel with DAS28 response across the full spectrum of response.
Conclusions
This study has discovered biomarkers that could potentially be used to identify patients destined not to respond adequately to adalimumab treatment at an early stage and to support treat-to-target approaches. Whilst patients can be withdrawn from a therapy according to a limited change in DAS28 at 3 months, in reality, many patients remain on ineffective treatment for longer periods. Biomarkers could therefore aid clinical decisions to rapidly switch non-responder patients at 3 months or earlier to an alternative therapy. Much larger studies are now needed to test the utility of the identified biomarkers as a classifier of future response.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Change history
08 May 2021
A Correction to this paper has been published: https://doi.org/10.1186/s13075-021-02519-6
Abbreviations
- ACR:
-
American College of Rheumatology
- APC:
-
Antigen-presenting cell
- BRAGGSS:
-
Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate
- DAS28:
-
28-joint count disease activity score
- DMARDs:
-
Disease modifying anti-rheumatic drugs
- EULAR:
-
European League Against Rheumatism
- FDR:
-
False discovery rate
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- HTA:
-
Human transcriptome array
- IPA:
-
Ingenuity Pathway Analysis
- IQR:
-
Interquartile range
- LIMS:
-
Laboratory information management system
- PCA:
-
Principal component analysis
- RA:
-
Rheumatoid arthritis
- RIN:
-
RNA integrity number
- RQ:
-
Relative quantification
- RT-qPCR:
-
Real-time quantitative polymerase chain reaction
- SD:
-
Standard deviation
- TNF:
-
Tumour necrosis factor
- TNFi:
-
TNF-inhibitor
- ∆∆Crt:
-
Change in relative cycle threshold
References
Finckh A, Liang MH, van Herckenrode CM, de Pablo P. Long-term impact of early treatment on radiographic progression in rheumatoid arthritis: a meta-analysis. Arthritis Rheum. 2006;55(6):864–72.
Scirè CA, Lunt M, Marshall T, Symmons DPM, Verstappen SMM. Early remission is associated with improved survival in patients with inflammatory polyarthritis: results from the Norfolk Arthritis Register. Ann Rheum Dis. 2014;73(9):1677–82.
Scirè CA, Verstappen SMM, Mirjafari H, Bunn DK, Lunt M, Montecucco C, et al. Reduction of long-term disability in inflammatory polyarthritis by early and persistent suppression of joint inflammation: results from the Norfolk Arthritis Register. Arthritis Care Res (Hoboken). 2011;63(7):945–52.
Hyrich KL, Watson KD, Silman AJ, Symmons DPM. British Society for Rheumatology Biologics Register. Predictors of response to anti-TNF- therapy among patients with rheumatoid arthritis: results from the British Society for Rheumatology Biologics Register. Rheumatology. 2006;45(12):1558–65.
Kourbeti IS, Ziakas PD, Mylonakis E. Biologic therapies in rheumatoid arthritis and the risk of opportunistic infections: a meta-analysis. Clin Infect Dis. 2014;58(12):1649–57.
Hyrich KL, Lunt M, Dixon WG, Watson KD, Symmons DPM, BSR Biologics Register on behalf of the BB. Effects of switching between anti-TNF therapies on HAQ response in patients who do not respond to their first anti-TNF drug. Rheumatology (Oxford). 2008;47(7):1000–5.
Potter C, Hyrich KL, Tracey A, Lunt M, Plant D, Symmons DPM, et al. Association of rheumatoid factor and anti-cyclic citrullinated peptide positivity, but not carriage of shared epitope or PTPN22 susceptibility variants, with anti-tumour necrosis factor response in rheumatoid arthritis. Ann Rheum Dis. 2009;68(1):69–74.
Oliver J, Plant D, Webster AP, Barton A. Genetic and genomic markers of anti-TNF treatment response in rheumatoid arthritis. Biomark Med. 2015;9(6):499–512.
Maier R, Moser G, Chen G-B, Ripke S, Coryell W, Potash JB, et al. Joint analysis of psychiatric disorders increases accuracy of risk prediction for schizophrenia, bipolar disorder, and major depressive disorder. Am J Hum Genet. 2015;96(2):283–94.
Zarringhalam K, Degras D, Brockel C, Ziemek D. Robust phenotype prediction from gene expression data using differential shrinkage of co-regulated genes. Sci Rep. 2018;8(1):1237.
Zhang W, Wan Y, Allen GI, Pang K, Anderson ML, Liu Z. Molecular pathway identification using biological network-regularized logistic models. BMC Genomics. 2013;14(Suppl 8):S7.
Smith SL, Plant D, Eyre S, Barton A. The potential use of expression profiling: implications for predicting treatment response in rheumatoid arthritis. Ann Rheum Dis. 2013;72(7):1118–24.
van Gestel AM, Prevoo ML, van ’t Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism Criteria. Arthritis Rheum. 1996;39(1):34–40.
Bluett J, Morgan C, Thurston L, Plant D, Hyrich KL, Morgan AW, et al. Impact of inadequate adherence on response to subcutaneously administered anti-tumour necrosis factor drugs: results from the Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate cohort. Rheumatology (Oxford). 2015;54(3):494–9.
Jani M, Chinoy H, Warren RB, Griffiths CEM, Plant D, Fu B, et al. Clinical utility of random anti-tumor necrosis factor drug-level testing and measurement of antidrug antibodies on the long-term treatment response in rheumatoid arthritis. Arthritis Rheumatol. 2015;67(8):2011–9.
Avci AB, Feist E, Burmester G-R. Targeting GM-CSF in rheumatoid arthritis. Clin Exp Rheumatol. 2016;34(4 Suppl 98):39–44.
Peres RS, Liew FY, Talbot J, Carregaro V, Oliveira RD, Almeida SL, et al. Low expression of CD39 on regulatory T cells as a biomarker for resistance to methotrexate therapy in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2015;112(8):2509–14.
Spiliopoulou A, Colombo M, Plant D, Nair N, Cui J, Coenen MJ, et al. Association of response to TNF inhibitors in rheumatoid arthritis with quantitative trait loci for CD40 and CD39. Ann Rheum Dis. 2019;78(8):1055–61.
Guo Y, Walsh AM, Fearon U, Smith MD, Wechalekar MD, Yin X, et al. CD40L-dependent pathway is active at various stages of rheumatoid arthritis disease progression. J Immunol. 2017;198(11):4490–501.
Wright HL, Thomas HB, Moots RJ, Edwards SW. Interferon gene expression signature in rheumatoid arthritis neutrophils correlates with a good response to TNFi therapy. Rheumatology. 2015;54(1):188–93.
Plant D, Maciejewski M, Smith S, Nair N, Hyrich K, Ziemek D, et al. Profiling of gene expression biomarkers as a classifier of methotrexate nonresponse in patients with rheumatoid arthritis. Arthritis Rheumatol. 2019;71(5):678–84.
Oswald M, Curran ME, Lamberth SL, Townsend RM, Hamilton JD, Chernoff DN, et al. Modular analysis of peripheral blood gene expression in rheumatoid arthritis captures reproducible gene expression changes in tumor necrosis factor responders. Arthritis Rheumatol. 2015;67(2):344–51.
Acknowledgements
We thank the NIHR Manchester and NIHR Newcastle Biomedical Research Centres for their support. AB is in receipt of an NIHR Senior Investigator award.
Funding
We thank Versus Arthritis (previously Arthritis Research UK) for their support (grant ref. 21754). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Author information
Authors and Affiliations
Consortia
Contributions
JO—acquisition, analysis, and interpretation of data and drafting manuscript. NN—analysis and interpretation of data and drafting manuscript. GO—conception/design of study and interpretation of data. SS—analysis and interpretation of data. KLH, AM, JI, and AGW—BRAGGSS co-ordinators. AB—conception/design of study, interpretation of data, and drafting manuscript. DP—conception/design of study, analysis and interpretation of data, and drafting manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All patients gave informed written consent for their samples to be analysed as part of this study. Ethics was approved by the North West 6 Central Manchester South Research Ethics Committee (COREC 04/Q1403/37).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: the authors reported an error in the spelling of the first author's lastname.
Supplementary Information
Additional file 1: Table S1.
Differentially expressed transcripts in adalimumab good-responders between baseline (pre-treatment) and 3-months of adalimumab treatment. The annotation for each probe was retrieved from the Affymetrix array manifest [MacDonald JW (2017). pd.hta.2.0: Platform Design Info for Affymetrix HTA-2_0. R package version 3.12.2]. Transcripts with a positive fold-change exhibited increased expression at 3-months compared to baseline. Transcripts with a negative fold-change exhibited reduced expression at 3-months compared to baseline.
Additional file 2: Table S2.
The differentially expressed genes in adalimumab good-responders (baseline versus 3-months) as analysed using Ingenuity Pathway Analysis (IPA) software. The -log(p-value) for each pathway association, the ratio of the number of differentially expressed genes within each pathway, and a list of the differentially expressed genes (molecules) within each pathway are shown.
Additional file 3: Figure S1.
Correlation between change in transcript expression level and change in DAS28 score in good responders for the top 10 differentially expressed transcripts according to p-value. Figure S2. Correlation between change in transcript expression level and change in swollen joint count (SJC) in good responders for the top 10 differentially expressed transcripts according to p-value. Figure S3. Correlation between change in transcript expression level and change in tender joint count (TJC) in good responders for the top 10 differentially expressed transcripts according to p-value. Figure S4. Correlation between change in transcript expression level and change in C-reactive protein (CRP) levels in good responders for the top 10 differentially expressed transcripts according to p-value.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Oliver, J., Nair, N., Orozco, G. et al. Transcriptome-wide study of TNF-inhibitor therapy in rheumatoid arthritis reveals early signature of successful treatment. Arthritis Res Ther 23, 80 (2021). https://doi.org/10.1186/s13075-021-02451-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-021-02451-9