
Abstract
Background
Sandoz etanercept (SDZ ETN; GP2015) is an etanercept biosimilar with equivalent efficacy and comparable safety and immunogenicity to reference etanercept (ETN) in patients with moderate-to-severe chronic plaque-type psoriasis.
Methods
EQUIRA was a phase III, double-blind study conducted in patients with moderate-to-severe rheumatoid arthritis and inadequate response to disease-modifying anti-rheumatic drugs. Eligible patients were randomized 1:1 to receive subcutaneous 50 mg SDZ ETN or ETN, once-weekly, for 24 weeks. At week 24, patients with at least moderate EULAR response in the SDZ ETN group continued SDZ ETN treatment, and those in the ETN group were switched to receive 50 mg SDZ ETN, for up to 48 weeks. Patients received concomitant methotrexate at a stable dose (10–25 mg/week) and folic acid (≥ 5 mg/week). Equivalence between SDZ ETN and ETN for change from baseline in disease activity score including 28 joint count C-reactive protein (DAS28-CRP) at week 24 (primary endpoint) and comparable safety and immunogenicity profile of SDZ ETN and ETN have previously been demonstrated at week 24. Herein, we present the 48-week results of the study after a single switch from ETN to its biosimilar at week 24.
Results
The least squares mean (standard error) change in DAS28-CRP from baseline up to week 48 was comparable between “continued SDZ ETN” (− 2.90 [0.12], n = 148) and “switched to SDZ ETN” (− 2.78 [0.13], n = 131) groups. The proportion of patients achieving EULAR good/moderate responses based on DAS28-erythrocyte sedimentation rate and ACR20/50/70 response rates were comparable between the two groups. The proportion of patients with at least one treatment-emergent adverse event was 42.9% in the “continued SDZ ETN” and 38.0% in the “switched to SDZ ETN” groups. Serious adverse events occurred in 4 patients in each of the two groups. After week 24, none of the patients in the switched group developed anti-drug antibodies (ADAs), while 4 patients in the continued SDZ ETN group had single-event, very low titer, non-neutralizing ADAs detected.
Conclusions
The 48-week results from the EQUIRA study demonstrate that switch from ETN to SDZ ETN in patients with moderate-to-severe rheumatoid arthritis does not impact the efficacy, safety, or immunogenicity of etanercept.
Trial registration
EudraCT number 2012-002009-23, Registered 19 April 2012—prospectively registered.
Similar content being viewed by others


Background
Etanercept, a tumor necrosis factor inhibitor, has been used successfully for the treatment of multiple immune-mediated inflammatory diseases including moderate-to-severe rheumatoid arthritis (RA) [1].
Sandoz etanercept (SDZ ETN; development name GP2015, Erelzi® [Sandoz Inc., Princeton, NJ 08540]) is an etanercept biosimilar. Pharmacokinetic equivalence and comparable safety for SDZ ETN and reference etanercept (ETN; Enbrel® [European Union-authorized]) was demonstrated in a phase I study in healthy subjects [2]. The phase III EGALITY study demonstrated equivalent efficacy and comparable safety and immunogenicity of SDZ ETN and ETN in patients with moderate-to-severe chronic plaque-type psoriasis [3].
The randomized, double-blind, EQUIRA study demonstrated similar efficacy and comparable safety and immunogenicity profile of SDZ ETN to ETN at week 24 in patients with moderate-to-severe RA who had an inadequate response to either conventional synthetic (cs) and/or biologic (b) disease-modifying anti-rheumatic drugs (DMARDs) [4]. Herein, we present the 48-week results from the study on the effects of a single switch between ETN and SDZ ETN at week 24 on efficacy, safety, and immunogenicity of etanercept.
Methods
Study population
Patients, aged ≥ 18 years, were included if they met the following criteria: (1) RA diagnosed according to the American College of Rheumatology (ACR) 1987 or ACR/European League Against Rheumatism (EULAR) 2010 criteria [5] for ≥ 6 months before baseline; (2) active disease defined as disease activity score including 28 joint count (DAS28)-C-reactive protein (CRP) ≥ 3.2; (3) CRP > 5 mg/L or erythrocyte sedimentation rate (ESR) ≥ 28 mm/h; (4) inadequate clinical response to methotrexate (MTX) at a dose of 10–25 mg/week after optimal dose escalation according to local standards (those who had failed a csDMARD other than MTX, and any other csDMARD used in combination with MTX prior to baseline, were allowed after an appropriate wash-out period of 4 weeks); (5) MTX therapy for ≥ 3 months and on a stable dose for ≥ 28 days prior to baseline; (6) stable dose of folic acid (≥ 5 mg per week) for ≥ 28 days before baseline.
The key exclusion criteria included (1) any previous exposure to ETN; (2) treatment with any other bDMARD therapy for RA, including tumor necrosis factor (TNF) inhibitors, anti-CD20, immune-modulator drug(s), other investigational drug(s), and/or device(s) within 3 months or 5 half-lives at the time of enrollment, whichever was longer; (3) previous use of > 2 bDMARDs (patients in whom bDMARDs were efficacious but withdrawn because of reasons other than efficacy failure or safety issues were not excluded); (4) functional status class IV according to the ACR 1991 revised criteria [6]; (5) systemic manifestations of RA, with the exception of Sjögren’s syndrome; (6) any active inflammatory or autoimmune diseases other than RA; and (7) tuberculosis or latent tuberculosis detected by imaging and/or by the QuantiFERON®-TB Gold test at screening.
Study design
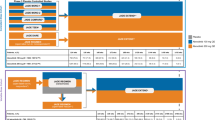
The EQUIRA study was conducted from 27 November 2015 to 12 June 2017 at 83 study centers across 16 countries (NCT02638259). Eligible patients were randomized 1:1 to self-administer 50 mg SDZ ETN or ETN (Enbrel® [European Union-authorized]), provided as pre-filled syringes, subcutaneously, once-weekly, for 24 weeks (treatment period 1 [TP1]). At week 24, patients achieving at least a moderate treatment response according to EULAR response criteria [7] in the SDZ ETN group continued SDZ ETN (defined as “continued SDZ ETN” group), and those in the ETN group were switched to SDZ ETN (defined as “switched to SDZ ETN” group), for up to 48 weeks (TP2). The initial randomization schedule and blinding have been described previously [4].
This study was conducted in accordance with the ethical principles derived from the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practices and in compliance with local regulatory requirements. The study protocol was approved by the Independent Ethics Committee or Institutional Review Board for each center. All patients provided written informed consent before entering the study.
Study assessments
The primary endpoint of the study was the change from baseline in DAS28-CRP up to week 24. The secondary endpoints, assessed up to week 24 and week 48 included (i) change from baseline in DAS28-CRP scores [8]; (ii) proportion of patients achieving good and moderate EULAR response based on DAS28-ESR [9]; (iii) proportion of patients achieving an improvement in the ACR20/50/70 response rates; (iv) physical function assessed by the health assessment questionnaire disability index (HAQ-DI) score [10, 11], and proportion of patients achieving HAQ index in normal range (≤ 0.5); and (v) impact of fatigue on patients assessed by the Functional Assessment of Chronic Illness Therapy (FACIT) fatigue scale [12, 13].
Safety assessments included evaluation of the adverse events (AEs) as well as the local tolerability of injection sites of both medications as assessed by the investigator during the study. Immunogenicity assessment included analysis of anti-drug antibodies (ADAs) up to 48 weeks using a validated screening, confirmatory, and titer determination electrochemiluminescence bridging assay [14].
Statistical analysis
The sample size determination has been described previously [4]. The TP2 full analysis set (FAS) consisted of all patients who continued to TP2 and had at least one study assessment documented in TP2. The TP2 per-protocol set (PPS) consisted of all patients completing the study until week 48 without major protocol deviations (major protocol deviations are listed in Additional file 1: Table S1); patients who prematurely withdrew from the study were also not included, although this was not a protocol deviation. The TP2 safety set (SAF) consisted of all patients who received at least one dose of study treatment during TP2. All efficacy analyses were performed on the TP2 PPS and repeated on the TP2 FAS. TP2 SAF was used for the safety summaries in TP2.
During TP2, the “continued SDZ ETN” group was compared with the “switched to SDZ ETN” group. A repeated measures analysis of (co)variance with treatment and time as factors up to week 48 was performed for DAS28-CRP change from baseline. Change from baseline in DAS28-CRP was estimated using values up to week 48 from the same mixed-model repeated measures analysis used to analyze the primary endpoint.
Results
Patient disposition and baseline characteristics
Of the 376 patients randomized in the study, 353 (SDZ ETN, n = 181; ETN, n = 172) completed TP1. A total of 341 patients entered TP2, of whom 324 completed TP2 (Fig. 1). The reasons for discontinuation during TP2 were AEs (SDZ ETN, n = 5 [2.9%]; ETN, n = 4 [2.4%]) and withdrawal of patient consent (SDZ ETN, n = 1 [0.6%]; ETN, n = 7 [4.2%]). All patients who entered TP2 were included in the TP2 FAS and TP2 SAF. The TP2 PPS included 279 patients (148 [84.6%] and 131 [78.9%] patients in the SDZ ETN and ETN groups, respectively).

Patient disposition. †The primary reason for not completing screening phase included: inclusion/exclusion criteria not fulfilled (n = 169), withdrawal of consent (n = 12), adverse event (n = 1), lost to follow-up (n = 1), and other (n = 8); *Four patients in each treatment group were not eligible for TP2; 2 patients in each treatment group were eligible but did not enter TP2; ETN, reference etanercept; SDZ ETN, Sandoz etanercept; TP, treatment period
In the overall population who entered TP2, mean (SD) age was 53.7 (12.0) years, most (80.4%) patients were in the 18–64 age category, and majority (82.1%) were females. The mean (SD) duration of RA was 8.4 (7.6) years and most (71.3%) patients were categorized as functional RA class II. The most common prior medications other than MTX and folic acid were methylprednisolone (28.2%), meloxicam (15.2%), and ibuprofen (7.6%). Patient demographics and baseline characteristics were well-balanced and comparable between TP2 treatment groups (Table 1).
Efficacy
The primary objective of the study to demonstrate that therapeutic equivalence between SDZ ETN and ETN was met, as the 95% confidence interval (CI) for the least squares mean difference between SDZ ETN and ETN for change from baseline in DAS28-CRP at week 24 was contained within the pre-specified equivalence margin of [− 0.6, 0.6] [4].
Efficacy up to week 48
The least squares mean (SE) DAS28-CRP change from baseline to week 48 was comparable between “continued SDZ ETN” (− 2.90 [0.12]) and “switched to SDZ ETN” (− 2.78 [0.130]) groups (Fig. 2). The proportion of patients achieving EULAR “good” and “moderate” responses based on DAS28-ESR was similar between “continued SDZ ETN” and “switched to SDZ ETN” groups (week 48: “continued SDZ ETN” group vs “switched to SDZ ETN” group: EULAR good response, 54.4% vs 51.9%; EULAR moderate response, 41.5% vs 44.2%; Fig. 3). The proportion of patients achieving ACR20, ACR50, and ACR70 response was generally comparable between “continued SDZ ETN” and “switched to SDZ ETN” groups; ACR50 and ACR70 response rates were numerically higher in the “switched to SDZ ETN” group at all time-points, but not clinically relevant (Fig. 4). At week 48, ACR20, ACR50, and ACR70 response rates were 89.1%, 63.3%, and 36.7%, respectively, in the “continued SDZ ETN” group and 82.4%, 65.6%, and 42.0% in the “switched to SDZ ETN” group.

DAS28-CRP change from baseline up to week 48 (TP2 per-protocol set). Analyzed using a repeated measures analysis of (co)variance with treatment and time as factors up to week 48. TP2 PPS comprised all patients completing the study until week 48 without major protocol deviations. CRP, C-reactive protein; DAS28, disease activity score including 28 joint count; PPS, per-protocol set; SE, standard error; TP, treatment period

EULAR good and moderate responses up to week 48 (TP2 per-protocol set). Response rate of at least moderate response = good response + moderate response; EULAR good response: DAS28 ≤ 3.2 and DAS28 improvement from baseline > 1.2; EULAR moderate response: DAS28 ≤ 3.2 and DAS28 improvement > 0.6 and ≤ 1.2, or DAS28 >3.2 and ≤5.1 and DAS28 improvement > 0.6 or DAS28 >5.1 but DAS28 improvement > 1.2. TP2 PPS comprised all patients completing the study until week 48 without major protocol deviations. EULAR, European League Against Rheumatism; PPS, per-protocol set; TP, treatment period

ACR20, ACR50, and ACR70 response rates up to week 48 (TP2 per-protocol set). TP2 PPS comprised of all patients completing the study until week 48 without major protocol deviations. ACR, American College of Rheumatology; PPS, per-protocol set; TP, treatment period
At week 48, the mean (SD) change from baseline in HAQ-DI score was − 0.62 (0.55) in the “continued SDZ ETN” group and − 0.66 (0.55) in the “switched to SDZ ETN” group, and the mean (SD) change from baseline in FACIT-fatigue score was 11.6 (9.7) in the “continued SDZ ETN” group and 10.6 (9.7) in the “switched to SDZ ETN” group (Additional file 1: Table S2). The proportion of patients achieving HAQ-DI in normal range (≤ 0.5) up to week 48 was comparable between “continued SDZ ETN” group (34.7%) and “switched to SDZ ETN” group (39.5%).
Safety during TP2
The median (min, max) duration of exposure to study drug was similar in both treatment groups (“continued SDZ ETN”, 162 days [8–174]; “switched to SDZ ETN”, 162 days [1–169]). The proportion of patients with at least one treatment-emergent AE (TEAE) was comparable in the “continued SDZ ETN” (n = 75, 42.9%) and “switched to SDZ ETN” (n = 63, 38.0%) groups. Nasopharyngitis was the TEAE with highest incidence in the “continued SDZ ETN” and “switched to SDZ ETN” groups (7.4% vs 5.4%) followed by upper respiratory tract infection (5.1% vs 5.4%; Table 2). Injection site reaction as a TEAE was only reported in the “switched to SDZ ETN” group (3.6%; Table 2), and all were considered by the investigators to be treatment-related.
No deaths were reported. The proportion of patients with at least one serious adverse event (SAE) was low and comparable between the two treatment groups (n = 4 in each group): “continued SDZ ETN” group: pneumonia, salivary gland cyst, tibia fracture and cystitis hemorrhagic in 1 patient [0.6%] each; “switched to SDZ ETN” group: osteomyelitis, breast cancer, colon adenoma, cardiac failure, and acute cholecystitis in 1 patient [0.6%] each. The SAEs of acute cholecystitis and osteomyelitis reported in the “switched to SDZ ETN” group were suspected to be related to the study drug by the investigator. Treatment-related TEAEs occurred in 23 (13.1%) patients in the “continued SDZ ETN” group and in 19 (11.4%) patients in the “switched to SDZ ETN” group. The treatment-related TEAEs with the highest incidence were nasopharyngitis (2.9%) in the “continued SDZ ETN” group" and injection site reactions (3.6%) in the “switched to SDZ ETN” group (Table 2).
Four (2.3%) patients in the “continued SDZ ETN” group (benign breast neoplasm, genitourinary tract neoplasm, pneumonia, cystitis hemorrhagic; 1 patient [0.6%] each) and 4 (2.4%) patients in the “switched to SDZ ETN” group (breast cancer, injection site reaction and alanine aminotransferase increase, acute cholecystitis, skin hyperpigmentation; 1 patient [0.6%] each) discontinued due to TEAEs. TEAEs of special interest were reported in 9 (5.1%) patients in the “continued SDZ ETN” group and 12 (7.2%) in the “switched to SDZ ETN” group (Additional file 1: Table S3).
Immunogenicity
Over 48 weeks, the proportion of ADA positive patients was small (< 3%) and comparable in the SDZ ETN/continued SDZ ETN groups and ETN/switched to SDZ ETN groups. After week 24, none of the patients in the switched group developed ADAs, while 4 patients in the continued SDZ ETN group had single-event, very low titer, non-neutralizing ADAs detected (Additional file 1: Table S4).
Discussion
The advent of biosimilars has increased the possibility for switching between the reference medicine and its biosimilars, and this process is being evaluated in several countries [15,16,17,18]. The 48-week results from the EQUIRA study demonstrates that switching patients from ETN to SDZ ETN did not impact the efficacy, safety, or immunogenicity of etanercept in patients with moderate-to-severe RA. All efficacy parameters including DAS28-CRP change from baseline, EULAR good/moderate response rates based on DAS28-ESR, ACR 20/50/70 response rates and all other efficacy parameters, assessed up to 48 weeks, were comparable between the two treatment groups. Although numerical differences between the two groups were observed in the ACR response rates at week 48, these differences were not clinically relevant.
Sandoz etanercept was well tolerated, and no new or unexpected safety signals were detected in this study. Overall, the incidence of TEAEs and SAEs up to week 48 were comparable between the “continued SDZ ETN” and “switched to SDZ ETN” treatment groups.
For biosimilars, immunogenicity is an important aspect for the evaluation of clinical comparability [19]. In this study, a validated state-of-the-art technique comprising a high sensitivity and drug tolerance, which enables the detection of low titer and transient ADAs was used for the detection of ADAs. A false-positive rate for confirmatory assay of 1% was applied, as recommended recently [20], instead of the commonly used 0.1% rate [21]. Previous reports have shown that applying a sensitive as well as a drug-tolerant assay may lead to a higher reported incidence of ADA compared with historical data [14].
During TP2, 4 (2.4%) patients in the continued SDZ ETN group had single event. All the measured ADAs were of low titer, near the detection limit of the applied highly sensitive method. The detected ADAs were transient and non-neutralizing, which is in line with published data showing that ETN has low immunogenicity, and ADAs, if any, appear most prominently at week 4 and disappear afterwards [22]. In addition, the ADAs detected in this study were not clinically relevant as no correlation was observed between the immunogenicity outcome and patients’ efficacy and safety.
The results are consistent with the findings from the EGALITY study in patients with plaque-type psoriasis, which also showed that switching between ETN and SDZ ETN does not have an impact on efficacy, safety, or immunogenicity of etanercept [23]. In addition, the results also support data on switching from other clinical trials in different indications [3, 15, 17, 23]. Future patient registry studies would help to further confirm the effect of switching on long-term efficacy, safety, and immunogenicity.
Conclusions
The 48-week results from the EQUIRA study demonstrate that switch from ETN to the biosimilar SDZ ETN in patients with moderate-to-severe RA did not impact the efficacy, safety, or immunogenicity of a continuous etanercept therapy.
Abbreviations
- ACR:
-
American College of Rheumatology
- ADAs:
-
Anti-drug antibodies
- AEs:
-
Adverse event
- CRP:
-
C-reactive protein
- DAS28:
-
Disease activity score including 28 joint count
- DMARDs:
-
Disease-modifying anti-rheumatic drugs
- ESR:
-
Erythrocyte sedimentation rate
- EULAR:
-
European League Against Rheumatism
- FACIT:
-
Functional Assessment of Chronic Illness Therapy
- FAS:
-
Full analysis set
- HAQ-DI:
-
Health assessment questionnaire disability index
- MedDRA:
-
Medical dictionary for regulatory activities
- PPS:
-
Per-protocol set
- RA:
-
Rheumatoid arthritis
- SAE:
-
Serious adverse event
- SDZ ETN:
-
Sandoz etanercept
- TEAE:
-
Treatment-emergent adverse event
- TP:
-
Treatment period
References
Amgen. Enbrel US Prescribing Information, 1998. Available from http://pi.amgen.com/united_states/enbrel/derm/enbrel_pi.pdf Accessed 17 Aug 2018.
von Richter O, Skerjanec A, Afonso M, Sanguino Heinrich S, Poetzl J, Woehling H, et al. GP2015, a proposed etanercept biosimilar: pharmacokinetic similarity to its reference product and comparison of its autoinjector device with prefilled syringes. Br J Clin Pharmacol. 2017;83:732–41.
Griffiths CEM, Thaci D, Gerdes S, Arenberger P, Pulka G, Kingo K, et al. The EGALITY study: a confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. the originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br J Dermatol. 2017;176:928–38.
Matucci-Cerinic M, Allanore Y, Kavanaugh A, Buch M, Schulze-Koops H, Kucharz EJ, et al. Efficacy, safety, and immunogenicity of GP2015, an etanercept biosimilar, compared to the reference etanercept in patients with moderate-to-severe rheumatoid arthritis: 24-week results from the comparative phase III, randomised, double-blind, EQUIRA study. RMD Open J. 2018; in press.
Kay J, Upchurch KS. ACR/EULAR 2010 rheumatoid arthritis classification criteria. Rheumatology (Oxford). 2012;51:5–9.
Hochberg MC, Chang RW, Dwosh I, Lindsey S, Pincus T, Wolfe F. The American College of Rheumatology 1991 revised criteria for the classification of global functional status in rheumatoid arthritis. Arthritis Rheum. 1992;35:498–502.
Fransen J, van Riel PLCM. The Disease Activity Score and the EULAR response criteria. Clin Exp Rheumatol. 2005;23:S93–9.
Prevoo ML, van ‘t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL, et al. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995;38:44–8.
Van Gestel AM, Prevoo MLL, Van ‘t Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL. Development and validation of the European league against rheumatism response criteria for rheumatoid arthritis. Arthritis Rheum. 1996;39:34–40.
Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980;23:137–45.
Bruce B, Fries JF. The Health Assessment Questionnaire (HAQ). Clin Exp Rheumatol. 2005;23:S14–8.
Cella D, Yount S, Sorensen M, Chartash E, Sengupta N, Grober J. Validation of the Functional Assessment of Chronic Illness Therapy Fatigue Scale relative to other instrumentation in patients with rheumatoid arthritis. J Rheumatol. 2005;32(5):811–9.
Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13:63–74.
Poetzl J, Arlt I, von Richter O, Wöhling H, Afonso M, Schaffar G. State-of-the-art immunogenicity evaluation in phase 3 confirmatory study (EGALITY) with etanercept biosimilar GP2015. J Eur Acad Dermatol Venereol. 2018;32:e130–2.
Glintborg B, Sorensen IJ, Loft AG, Lindegaard H, Linauskas A, Hendricks O, et al. A nationwide non-medical switch from originator infliximab to biosimilar CT-P13 in 802 patients with inflammatory arthritis: 1-year clinical outcomes from the DANBIO registry. Ann Rheum Dis. 2017;76:1426–31.
Tweehuysen L, van den Bemt BJF, van Ingen IL, de Jong AJL, van der Laan WH, van den Hoogen FHJ, et al. Subjective complaints as the main reason for biosimilar discontinuation after open-label transition from reference infliximab to biosimilar infliximab. Arthritis Rheumatol. 2018;70:60–8.
Jørgensen KK, Olsen IC, Goll GL, Lorentzen M, Bolstad N, Haavardsholm EA, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017;389:2304–16.
De Cock D, Kearsley-Fleet L, Watson K, Hyrich KL. Switching from RA originator to biosimilar in routine clinical care: early data from the British Society for Rheumatology biologics register for rheumatoid arthritis. Arthritis Rheumatol. 2017;69:3489–91.
FDA. Scientific Considerations in demonstrating biosimilarity to a reference product. Guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm291128.pdf. Accessed 20 Aug 2018
FDA. Assay development and validation for immunogenicity testing of therapeutic protein products (draft guidance, 2016). https://www.fda.gov/downloads/Drugs/Guidances/UCM192750.pdf. Accessed 31 Jan 2018
Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48:1267–81.
Emery P, Vencovský J, Sylwestrzak A, Leszczyński P, Porawska W, Baranauskaite A, et al. A phase III randomised, double-blind, parallel-group study comparing SB4 with etanercept reference product in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2017;76:51–7.
Gerdes S, Thaçi D, Griffiths CEM, Arenberger P, Poetzl J, Wuerth G, et al. Multiple switches between GP2015, an etanercept biosimilar, with originator product do not impact efficacy, safety and immunogenicity in patients with chronic plaque-type psoriasis: 30-week results from the phase 3, confirmatory EGALITY study. J Eur Acad Dermatol Venereol. 2018;32:420–7.
Acknowledgements
The authors thank all investigators (Clinicaltrials.gov: NCT02638259) and participating patients who contributed to the successful conduct of this study, and Divya Chandrasekhar and Lakshmi Venkatraman (Product Lifecycle Services-NBS, Novartis Healthcare Pvt. Ltd., Hyderabad, India) for the medical writing and editorial assistance.
Funding
This study was funded by Hexal AG, a Sandoz company.
Availability of data and materials
All data generated or analyzed are included in this article and the supplementary information files.
Author information
Authors and Affiliations
Contributions
JJ contributed to the conception of the study and interpretation of the results; MM-C contributed to the conception of the study and interpretation of the results; HS-K contributed to the conception of the study and interpretation of the results; MB contributed to the conception of the study and interpretation of the results; EJK contributed to the conception of the study and interpretation of the results; YA contributed to the conception of the study and interpretation of the results; AK contributed to the conception of the study and interpretation of the results; PY contributed to the statistical planning of the study and the interpretation of the results; GB contributed to the study design, the study conduct, and the interpretation of the results. All authors agree to be accountable for all aspects of the work and in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonization Guidelines for Good Clinical Practice and local regulations. An institutional review board or independent ethics committee at each site approved the protocol, consent form, and any other written information provided to patients or their legal representatives. All patients or their legal representatives gave written informed consent prior to study entry.
Consent for publication
Not applicable.
Competing interests
Janusz Jaworski reports trial payment for this research study and lecture fees from Sandoz. Marco Matucci-Cerinic reports no conflicts of interest. Hendrik Schulze-Koops reports personal fees from Sandoz/Hexal, during the conduct of the study, and grants and personal fees from Novartis, outside the submitted work. Maya Buch reports personal fees from Sandoz, during the conduct of the study. Eugeniusz J. Kucharz reports no conflict of interest. Yannick Allanore reports personal fees from Sandoz, grants and personal fees from Pfizer, and grants and personal fees from Roche, during the conduct of the study. Arthur Kavanaugh reports personal fees from Sandoz, outside the submitted work. Philip Young and Goran Babic are employees of Hexal AG.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Protocol deviations defined as major by category. Table S2. HAQ-DI and FACIT-fatigue scores over 48 weeks (TP2 per-protocol set). Table S3. TEAEs of special interest (TP2 safety set). Table S4. Summary of anti-drug antibodies up to week 48 using a 1% false-positive cut-point (Safety set). (DOCX 37 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jaworski, J., Matucci-Cerinic, M., Schulze-Koops, H. et al. Switch from reference etanercept to SDZ ETN, an etanercept biosimilar, does not impact efficacy, safety, and immunogenicity of etanercept in patients with moderate-to-severe rheumatoid arthritis: 48-week results from the phase III, randomized, double-blind EQUIRA study. Arthritis Res Ther 21, 130 (2019). https://doi.org/10.1186/s13075-019-1907-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-019-1907-x