
Abstract
Introduction
Takayasu arteritis (TA) is an idiopathic large-vessel vasculitis affecting the aorta and its major branches. Although the disease rarely affects children, it does occur, even in infants. The objective of this study was to evaluate the clinical features, disease activity, treatment and outcome of childhood TA in a tertiary UK centre.
Methods
We analysed a retrospective case series of children fulfilling the TA classification criteria of the European League against Rheumatism, the Paediatric Rheumatology European Society and the Paediatric Rheumatology International Trials Organisation. Data regarding demographics, clinical features, treatments and outcomes were recorded. Descriptive statistics are expressed as median and range. Fisher’s exact test was used for group comparisons. The Paediatric Vasculitis Activity Score (PVAS), Paediatric Vasculitis Damage Index (PVDI), Disease Extent Index-Takayasu (DEI.Tak) and Indian Takayasu Arteritis Activity Score (ITAS2010) were calculated retrospectively.
Results
A total of 11 children (64% female) with age at diagnosis of 11.8 (1.3 to 17) years were identified over a 23-year period. The median time to diagnosis was 17 (0 to 132) months. The most common clinical features at presentation were arterial hypertension (72.7%), systemic features (36%) and cardiovascular (45%), neurological (36%), pulmonary (27%), skin (9%), renal (9%) and gastrointestinal (9%) involvement. At presentation, PVAS was 5/63 (1 to 13); DEI.Tak was 7/81 (2 to 12) and ITAS2010 was 9/57 (6 to 20). Treatment included corticosteroids (81.8%), combined with methotrexate in most cases (72.7%). Cyclophosphamide (36.4%) and biologic agents (45.5%) were reserved for severe and/or refractory cases. PVDI at latest follow-up was 5.5/72 (3 to 15). Mortality was 27%. Young age at disease onset (<5 years old) and permanent PVDI scores ≥3 were significantly associated with mortality risk (P = 0.024).
Conclusion
TA is a rare and potentially life-threatening large-vessel vasculitis. Improved awareness of TA is essential to secure a timely diagnosis. Although the evidence base for the treatment of TA in children is weak, we found that it is essential to treat it aggressively because our data emphasise that the mortality and morbidity in the paediatric population remains high.
Similar content being viewed by others
Introduction
Takayasu arteritis (TA) is an idiopathic large-vessel vasculitis (LVV) affecting the aorta and its major branches [1]. TA has a worldwide distribution, with a reported incidence of 1.2 to 2.6 per 1 million per year in Caucasians and a 100-fold higher incidence in East Asians [2-4]. Although the disease rarely affects children, it does occur, even in infants [5-7]. Diagnostic delay is unfortunately common [5], sometimes occurring years after the acute onset of TA. Untreated TA commonly leads to cardiovascular injuries, such as aneurysm formation, concentric arterial wall fibrosis and thrombotic complications [8,9]. Because large artery biopsies are usually infeasible and laboratory investigations are neither sensitive nor specific, the diagnosis of TA is based on clinical features, including a history compatible with systemic inflammation (with or without documentation of acute-phase response) and angiographic features compatible with LVV [7,8]. Angiography refers to either formal catheter, computed tomography angiography (CTA) or magnetic resonance angiography (MRA) [10,11]. Other imaging modalities, such as 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography-computed tomography (PET-CT) [12] or, more recently, 18F-FDG PET magnetic resonance imaging (18F-FDG-PET-MRI) [10] can provide important adjunctive information, although their sensitivity and specificity for TA disease activity are not well defined, particularly in children.
Notably, there is a lack of high-quality evidence on which to base treatment recommendations in adults, and there is even less in children [13]. Hence, there is an ongoing need to describe the limited paediatric TA experience available because formal clinical trials are unlikely to come to fruition within the next decade, if ever. The majority of currently published studies describe childhood-onset disease in countries where TA is most prevalent [7,11,14]. The clinical phenotype and disease burden in the UK paediatric population are currently not well defined, even though case reports have raised concerns that TA may have the worst long-term prognosis of all the paediatric vasculitides in the United Kingdom [5,8,9]. To address this issue, we established the following aims in the present study: (1) to describe the presenting clinical, histopathological and radiological findings; (2) to explore the utility of currently available disease activity and damage instruments for evaluating TA; (3) to describe the treatments used; and (4) to describe outcomes for children with TA in a UK paediatric rheumatology tertiary referral centre.
Methods
Patient identification and ethical approval
We identified, by searching clinical and radiological databases, all the patients with a clinical diagnosis of TA seen at Great Ormond Street Hospital (GOSH) in London between January 1990 and May 2013. One patient was referred to GOSH from Sheffield Children’s Hospital; the team there (AM and MO) continued to provide shared care for this child. Case notes were reviewed retrospectively, and patients fulfilling the following classification criteria of the European League against Rheumatism, the Paediatric Rheumatology European Society and the Paediatric Rheumatology International Trials Organisation for childhood TA were included in the study [15]: angiographic abnormalities of the aorta or its main branches (mandatory criterion) plus at least one of the following four features—(1) decreased peripheral artery pulse(s) and/or claudication of extremities; (2) blood pressure difference >10 mmHg; (3) bruits over the aorta and/or its major branches; and/or (4) hypertension (related to childhood normative data) [15]. Ethical approval was granted by the Institute of Child Health/GOSH Ethics Committee for a retrospective case notes review, and no individual patient consent was therefore required.
Data gathered
Data recorded at diagnosis included sex, age, ethnicity, blood pressure and organ involvement. Disease activity was assessed retrospectively using a number of tools: the Paediatric Vasculitis Activity Score (PVAS) [16], the Disease Extent Index-Takayasu (DEI.Tak) [17] and the Indian Takayasu Arteritis Activity Score (ITAS2010) (Additional files 1, 2, 3, 4 and 5) [18]. The PVAS is a validated disease activity tool specific to paediatric vasculitis and is the only tool currently used as an outcome measure in paediatric vasculitis clinical trials. Despite that, data related to PVAS and LVV are limited. The ITAS2010 and DEI.Tak are TA-specific disease activity tools. The DEI.Tak was designed to record disease extent, including both activity and damage. The ITAS2010 provides a quantitative score of new active disease. The use of these two TA-specific tools has been validated in large adult studies for interrater reliability, convergence with Birmingham Vasculitis Activity Score (BVAS), correlation with the Physician’s Global Assessment and erythrocyte sedimentation rate (ESR) and serum C-reactive protein (CRP), ability to discriminate between active and inactive disease state at first visit, and sensitivity to change. Neither of the two TA-specific tools is currently used in routine paediatric practice, however. Therefore, the purpose of using all three in the present study was to compare and contrast them to provide insight into their relative clinical utility and to inform future studies regarding their refinement. ESR, serum CRP, cardiac enzymes (troponin where available), levels of antineutrophil cytoplasmic antibodies (ANCA) and antinuclear antibodies (ANA), and histopathological findings were reviewed. Other investigation results recorded, when available, included electrocardiograms (ECGs), echocardiograms, plain radiographs, ultrasonograms, CTA scans, MRA and MRI scans, digital subtraction angiography (DSA) scans, 18F-FDG-PET scans and dual-energy x-ray absorptiometry (DEXA) scans. The time to diagnosis and medical and surgical treatments were also recorded. Outcome measures included changes in acute-phase reactants, PVAS, ITAS2010, DEI.Tak, z-scores for height and weight at latest follow-up, cause of death and contributing factors, disease and treatment-related adverse events, and frequency of osteopenia, defined as a z-score for bone density more than 2 standard deviations below the mean for age identified with a DEXA scan. The Paediatric Vasculitis Damage Index (PVDI) [19] was calculated retrospectively. PVDI is based on a modified version of the adult Vasculitis Damage Index tool [19]. Despite the fact that the paediatric version is not yet validated, we explored its utility in the present study because it provides a systematic approach to the documentation of damage in the context of vasculitis.
Statistical analysis
Continuous variables were summarized as median and range. Categorical variables are presented as percentages and frequencies. Parameters between groups were compared using Fisher’s exact test. Wilcoxon matched-pairs signed-rank tests were used for comparisons before and after biologic therapy. Correlations between disease activity tools were assessed using Spearman’s correlation coefficient. A P-value (two-sided) <0.05 was considered significant. Statistical analyses were performed using IBM SPSS version 18 software (IBM, Armonk, NY, USA). Growth standard deviation scores (z-scores) were calculated based on UK normative data using a free downloadable Microsoft Excel add-on [20].
Results
Demographics and clinical manifestations at presentation
A total of 11 children (64% female, n = 7) aged 11.8 (1.3 to 17.0) years who fulfilled the classification criteria for TA were identified (Table 1). Eight (72%) of eleven were Caucasian, two (18%) were Asian and one (9%) was Afro-Caribbean. The median interval from first symptom onset to diagnosis was 17 (0 to 132) months. PVAS at presentation was 5/63 (1 to 13); DEI.Tak was 7/81; and ITAS2010 was 9/57 (6 to 20). The classification of TA was based on the presence of angiographic abnormalities of the aorta or its main branches (present in all 11 patients), plus at least one of the following four features: decreased peripheral artery pulse(s) (3/11, 27%), blood pressure difference >10 mmHg (3/11, 27%), bruits over the aorta and/or its major branches (6/11, 54%) and/or hypertension related to childhood normative data (8/11, 72%).
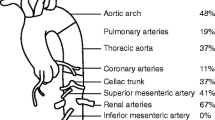
The main presenting clinical features are shown in Table 2. Arterial hypertension was the commonest presenting feature.
Established cardiac and/or large arterial injury was common at presentation. A total of 5 (45%) of 11 patients presented with bruits over the aorta and/or its major branches. Three patients (27%) had cardiomyopathy. Loss of peripheral pulses was documented in two patients (18%). Ischaemic cardiac pain was confirmed on ECGs and with elevated troponin in two patients (18%). Lower-limb claudication was present in one patient (9%), pericarditis in one patient (9%) and valvular heart disease in one patient (9%).
Two patients had major cardiac involvement at initial presentation. Patient 1 presented with a giant aneurysm of the left ventricle (LV). The aneurysm was considered traumatic because his symptoms were preceded by direct (albeit mild) trauma to his chest, and he underwent surgical repair. Over the course of the next 4 years, he had a number of surgical procedures, including surgery for LV aneurysm recurrence. Histology of the LV and aortic wall tissue indicated the presence of inflammatory cells and laminated thrombus compatible with a false aneurysm, and the aortic wall intima showed necrosis with surface fibrinous exudate. On the basis of these cardiac histological findings, he was eventually diagnosed with TA. Patient 2 presented in infancy, when she was admitted acutely to the cardiac intensive care unit with pulmonary oedema secondary to severe acute aortic and mitral valve regurgitation; the full details of this case are published elsewhere [5].
Laboratory parameters
At diagnosis for all 11 patients, the median ESR was 72 (12 to 108) mm/hr and CRP was 53 (0 to 237) mg/L. ANCA were negative for four patients tested. ANA were modestly positive (≤1:320) in two of six patients tested. For two (18%) of eleven patients who presented with ischaemic cardiac pain, there was an increase in troponin levels 8.7 (range 5.40-12; normal levels <0.04 microgram/L).
Histology
Histology results were available for five (45%) of eleven patients. Four were obtained at diagnosis: mitral valve (n = 1), left ventricle myocardium (n = 1), pericardium (n = 1) and intestinal biopsy (n = 1). One was obtained from a postmortem autopsy for one of the deceased patients (renal and cardiac tissue; patient 7). Histology showed florid vasculitis in three of the five patients, fibrous intimal thickening in four patients (periadventitial and adventitial), calcification in one, small vessel thrombosis in two, granulomas in one and neovascularisation in two. Intestinal biopsy was normal in the one single patient that had an intestinal biopsy.
Imaging studies
MRI with gadolinium contrast enhancement and/or MRA imaging studies (performed in all patients at diagnosis) revealed abnormalities in all patients as follows: arterial narrowing and stenosis (10/11; 90%), arterial dilatation or aneurysm formation (7/11; 63%), arterial wall thickening (9/11; 81%) and collateral vessels or other calibre variations (10/11; 90%). A total of eight (73%) of eleven patients had positive gadolinium contrast arterial wall enhancement, consistent with active LVV at the time the study was undertaken. CTA was performed in six patients, which revealed pseudoaneurysms and/or aneurysms (4/6), reduced and/or absent blood flow (3/6), narrowing and/or stenosis (4/6), arterial calcifications (2/6) and occlusion (1/6). DSA was performed in four (36%) of eleven patients and showed stenosis of thoracic and abdominal aortas in two patients, collaterals in two, stenosis of renal arteries in one and cerebral aneurysms in one. 18F-FDG-PET was performed in four (36%) of eleven patients and revealed avid uptake, mostly in the ascending aorta, in two of four studies.
The distribution of involved arteries according to the angiographic classification of TA [21] showed that the most common type of vessel involvement was type V (Table 3). The most common type of lesion was stenosis, which was present in nine (81%) of eleven patients, followed by aneurysms in five (45%), dilatation in three (27%) and occlusion in two (18%).
Treatment
The general therapeutic approach in our cohort was that of induction of remission (high-dose corticosteroid combined with another immunosuppressant), followed by maintenance of remission therapy (lower-dose corticosteroid combined with a maintenance immunosuppressive agent, usually methotrexate (MTX)) or institution of second-line therapy for failed induction. The medical treatments received by the patients are summarized in Additional file 6: Table S1.
Corticosteroids
Nine (81%) of eleven patients received corticosteroids as induction therapy. Five of these nine patients received prednisolone at 1 or 2 mg/kg/day; the remaining four received intravenous methylprednisolone 30 mg/kg for 3 days and were then switched to oral prednisolone 1 or 2 mg/kg/day. Two patients (patients 5 and 7) did not receive corticosteroids (or any other immunosuppressive agent), because the diagnosis of TA was made in the late stenotic phase of the disease (132 and 123 months, respectively, after the acute onset of the disease), and clinically and/or radiologically these patients had absence of disease activity.
Adjunctive immunosuppressive therapy
First-line therapies were as follows: Four (36%) of eleven patients received weekly subcutaneous MTX combined with corticosteroids three (27%) received cyclophosphamide at induction, two (18%) received intravenous immunoglobulin (2 g/kg) and one (9%) received azathioprine (2 to 3.7 mg/kg/day).
Regarding second-line therapy, five (45%) of eleven patients were switched to MTX at a median of 8 (5 to 132) months from diagnosis. Overall, there was evidence of ongoing disease activity at that time as indicated by all three disease activity measures: PVAS of 7/63 (2 to 11), ITAS2010 of 11/57 (5 to 14/), DEI.Tak of 8/81 (8 to 15/81). At that time point, median ESR was 11 (2 to 56) mm/hr and median CRP was 9.5 (1 to 20) mg/L. The median dose of oral prednisolone for these patients was 0.3 (0.2 to 0.75) mg/kg/day. Patient 11, who had been on MTX for 7 months as a first-line therapy, was switched to cyclophosphamide because she had progressive disease detected by MRA.
Biologic therapies
Biologic therapy was initiated for 6 (54%) of 11 patients at 11 (4 to 156) months from diagnosis. The median ESR was 63 (3 to 115) mm/hr, and the median CRP was 37 (3 to 140) mg/L. Anti–tumour necrosis factor alpha (anti-TNFα) therapy was the first class of biologic agent chosen in all six cases. Three patients received adalimumab (24 mg/m2 fortnightly if body weight was <30 kg, 40 mg fortnightly if >30 kg) as the first biologic. Three patients received infliximab (6 mg/kg intravenously monthly) as the first biologic, including patient 2, who was later converted to adalimumab because of difficulties with venous access. Patient 3 was switched from adalimumab to tocilizumab therapy at 14 months from diagnosis because he developed severe myalgia and arthralgia with raised inflammatory markers. The median dose of prednisolone at time of biologic therapy initiation was 0.2 (0.1 to 2) mg/kg/day, and, at a median of 2.5 (2 to 28 months) of biologic therapy, it was 0 (0 to 0.6) mg/kg/day (P = 0.41). In addition, in response to this biologic therapy, PVAS changed from 9.5/63 (4 to 24) to 0/63 (0 to 21) (P = 0.312), ITAS2010 changed from 6/57 (4 to 18) to 0/57 (0 to 18) (P = 0.104) and DEI.Tak changed from 3/81 (1 to 20) to 0/82 (0 to 20) (P = 0.187). Of note, in patient 3, the PVAS score was 0/63 at the time of initiation of biologic therapy, indicating absence of clinical disease activity; however, the ITAS2010 and DEI.Tak scores were 4/57 and 2/81, respectively. Regarding patient 3, following an initially promising response to tocilizumab, the disease relapsed 12 months later, and, at the time of this writing, this patient was receiving rituximab.
Other medical therapy
Additional file 6: Table S1 (available online) summarizes the adjunctive medical therapy received. Seven (63%) of eleven patients required a median of three (range, from two to six) antihypertensive medications, five received antiplatelet doses of aspirin and two received anticoagulation (warfarin after initial heparinisation). Patient 11 required treatment with sildenafil and epoprostenol infusion for pulmonary arterial hypertension that developed 4 months following diagnosis.
Surgical and endovascular management
Five patients required surgical and/or endovascular treatment. These procedures are summarized in Table 1.
Outcomes
The median length of follow-up was 16 (6 to 168) months. At the last follow-up, PVAS was 0/63 (0 to 21), ITAS2010 was 0/57 (0 to 18) and DEI.Tak was 0/81 (0 to 20). ESR was 5 (2 to 20) mm/hr, and CRP was 5 (range 1 to 12) mg/L. Seven of eleven patients were in remission at latest follow-up, as indicated by normal CRP and ESR values and a PVAS of 0 on two consecutive measurements 1 month apart [16]. The following corticosteroid-associated side effects were documented in 72.7% (8/11) of the patients: Cushing’s syndrome (5/8; 62%), osteoporosis (2/8; 25%), skeletal fracture (1/8; 12.5%) and hip avascular necrosis (1/8; 12.5%).
The median z-score for height at diagnosis was −0.10 (−1.39 to 3.79), and at the last follow-up it was −0.89 (−5.6 to 1.58) (P = 0.019). The median z-score for weight at diagnosis was +1.13 (−0.96 to 3.46), and it was +0.47 (−9.21 to 2.57) at the last follow-up (P = 0.019).
Two patients had pancytopenia, which was related to treatment with MTX and cyclophosphamide, respectively. Both recovered with cessation of therapy. Patient 11 developed Candida and Pseudomonas sepsis whilst in intensive care for multiorgan failure.
The median PVDI at latest follow-up was 5.5/72 (3 to 15). When the items that had been present for more than 3 months at one time but had resolved at latest follow-up [19] were removed, the PVDI changed to 3/72 (0 to 10).
Three of eleven patients died during follow-up (27% mortality). Patient 7 presented with acute cardiomyopathy, and no clear diagnosis was made at initial presentation. Approximately 6 years later, the diagnosis of TA was made based on the loss of peripheral pulses and lower-limb claudication. Patient 7 did not receive any immunosuppressive treatment, as he was considered to be in the late stenotic phase of the disease. Four years after receiving his diagnosis, he collapsed suddenly and died. The postmortem examination revealed severe LV hypertrophy and 70% occlusion of the coronary arteries. His abdominal aorta was narrowed and had a patchy and sparse chronic inflammatory cell infiltrate and evidence of active inflammation within the vasa vasorum of the distal aorta. His final PVDI was 5/72.
Patient 6 was diagnosed with TA at screening after her sister had died as a result of TA [9]. Patient 6 received treatment with corticosteroids, azathioprine, MTX and adalimumab. She accrued significant damage over time. Her final PVDI score was 10/72. She died suddenly as a result of acute aortic rupture 15 years following diagnosis.
Patient 11 was diagnosed with coarctation of the aorta during her first 5 years of life and had undergone surgical repair with patch augmentation. She was followed up for severe arterial hypertension for the next 3 years. At 3 years after her diagnosis, she presented with acute respiratory failure, pericarditis, no palpable pulses on the lower limbs due to critical stenosis of the whole aorta and the main branches, anuria and hypertension. She died as a result of progressive multiorgan failure, despite receiving escalating immunosuppression (high-dose intravenous methylprednisolone, MTX, cyclophosphamide, infliximab). Her final PVDI was 12/72.
Correlations between Takayasu arteritis vasculitis disease activity tools
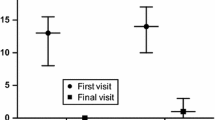
Figure 1 shows the correlations among the various TA disease assessment instruments at the time of diagnosis and follow-up. PVAS correlated strongly with both ITAS2010 (r = 0.66; P = 0.030) and DEI.Tak (r = 0.86; P = 0.0012) at diagnosis and latest follow-up (ITAS2010: r = 0.98, P = 0.0001; DEI.Tak: r = 1, P = 0.0001).

Correlations between Takayasu arteritis disease activity measure tools. (A) Correlations at time of diagnosis. (B) Correlations at time of latest follow-up. DEI.Tak, Disease Extent Index-Takayasu; ITAS2010, Indian Takayasu Arteritis Activity Score; PVAS, Paediatric Vasculitis Activity Score. Total possible scores: PVAS: 0 to 63, ITAS2010: 0 to 57 and DEI.Tak: 0 to 81. Higher scores reflect higher disease activity for all tools [16-18].
Factors associated with mortality
Age at disease onset ≤5 years was significantly associated with mortality (P = 0.024). Permanent damage items on PVDI scoring at latest follow-up ≥3/72 were also significantly associated with mortality (P = 0.024). There was no statistical association between mortality and time to diagnosis >12 months (P = 0.545), cardiac involvement (P = 0.18), endovascular procedures within 12 months of diagnosis (P = 0.545) or need for biologic therapy (P = 1) (Additional file 7: Table S2).
Discussion
In our present retrospective study, we report the largest UK cohort so far of paediatric patients with TA. This study is the first to systematically apply three different vasculitis disease activity scores and to explore damage accrual using the PVDI. We observed a significant delay in diagnosis (up to 132 months) due to the insidious and non-specific presentation of TA. The burden of disease and its therapy was high, as depicted by PVDI scores at latest follow-up. Biologic therapies were used in 54% of patients for therapy-resistant disease. Mortality was high at 27%. Disease onset before 5 years of age and persistent damage items providing PVDI scores ≥3 were significantly associated with mortality.
Similarly to other case series, the most common presenting feature of TA in our study was arterial hypertension (70%), but notably in the absence of other systemic symptoms, which may partly explain the diagnostic delay [6-8,14]. We report a higher frequency of cardiac and neurological involvement in our sample compared with other published paediatric TA cohorts, perhaps reflecting the severity of the cases referred to our tertiary care centre. The frequency of cutaneous and musculoskeletal symptoms were similar to other reports, however [6-8,14]. We emphasize that severe cardiac involvement can be the first presenting feature of LVV in children (3 of 11 patients in our study).
Histology for the majority of our cases revealed bland lymphocytic inflammation with some neovascularisation, but absence of granulomas, in contrast to histological studies of adult-onset TA, where granulomas are often a major feature [22]. Because mononuclear infiltration of the adventitia with perivascular cuffing of the vasa vasorum is common in the early phase of TA [22,23], we suggest that one reason why granulomas may not be so conspicuous in children is that these patients are likely to have had shorter disease duration at the time of tissue sampling.
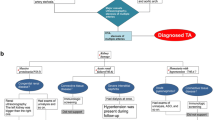
Assessment of disease activity in TA remains challenging, particularly in children [8]. Acute-phase reactants lack sufficient sensitivity to be clinically reliable; most clinical manifestations are non-specific, and vascular injury progresses insidiously [8]. Active vasculitis may still be present, even in the absence of an acute-phase response and with negative 18F-FDG PET-CT scans as in patient 7, in whom autopsy results demonstrated chronic active LVV. On the basis of our experiences and those reported by others [10], we provide an approach to the use of imaging for the diagnosis and monitoring of TA in paediatric patients (Figure 2). In particular, MRI techniques coregistered with PET have the potential to improve evaluation of TA, both at diagnosis and during follow-up. The role of circulating biomarkers such as pentaxin 3 (PTX3) level is currently unclear [24], but PTX3 could have a role if validated in a clinical context.

Recommended imaging algorithm for children with Takayasu arteritis. CT, Computed tomography; CTA, Computed tomography angiography; MRA, Magnetic resonance angiography; MRI, Magnetic resonance imaging; PET, Positron emission tomography; TA, Takayasu arteritis; USS, Ultrasound.
Two specific tools to assess TA disease activity and extent have been validated in adults: ITAS2010 and DEI.Tak [17,18]. These tools were derived from the BVAS, but with particular emphasis on cardiovascular manifestations because these predominate in TA. Although they are not validated in children, we explored their utility in comparison with the PVAS, the only validated vasculitis activity score for children. Perhaps unsurprisingly, activity scores using both TA-specific tools were consistently higher than those with PVAS, as the former allow more detailed assessment of the extent of vascular involvement. We note, however, the strong correlation of measurements between the PVAS versus ITAS2010 and DEI.Tak tools at both diagnosis and follow-up, suggesting that, overall, all three tools consistently reflect TA disease activity. Development of these TA-specific tools to include modifications for age-specific items such as body weight and arterial hypertension, as well as prospective validation in children with TA, is now warranted. In addition, using standard definitions of remission employed in clinical trials for other vasculitides, a total of 7 of 11 patients fulfilled remission criteria at their latest follow-up examination. We acknowledge, however, that grumbling disease activity may not be sufficiently captured by these clinical indices. Further studies designed to refine and validate reliable definitions of disease remission and response to therapy in TA are now needed.
We also explored the utility of the prototypic PVDI [19] for the first time in an attempt to systematically assess damage accrual. All patients scored high for items related to cardiovascular morbidity, and 8 of 11 scored positively for treatment-related morbidity items. Notably, only permanent damage items detected using PVDI were associated with mortality.
Corticosteroids combined with MTX or cyclophosphamide were the standard induction therapy for our patients. More than one-half (54%) required addition of biologic therapy (within the latter 10 years of the study, when these treatments became available) because of a severe and refractory disease course. In addition to anti-TNFα agents, promising results have been reported with anti-interleukin-6 therapy (tocilizumab) for adults with TA [25-27]. Tocilizumab was used for one of our patients, who went on to receive rituximab for inadequate disease control. There has never been a controlled trial for the treatment of TA. Rare disease trial designs that have recently been adapted for other rare childhood vasculitides (for example, the open-label randomised controlled trial of mycophenolate mofetil versus cyclophosphamide for the induction of remission of childhood polyarteritis nodosa (ISRCTN 75434563)) should also be considered for children with TA [28].
We report a mortality rate of 27%, which is in line with mortality rates reported by others (ranging from 16% to 40%) [6,7,14]. Disease mortality was related to younger age at presentation (P = 0.024). This finding is unsurprising, given that these children had a prolonged, severe disease course and extensive stenotic disease well before adolescence. Whether these patients represent a distinct group of early-onset severe disease due to an as yet undefined genetic disorder remains to be established [9], and this is an area of ongoing study by our group.
Our study is limited by all the confounding factors associated with any retrospective case series. We acknowledge the small sample size and lack of heterogeneity of our cohort. There is also a possibility that referral bias resulted in the more severe spectrum of the disease observed in our patients.
Conclusions
TA in children is a rare but potentially life-threatening condition. Improved awareness of TA is essential to securing an early diagnosis. LVV should be considered in any young child presenting with hypertension with or without elevated acute-phase reactants. Other clinical features are unfortunately non-specific, but the case for suspected TA is even stronger in a patient with hypertension in the presence of fever, weight loss, headaches, arthralgia, fatigue and/or bruits. In the future, use of the currently available clinical scores explored herein may be beneficial for monitoring response to therapy. Undoubtedly, however, imaging is particularly important for the diagnosis and monitoring of LVV (Figure 2). Although the evidence base for the treatment of TA in children is weak, it is essential to treat TA aggressively as soon as the diagnosis is secured to reduce mortality and morbidity.
Abbreviations
- 18F-FDG:
-
18F-fluorodeoxyglucose
- ANA:
-
Antinuclear antibodies
- ANCA:
-
Antineutrophil cytoplasmic antibodies
- BVAS:
-
Birmingham Vasculitis Activity Score
- CA:
-
Coronary artery
- CRP:
-
C-reactive protein
- CT:
-
Computed tomography
- CTA:
-
Computed tomography angiography
- DEI.Tak:
-
Disease Extent Index-Takayasu
- DEXA:
-
Dual-energy X-ray absorptiometry
- DSA:
-
Digital subtraction angiography
- ECG:
-
Electrocardiogram
- ESR:
-
Erythrocyte sedimentation rate
- GFR:
-
Glomerular filtration rate
- GOSH:
-
Great Ormond Street Hospital
- hpf:
-
High-powered field
- ITAS2010:
-
Indian Takayasu Arteritis Activity Score
- LIMA:
-
Left internal mammary artery
- LV:
-
Left ventricle
- LVV:
-
Large-vessel vasculitis
- MRA:
-
Magnetic resonance angiography
- MRI:
-
Magnetic resonance imaging
- MTX:
-
Methotrexate
- PET:
-
Positron emission tomography
- PVAS:
-
Paediatric Vasculitis Activity Score
- PVDI:
-
Paediatric Vasculitis Damage Index
- RBC:
-
Red blood cells
- SMA:
-
Superior mesenteric artery
- TA:
-
Takayasu arteritis
- TNF-α:
-
Tumour necrosis factor alpha
- USS:
-
Ultrasound
- VDI:
-
Vasculitis Damage Index
References
Arend WP, Michel BA, Bloch DA, Hunder GG, Calabrese LH, Edworthy SM, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990;33:1129–34.
Watts R, Al-Taiar A, Mooney J, Scott D, MacGregor A. The epidemiology of Takayasu arteritis in the UK. Rheumatology. 2009;48:1008–11.
Jain S, Kumari S, Ganguly NK, Sharma BK. Current status of Takayasu arteritis in India. Int J Cardiol. 1996;54:S111–6.
Hall S, Barr W, Lie JT, Stanson AW, Kazmier FJ, Hunder GG. Takayasu arteritis: a study of 32 North American patients. Medicine. 1985;64:89–99.
Singh N, Hughes M, Sebire N, Brogan P. Takayasu arteritis in infancy. Rheumatology. 2013;52:2093–5.
Morales E, Pineda C, Martínez-Lavin M. Takayasu’s arteritis in children. J Rheumatol. 1991;18:1081–4.
Cakar N, Yalcinkaya F, Duzova A, Caliskan S, Sirin A, Oner A, et al. Takayasu arteritis in children. J Rheumatol. 2008;35:913–9.
Watson L, Brogan P, Peart I, Landes C, Barnes N, Cleary G. Diagnosis and assessment of disease activity in Takayasu arteritis: a childhood case illustrating the challenge. Case Rep Rheumatol. 2014;2014:603171.
Morishita KA, Rosendahl K, Brogan PA. Familial Takayasu arteritis - a pediatric case and a review of the literature. Pediatr Rheumatol Online J. 2011;9:6.
Mavrogeni S, Dimitroulas T, Chatziioannou SN, Kitas G. The role of multimodality imaging in the evaluation of Takayasu arteritis. Semin Arthritis Rheum. 2013;42:401–12.
Aluquin VPR, Albano SA, Chan F, Sandborg C, Pitlick PT. Magnetic resonance imaging in the diagnosis and follow up of Takayasu’s arteritis in children. Ann Rheum Dis. 2002;61:526–9.
Webb M, Chambers A, Al-Nahhas A, Mason JC, Maudlin L, Rahman L, et al. The role of 18F-FDG PET in characterising disease activity in Takayasu arteritis. Eur J Nucl Med Mol Imaging. 2004;31:627–34.
Unizony S, Stone JH, Stone JR. New treatment strategies in large-vessel vasculitis. Curr Opin Rheumatol. 2013;25:3–9.
Jain S, Sharma N, Singh S, Bali HK, Kumar L, Sharma BK. Takayasu arteritis in children and young Indians. Int J Cardiol. 2000;75:S153–7.
Özen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, et al. Paediatric Rheumatology International Trials Organisation (PRINTO). EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: final classification criteria. Ann Rheum Dis. 2010;69:798–806.
Dolezalova P, Price-Kuehne FE, Özen S, Benseler SM, Cabral DA, Anton J, et al. Disease activity assessment in childhood vasculitis: development and preliminary validation of the Paediatric Vasculitis Activity Score (PVAS). Ann Rheum Dis. 2013;72:1628–33.
Aydin SZ, Yilmaz N, Akar S, Aksu K, Kamali S, Yucel E, et al. Assessment of disease activity and progression in Takayasu’s arteritis with Disease Extent Index-Takayasu. Rheumatology. 2010;49:1889–93.
Misra R, Danda D, Rajappa SM, Ghosh A, Gupta R, Mahendranath KM, et al. Development and initial validation of the Indian Takayasu Clinical Activity Score (ITAS2010). Rheumatology. 2013;52:1795–801.
Dolezalova P, Wilkinson N, Brogan PA, Anton J, Benseler SM, Brunner J, et al. Paediatric Vasculitis Damage Index: a new tool for standardised disease assessment [SAT0286]. Ann Rheum Dis. 2014;73 Suppl 2:696–7. doi:10.1136/annrheumdis-2014-eular.5893.
Pan H, Cole TJ. LMSgrowth [Microsoft Excel add-in to access growth references based on the LMS method]. Version 2.76. Harlow Healthcare, Tyne & Wear, UK. 2011. http://www.healthforallchildren.com/lmsgrowth-download/. Accessed 5 Mar 2015.
Moriwaki R, Noda M, Yajima M, Sharma BK, Numano F. Clinical manifestations of Takayasu arteritis in India and Japan new classification of angiographic findings. Angiology. 1997;48:369–79.
Hotchi M. Pathological studies on Takayasu arteritis. Heart Vessels. 1992;7:11–7.
Virmani R, Burke AP. Pathologic features of aortitis. Cardiovasc Pathol. 1994;3:205–16.
Tombetti E, Di Chio M, Sartorelli S, Papa M, Salerno A, Bottazzi B, et al. Systemic pentraxin-3 levels reflect vascular enhancement and progression in Takayasu arteritis. Arthritis Res Ther. 2014;16:479.
Bravo Mancheño B, Perin F, del Mar Rodríguez Vázquez del Rey M, García Sánchez A, Alcázar Romero PP. Successful tocilizumab treatment in a child with refractory Takayasu arteritis. Pediatrics. 2012;130:e1720–4.
Unizony S, Arias-Urdaneta L, Miloslavsky E, Arvikar S, Khosroshahi A, Keroack B, et al. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res (Hoboken). 2012;64:1720–9.
Salvarani C, Magnani L, Catanoso M, Pipitone N, Versari A, Dardani L, et al. Tocilizumab: a novel therapy for patients with large-vessel vasculitis. Rheumatology. 2012;51:151–6.
Hampson LV, Whitehead J, Eleftheriou D, Brogan P. Bayesian methods for the design and interpretation of clinical trials in very rare diseases. Stat Med. 2014;33:4186–201.
Acknowledgements
DE acknowledges research support from Arthritis Research UK (grant 20164). PD was supported by grant IGA NT/14149-3.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DE conceived the study and participated in its design and coordination, collected and analysed data and drafted the manuscript. GV participated in the design of the study, collected and analysed data and drafted the manuscript. PD, AM and MO participated in the design and coordination of the study, collected and analysed data and helped to draft the manuscript. PB conceived the study, participated in its design and coordination, collected data and analysed data and helped to draft the manuscript. All authors have agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors read and approved the final manuscript.
Despina Eleftheriou and Giulia Varnier contributed equally to this work.
Additional files
Additional file 1:
Disease Extent Index for Takayasu Arteritis tool.
Additional file 2:
Indian Takayasu Arteritis Activity Score tool.
Additional file 3:
Indian Takayasu Arteritis Activity Score tool glossary.
Additional file 4:
Paediatric Vasculitis Activity Score tool.
Additional file 5:
Paediatric Vasculitis Activity Score tool glossary.
Additional file 6: Table S1.
Treatment for 11 children with Takayasu arteritis in a UK-based centre. †In case of failed induction. Pred = oral prednisolone; mepred = methylprednisolone (30 mg/kg/day intravenously three times per day, unless stated otherwise); MTX = methotrexate (15 mg/m2wk, unless otherwise stated); CYC = cyclophosphamide; iv = intravenous; sc = subcutaneous; IVIG = intravenous immunoglobulin; PVAS = Paediatric Vasculitis Activity Score; DEI.TAK = Disease Extent Index-Takayasu; ITAS2010 = Indian Takayasu Arteritis Activity Score 2010; asp = aspirin (2 to 5 mg/kg/day); inflix = infliximab (6 mg/kg intravenously monthly unless stated otherwise); adalim = adalimumab (24 mg/m2; maximum dose 40 mg, sc every 2 weeks); NA = not applicable. For other treatments where dose is not stated, standard paediatric doses were used.
Additional file 7: Table S2.
Factors associated with mortality in a cohort of 11 children with Takayasu arteritis seen in a UK tertiary referral centre. Fisher’s exact test was used for comparison between groups. PVDI = Paediatric Vasculitis Damage Index [19]. P-values <0.05 (two-sided) were considered significant.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Eleftheriou, D., Varnier, G., Dolezalova, P. et al. Takayasu arteritis in childhood: retrospective experience from a tertiary referral centre in the United Kingdom. Arthritis Res Ther 17, 36 (2015). https://doi.org/10.1186/s13075-015-0545-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-015-0545-1