
Abstract
Background
Autism spectrum disorder (ASD) is a neurodevelopmental condition characterized by impaired social and communication skills, restricted interests, and repetitive behaviors. The prevalence of ASD among children in Qatar was recently estimated to be 1.1%, though the genetic architecture underlying ASD both in Qatar and the greater Middle East has been largely unexplored. Here, we describe the first genomic data release from the BARAKA-Qatar Study—a nationwide program building a broadly consented biorepository of individuals with ASD and their families available for sample and data sharing and multi-omics research.
Methods
In this first release, we present a comprehensive analysis of whole-genome sequencing (WGS) data of the first 100 families (372 individuals), investigating the genetic architecture, including single-nucleotide variants (SNVs), copy number variants (CNVs), tandem repeat expansions (TREs), as well as mitochondrial DNA variants (mtDNA) segregating with ASD in local families.
Results
Overall, we identify potentially pathogenic variants in known genes or regions in 27 out of 100 families (27%), of which 11 variants (40.7%) were classified as pathogenic or likely-pathogenic based on American College of Medical Genetics (ACMG) guidelines. Dominant variants, including de novo and inherited, contributed to 15 (55.6%) of these families, consisting of SNVs/indels (66.7%), CNVs (13.3%), TREs (13.3%), and mtDNA variants (6.7%). Moreover, homozygous variants were found in 7 families (25.9%), with a sixfold increase in homozygous burden in consanguineous versus non-consanguineous families (13.6% and 1.8%, respectively). Furthermore, 28 novel ASD candidate genes were identified in 20 families, 23 of which had recurrent hits in MSSNG and SSC cohorts.
Conclusions
This study illustrates the value of ASD studies in under-represented populations and the importance of WGS as a comprehensive tool for establishing a molecular diagnosis for families with ASD. Moreover, it uncovers a significant role for recessive variation in ASD architecture in consanguineous settings and provides a unique resource of Middle Eastern genomes for future research to the global ASD community.
Similar content being viewed by others

Background
Autism spectrum disorder (ASD) is a neurodevelopmental condition characterized by impaired social interactions, deficits in communication, restricted interests, and repetitive behaviors [1]. ASD often co-occurs with other conditions, including intellectual disability (ID), attention-deficit hyperactivity disorder (ADHD), epilepsy, and gastrointestinal (GI) problems [2]. Various factors, including genetic, epigenetic, environment, and hormonal changes contribute to the broad phenotypic spectrum of ASD. The high heritability of ASD (70–90% based on twin studies) [3] and the increased relative risk to siblings (10–20-fold) suggest that genetic factors play a prominent role in ASD etiology [4].
Advances in both genomic technologies and ASD phenotyping have improved our understanding of the genetic architecture of ASD. Studies of genomic data at scale have revealed over hundreds genes and variants to be associated with ASD, disrupting key biological processes such as neurotransmission, synapse function, chromatin remodeling, cortical development, and metabolism [4, 5].
De novo variation in coding regions, including SNVs, small insertions or deletions (indels), and structural variants (SVs), together account for 10–30% of simplex ASD cases [2, 6, 7]. Recently, other variant classes such as TREs and mitochondrial variants have been shown to contribute to ASD susceptibility in large population cohorts [8,9,10]. Furthermore, the use of statistical methods such as the transmission and de novo association analysis (TADA) helped identify risk genes by combining both de novo and transmitted SNVs/Indels [11]. A recent study applied TADA analysis and highlighted 134 dominant genes to be ASD-associated with false discovery rate < 0.1 [12].
There has been growing evidence implicating recessive variation in ASD susceptibility, especially in consanguineous settings (approximately 5% of all ASD cases) [13, 14]. Rare homozygous loss-of-function (LoF) variants have been described in several genes such as CA2, DDHD1, FEV, NSUN2, PAH, SLC1A1, and USH2A [15, 16]. Despite these discoveries, recessive causes of ASD generally form a minority of the overall genetic architecture of ASD among large cohorts published to date, estimated at around 1.1% in MSSNG and 0.3% in the SSC datasets [10]. Additionally, recent studies that focused on families with high consanguinity have demonstrated a higher rate of recessive causes, e.g., 39% [17], suggesting the recessive burden in ASD is yet to be explored among global consanguineous populations.
Successful molecular diagnosis of individuals with ASD brings several benefits allowing earlier behavioral interventions, assessment of familial recurrence risk (low in case of de novo mutation) as well as informing more precise interventions. Nevertheless, despite the improvements in understanding the genetics of ASD, most discoveries have been only produced in certain geographical areas, which limits the diversity of ethnic backgrounds that can benefit from research. In ASD research, for instance, people of non-European ancestry are still significantly underrepresented [10], with those of Middle Eastern origin being among the most underrepresented globally.
ASD research has recently received a lot of attention in Qatar. The incidence of ASD in Qatar is estimated to be 1 in 87 (1.1%) [18], which is relatively similar to the global estimates in different populations [19, 20]; however, the genetic architecture of ASD in Arab world remains poorly explored. The BARAKA study (Building a Resource for the Advancement of Knowledge of Autism in Qatar) aims to establish a national resource on ASD research, consisting of a biorepository of samples and data on patients at Sidra Medicine broadly consented for research. The repository hosts extensive clinical and questionnaire data on each individual including electronic health records (EHR), aliquots of whole blood, plasma, cells, RNA, saliva, and microbiome samples. Importantly, most patients were consented to be recontacted in the future. This resource is expected to be a valuable resource contributing to regional and global efforts investigating genetic and environmental determinants of ASD.
Herein, we describe the results of BARAKA-WGS analysis of 100 families (372 subjects), where we comprehensively investigate the genetic architecture (including dominant/recessive, nuclear/mitochondrial variants) contributing to ASD. Being the first comprehensive genomic study of ASD from the Middle East, this sets an important baseline for understanding the architecture of this complex condition in highly consanguineous populations.
Methods
Cohort description and phenotyping
A total of 100 families (372 total individuals, including 104 individuals with ASD plus their parents and unaffected siblings) were enrolled from Sidra Medicine’s various pediatric clinics (Developmental Pediatrics, Child and Adolescent Psychiatry, Adolescent Medicine) as part of the BARAKA-Qatar study cohort. Most of the families where simplex (98/100) and only two families where multiplex families both with 3 affected siblings each. The majority of families were of Arab descent (58%), followed by South Asian (25%), European (7%), African (5%), and other ethnicities. Children with known karyotyping abnormalities, Fragile X syndrome, and Rett syndrome were excluded. ASD diagnosis was made following standard autism diagnostic measures (DSM-V). The study was approved by the institutional review board (IRB) of Sidra Medicine (IRB No. 1500767), and written informed consent was obtained from all participants (the full description of the cohort phenotypes is presented in Additional file 1: Table S1 and Additional file 2: Figure S1). De novo SNVs/SVs and compound heterozygous variants analysis were performed only on complete trios (79% of families).
WGS and variant detection
Whole blood samples were collected from individuals with ASD and family members. Total genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen sciences LLC, Germantown, MD, USA) according to the manufacturer’s instructions. DNA samples were processed at Sidra Medicine as previously described [21]. Briefly, samples were sequenced (150 bp paired-end reads) using Illumina HiSeq X to a minimum depth of 30 × , and reads were aligned to GRCh37/hg19 using BWA version 0.7.10 [22]. Sequence-level variants were detected with GATK version 3.3 using the best practices pipeline [23]. VCF files were annotated using the SnpEff/SnpSift tool [24] by adding allele frequencies from variant databases (1000 Genomes Project [25], gnomAD [26], and ExAC [27], and Qatar-genome project (QGP)). De novo variants were detected in complete trios (n = 79) using a combination of three tools (VarScan [28], RUFUS [29], and FreeBayes [30]) as previously described [31]. All variants reported in this study were lifted over to GRCh38/hg38 using Broad Institute liftover tool (https://liftover.broadinstitute.org) [32].
SNV and indel analysis
Quality filtration
We retained variants that met all the following criteria: (i) flagged as “PASS” all GATK filters, (ii) genotype quality (GQ) ≥ 10, (iii) read depth ≥ 20, (iv) allele fraction between 0.2 and 0.8 (for heterozygous variants), and (v) not present in low-complexity regions. Rare variants were defined as those with minor allele frequencies (MAF) < 1% in all general databases such as 1000G, gnomAD, ExAC, QGP, and an internal database of > 35,000 alleles sequenced as part of various projects at Sidra Medicine. To determine the level of consanguinity from our cohort, we used KING for pair-wise measurement of relationships (–-kinship command, with a cutoff of ≥ 0.044) (Additional file 2: Figure S2) [33] and calculated inbreeding coefficient (F) for per-sample using plink1.9 (–het command with cutoff > 0.1) (Additional file 2: Figure S3) [34].
Variant prioritization
De novo, homozygous, compound heterozygous, and X-linked recessive variants that are rare and coding were considered to be potentially pathogenic if they met the following criteria: (i) LoF effect on the protein (stop gain, frameshift deletion, frameshift insertion, or canonical splice site variation) or (ii) damaging missense variants (Dmiss), defined as variants deemed deleterious by at least 5 in silico prediction tools. These tools included CADD (threshold for deleteriousness ≥ 10) [35], SIFT (deleterious) [36], PolyPhen2-HDIV (probably-damaging or possibly damaging) [37], PolyPhen2-HVAR (probably-damaging or possibly damaging) [37], LRT (deleterious) [38], MutationAssessor (high or medium) [39], MutationTaster (deleterious) [40], MPC score (≥ 1) [41], and PROVEAN (deleterious) [42].
Gene constraint was assessed using the gnomAD pLI score for dominant variants and pRec score for recessive variants. Variants were also screened for any phenotypic association in the database of Online Mendelian Inheritance in Man (OMIM) [43]. Variants found in genes causing phenotypes relevant to ASD (such as developmental delay (DD), intellectual disability, etc.) were curated based on American College of Medical Genetics (ACMG) guidelines [44] using Franklin and InterVar (Available online: https://franklin.genoox.com, [45]). (Note: For all de novo variants, PS2 criteria were manually adjusted).
Known ASD/NDD panel genes/regions
To further prioritize likely ASD-associated variants, we identified variants impacting genes in a list of known neurodevelopmental disorder (NDD)/ASD genes, which included the Genomics England NDD/autism panel genes and Simons Foundation Autism Research Initiative (SFARI) genes with a score of 1. This panel contained 1714 genes (634 dominant, 942 biallelic, and 138 X-linked; Additional file 1: Table S2). CNVs that overlap previously published list of genes/regions described as pathogenic to ASD [12] or known NDD/ASD genes were defined as “known” CNVs. In addition, we investigated TREs that affect known ASD genes from the recently reported list (57 genes) in ASD [8].
Novel genes/regions associated with ASD/NDD
In addition to identifying damaging variants in known genes, we flagged damaging de novo and rare homozygous variants (LoF, Dmiss) in novel candidate genes. For de novo variants, we leveraged other ASD cohorts (MSSNG, SSC, and SPARK) to look for additional individuals with evidence in these same genes. For homozygous variants, we used an additional filter of genes with high pRec scores (> 0.9). We also considered de novo or homozygous CNVs in novel genes/regions. In both cases, we also looked in other ASD cohorts for additional individuals with variants of the same category and inheritance patterns in the same gene to strengthen evidence for causality.
CNV detection and analysis
CNV detection was performed using a pipeline comprising multiple algorithms: CNVnator [46], DELLY [47], ERDS [48], Manta [49], Speedseq [50], and SvABA [51]. We retained only CNVs detected by at least two tools to increase specificity. We then merged CNVs detected by the 6 tools if they were of the same type and their start and end coordinates were within 500 bp window. First we merged CNVs within each individual to generate a unique set of CNVs per-sample and subsequently across individuals to create a population-level variant file using Survivor (version 1.0.7) [52], which was then annotated using AnnotSV (version 2.2) [53]. De novo and homozygous CNVs were identified using custom scripts with the following additional allele frequency filters (allele frequency < 0.1% for de novo and < 1% for homozygous) from global biobank SVs studies [54,55,56]. After filtering, we visualized CNVs using samplot (version 1.0.17) [57].
Variant validation
We selected 12 de novo variants to confirm using Sanger sequencing as previously described [58]. As a further quality check, we used digital-droplet PCR (ddPCR) to validate a subset of CNVs, as described previously [59]. We successfully confirmed all de novo SNVs and CNVs (Additional file 2: Figure S4)
Calling of tandem repeats and expansions
Genome-wide detection of tandem repeats expansions (TREs) was performed using ExpansionHunter Denovo (EHdn) [60], which uses anchored in-repeat reads to estimate the size and location of tandem repeats, using the same pipeline as previously described [8].
Mitochondrial variant calling
Variant calling in mitochondrial DNA was performed using Mutect2 (GATK v4.1.2.0) [23] using the newly implemented –mitochondria option. We only kept properly mapped reads for variant calling and filtered these using the FilterMutectCalls options. Left alignment and trimming were performed on variants and only variants with the PASS filter were retained for further analysis.
Results
Cohort description
All individuals with ASD in the BARAKA Study met diagnostic criteria according to the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) (American Psychiatric Association, 2013). A total of 104 affected individuals from 100 families (79% complete trios) were analyzed, including 98 simplex and 2 multiplex families (both with 3 affected siblings each), with a male to female ratio of 5.5 (88 males and 16 females). The most common comorbidities among the BARAKA cohort were ADHD (35.6%), ID (29.8%), DD (28.8%), GI problems (19.2%), learning disabilities (10.6%), and seizures (6.7%) (Table 1, Additional file 2: Figure S1). Consistent with the demographic breakdown of Qatar, the majority of families were of Arab descent (58%), followed by South Asian (25%), European (7%), African (5%), and other ethnicities. In total, 44 out of 100 families (44%) were consanguineous (Additional file 2: Figure S2 and Figure S3).
WGS and variant discovery
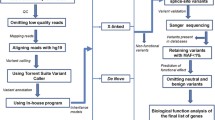
All children and their families (n = 372 individuals) underwent WGS to an average read depth of 36 × ,while almost 96% of bases were covered at a mean depth of 20. Individuals had, on average, 4,206,499 SNVs and 110,600 indels per genome. After filtering variants based on MAF < 1% in general population databases such as 1000G, gnomAD, ExAC, and an extensive internal database of > 15,000 Qatari alleles, an average of 26,743 rare SNVs (95.3% heterozygous and 4.7% homozygous) and 67,292 rare indels (87.8% heterozygous and 12.2% homozygous) per genome remained for downstream analysis (Fig. 1). We then proceeded with a two-tier approach—first investigating variants of different classes in known ASD genes, and then transitioning genome-wide for putatively novel candidate genes causing ASD in this cohort.

Overview of WGS approach and variant prioritization
Pathogenic variants in known ASD-risk genes and regions
Small variants (SNVs + indels)
We first sought to identify (DN) or rare inherited LoF or Damaging missense (Dmiss) variants in 1714 known NDD and ASD genes (curated from multiple sources as described in “Methods”) and found 26 such variants in 24 genes in 24 individuals (Table 2). Nine families had nine DN variants in known ASD/NDD genes (STAG1, SCN2A, MTOR, WDR37, EIF5A, KCNMA1, KDM5B GRIN2B, and MYO5A). All of these variants were Dmiss except for one LoF in KCNMA1. Two variants (p.Arg373Gln in STAG1 and p.Ala1773Val in SCN2A) were already reported as pathogenic in ClinVar for complex neurodevelopmental disorders. Using ACMG classification, the seven remaining DN variants were scored as likely pathogenic. One paternally inherited heterozygous variant (p.Arg266Cys) in DNM1 was shared between three siblings with ASD and scored as VUS (Table 2).
In addition to DN variants, we found recessive variants (homozygous) in 8 ASD/NDD genes (TRAPPC9, NBN, TSEN2, UBR1, MED17, TIAM1, CTSA, and ZNF335) in 7 families. All of which were Dmiss variants except for one stop-gain (p.Arg570* in TRAPPC9). Out of the 7 families with recessive events, 6 were consanguineous families (85.7%). Manual curation, according to ACMG guidelines, classified all recessive variants as VUS except for the TRAPPC9 variant (p.Arg570*), which was already reported in ClinVar as pathogenic/likely-pathogenic. In addition, we identified five X-linked Dmiss variants in four genes (PTCHD1, DMD, WNK3, and SLC9A6) in 5 males with ASD, all of which were scored as VUS (Table 2).
Structural variants (CNVs + TREs)
Given the known association of ASD with genomic disorder regions, we investigated the overlap of CNVs detected within our patients with a list of regions where deletions and duplications were previously identified in individuals with ASD [12] (see “Methods”). We found two candidate variants: a de novo 1.4 Mb deletion in 22q11.21 and a 1.7 Mb maternally inherited duplication in 16p13.3 (Table 2). No other CNVs overlapped the known ASD/NDD gene list from our cohort. We further investigated TREs in known ASD genes [8] and found two matching TREs in SHANK2 and NCOR2 in two families (Table 2).
Mitochondrial variants
We investigated pathogenic mtDNA variants and heteroplasmy (where mutated mtDNA co-exist with unmutated mtDNA) that overlap previously reported variants (n = 15) associated with ASD [12]. We identified only one de novo variant (heteroplasmy of 2.1%) of the m.3243A > G variant associated with mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) in an individual with ASD (maternal heteroplasmy was undetectable) (Table 2). We also considered overlap with mtDNA variants causing homoplasmic disorders generally affecting vision and hearing (n = 6) and found two matches: one individual with ASD had a 2.3% load of 14484 T > C variant (maternal genotype was undetectable), and a father had a 59.2% load of 11778G > A variant. Both variants are associated with Leber Hereditary Optic Neuropathy (LHON) syndrome (Additional file 1: Table S3).
Altogether, 27 families (27%) had at least one damaging variant in a known ASD/NDD gene panel in this cohort.
Pathogenic variants in novel ASD-risk genes and regions
Small variants (SNVs + indels)
Beyond known genes, we searched genome-wide for damaging DN and homozygous variants (LoF, Dmiss) in novel candidate genes that could explain ASD in the remaining families.
For DN variants, we found 17 in as many genes (CHD9, STAB2, MOV10, HDAC7, DNAJC10, SYNE3, COPS5, B4GALT1, DCAF17, FCHO2, INCENP, ING5, PTOV1, PRRC2C, TLN1, RRN3, and STRIP2) in 14 families. Four were predicted LoF, all in genes, with pLI > 0.99 (MOV10, HDAC7, TLN1, and CHD9) and 13 were Dmiss variants. Three families had two damaging DN variants in two different genes each. All damaging DN variants in novel genes had additional carriers from ASD cohorts (MSSNG, SSC, and SPARK) (Additional file 1: Table S4).
We also looked for damaging homozygous variants (LoF and Dmiss) in genes with high pRec scores (> 0.9). Six novel genes (TRIM29, EIF2A, CDH23, NOC3L, KDM8, and IFT140) were identified in four families; five of which were affected by Dmiss variants and one by a LoF (splice acceptor variant, c.3236-1G > A) in CDH23 (Table 3). Three of the four families with homozygous variants (75%), were consanguineous. We found additional biallelic variant carriers in ASD cohorts (MSSNG and SSC) for CDH23 and IFT140.
Structural variants (CNVs)
A total of 5 ASD-associated CNVs were identified in 5 families. One was a de novo 7.7 kb deletion of exons 7 to 10 of CSNK1A1 (Fig. 2, Additional file 1: Table S4). The other four were homozygous deletions in four families (Table 3) as follows: a 2.33 kb deletion in ELOVL2 partially deleting exon 8 (Fig. 2), a 12.9 kb deletion overlapping exon 9 of FAM204A, a partial deletion of exon 11 (65 bp) in AFG3L1P, and a 47.6 kb deletion of full length long non-coding RNA gene (LINC00648) and complete deletion of a microRNA (MIR548Y). Most of these genes were novel in their association with ASD except for ELOVL2, which is reported in the SFARI Gene database (score 2). We checked if CNVs in these genes were found in additional individuals in global ASD cohorts and found a 6 kb deletion in ELOVL2 in one family, a large de novo deletion (> 4 Mb) including CSNK1A1 gene in one family, multiple large CNVs in six individuals that include AFG3L1P gene, and three individuals with deletions (> 12 kb) in FAM204A.

Examples of ASD-relevant CNVs. A Pedigree, IGV visualization, and UCSC genomic context of a 2.33 kb homozygous deletion comprising ~ 330 bp of exon 8 of ELOVL2 (see colored region of the UCSC panel, http://genome.ucsc.edu). B ddPCR results showing a copy number of zero in the proband (indicated by red star), equivalent to no reads detected from the inside primer. C Pedigree, IGV visualization, and UCSC genomic context of 7.7 kb de novo deletion from a simplex family comprising exon 7 to 10 of CSNK1A1 gene (see colored region of the UCSC panel, http://genome.ucsc.edu). D ddPCR results showing copy number calculation equals to one in proband, heterozygous status, (indicated by red star) equivalent to less reads detected from inside primer in the proband sample. OP1 outside primer 1, OP2 outside primer 2, IP inside primer
Altogether, we identify 28 candidate novel genes in 22 families (22%), of which 23 genes (82.1%) are supported by additional carriers in MSSNG and SSC, affected by variants in similar classes and zygosity.
Discussion
The past decade has seen rapid advances in the discovery of genetic and genomic variants underlying complex neurodevelopmental conditions, including ASD [10, 11, 15, 61, 62]. Recently, WGS has emerged as a comprehensive approach for genomic discovery, enabling the detection of pathogenic variants spanning all types and size classes, including SNVs, indels, CNVs,TREs, and mtDNA [12, 63]. In this study, we present a comprehensive evaluation of genetic risk factors detected by WGS in a cohort of 100 families with ASD from vastly under-represented Middle Eastern populations as part of the first release of the BARAKA-Qatar Study.
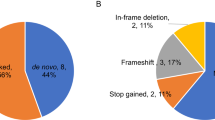
We discover at least one candidate pathogenic variant in known ASD/NDD genes/regions in 27 families (27%) (Fig. 3A). Despite the high heritability of ASD, the majority of previously identified genetic risk appears to be from de novo variation [11]. Our cohort identified dominant risk variants, including de novo and inherited variants, in 15 of 27 (55.6%) families (37.1% de novo and 18.5% inherited). In terms of variant classes, the majority of dominant risk factor was from SNVs/indels (66.7%), followed by CNVs (13.3%), TREs (13.3%), and mtDNA variants (6.7%).

Genetic risk variants in known ASD/NDD genes. A Percentage ASD probands having candidate causative rare variants, stratified by B type of variant
Notably, only two de novo SNVs (22.2%) were identified previously (p.Arg373Gln in STAG1 and p.Ala1773Val in SCN2A) underscoring the high allelic heterogeneity underlying ASD across global populations. We sought to manually curate novel alleles according to ACMG criteria and found that all de novo novel alleles were classified as likely pathogenic. These mainly included damaging missense variants and only one frameshift variant (p.Met1176fs) in KCNMA1. KCNMA1 encodes for potassium calcium-activated channel subfamily M Alpha 1 which are large conductance, voltage, and calcium-sensitive potassium channels fundamental to several physiological processes including smooth muscle contraction, neurotransmitter release, and neuronal excitability [64]. Mutations in this gene have been associated with a broad spectrum of neurological phenotypes and developmental disorders including cerebellar atrophy, DD, and seizures. A recent study reported KCNMA1 mutations in individuals with ASD [64].
One of the most distinguishing features of middle eastern populations is the high degrees of consanguinity. While public databases comprise mostly outbred individuals, the local population of Qatar, for example has consanguinity levels of > 54% [65], suggesting that recessive architecture may contribute to a sizeable fraction of ASD etiology in this population. There have only been a few studies today examining ASD in consanguineous settings. One looked only at homozygous deletions and reported seven exonic deletions from 123 consanguineous families (5.7%) [66]. A more recent study investigating biallelic SNVs in highly consanguineous families found recessive gene risk in known ASD/NDD genes in 9 out of 23 (39%) families [17]. Data from our study suggest a recessive burden somewhere in between (6 of 44 consanguineous families (13.6%)). This burden is almost sixfold higher than in non-consanguineous families in our cohort, where only 1 of 56 families (1.8%) had a candidate homozygous causative variant in a known ASD/NDD gene (p = 0.02) (Fig. 4).

Genetic variants in known ASD/NDD genes stratified by consanguinity status of families. Recessive burden was significantly higher (p-value = 0.02) in consanguineous families
Moreover, in comparison to the largest WGS study investigating > 7,000 families with ASD from MSSNG and SSC cohorts which represent largely outbred populations [12], recessive genetic risk, accounting for different criteria used to define recessive events (i.e., recessive events with only LoF on both alleles were included in MSSNG/SSC), contributed to higher risk in our cohort (1.1%, 0.3%, and 3.7% for MSSNG, SSC, and BARAKA, respectively). Although the sample size of the BARAKA cohort is relatively modest at this time, these results highlight the potential impact of recessive variants on the etiology of ASD in highly consanguineous populations.
In some cases, the high levels of consanguinity may lead to certain challenges not anticipated when studying largely outbred cohorts. Among these are examples where it may be difficult to differentiate between driver and passenger mutations on a given haplotype. For example, two homozygous putatively damaging missenses variants (p.Gly289Arg, p.Val1243Leu) were identified in, two known NDD genes, CTSA and ZNF335 a (neighboring genes on Chr 20) in a consanguineous male individual with ASD and ID. ZNF335 plays an essential role in neurogenesis and biallelic variants in ZNF335 have been associated with ASD-like phenotypes [67]. CTSA has been associated with an autosomal recessive form of Galactosialidosis (OMIM: 613111), for which intellectual disability is a common symptom.
We only had two multiplex families in the setting of parental consanguinity in our cohort (each with three affected siblings). While we expected to find recessive variants in these families, no candidate gene emerged with shared recessive variants across the three siblings. Instead, in one family (Family BRK-13), we found a paternally inherited heterozygous damaging missense variant (p.Arg266Cys) in DNM1 that segregated with all affected siblings. A re-evaluation of the father's phenotype showed a diagnosis of ADHD and features of ASD. DNM1 encodes dynamin 1, a GTP-binding protein mainly expressed in the central nervous system [68]. Pathogenic DNM1 variants affect brain development and function and cause epileptic encephalopathy associated with global DD [69, 70]. Pathogenic variants in DNM1 have also been reported in association with other clinical phenotypes such as hypotonia, movement disorder, ASD, cortical visual impairment, and microcephaly [69, 70]. The three affected siblings lacked epilepsy and showed symptoms of ADHD, although DNM1 has not yet been associated with ADHD. Such an example of a multiplex family highlights the importance of taking a comprehensive approach with variant identification in each family, regardless of consanguinity status.
In addition, X-linked recessive variants (Dmiss) were found in 5 of 27 families (18.5%), supporting the role of the X-chromosome in ASD susceptibility in males. Variants in the X chromosome greatly contributed to ID and ASD in males with more than 140 genes being involved [62]. Two damaging missense variants (p.Glu805Gln and p.Asn205Ser) in WNK3 were identified in two unrelated male probands. WNK3 encodes a cell volume-sensitive kinase that is highly expressed during early brain development [71]. Previously, multiple hemizygous, LoF, and pathogenic missense variants were identified in WNK3 in male individuals with sporadic and familial forms of ID [72]. Re-examination of comorbidities in the two probands in our cohorts reveals that neither had ID, and only one had ADHD, potentially representing an expansion of the WNK3-related phenotype.
In total, SNVs and Indels alone were present in 81.5% of our cohort, suggesting other variant classes could explain the missing heritability in the remaining families. Indeed, we employed WGS to enable the detection of CNVs and TREs associated with ASD. Our sample size was underpowered to detect significant enrichment of TREs in individuals with ASD compared to siblings without ASD. Only two families (7.4%) had TREs impacting known ASD genes. One of these was a (high functioning) female proband (Family BRK-51) with a TREs affecting intron 7 of SHANK2, a member of a family of scaffold proteins (comprising SHANK1, 2 and 3) that localize to the postsynaptic site of excitatory synapses in the central nervous system [73]. SHANK2 has been implicated in various brain disorders, including ASD, ID, DD, ADHD, schizophrenia, epilepsy, and obsessive–compulsive disorder [74]. Another female proband (Family BRK-89) was diagnosed with Down syndrome disintegrative disorder (DSDD) (a developmental regression that leads to loss of previously acquired cognitive and social functioning, and the development of features of ASD) [75]. The genetic implications of DSDD have not yet been associated with any gene. We identified a TREs affecting intron 18 of NCOR2, a nuclear receptor corepressor 2 as part of a multi-protein corepressor complex known as the NCOR complex [76]. The NCOR complex plays a vital role in neurocognition with implications for autism [77].
Altogether, SNVs/Indels were the major risks affecting 22 of 27 families (81.5%: dominant (45.5%), recessive (31.8%), and X-linked (22.7%)) compared to CNVs (7.4%), TREs (7.4%), and mtDNA variant (3.7%) (Fig. 3B).
As only 27% of families had genetic risk from known ASD/NDD genes/regions, we expanded our search genome-wide for putatively novel genes or regions that could contribute to the genetic risk of ASD in the remaining families. Using similarly strict criteria as with known genes but limiting only to damaging de novo or homozygous variants, we identified candidate genes in 22 of 100 families (22%), 15 (68.2%) with de novo variants (SNVs 63.6%; CNVs 4.6%), and 7 families (31.8%) with homozygous variants (SNVs 13.6%, CNVs 13.6%, and one family (Family BRK-83) with both SNV and CNV (4.6%)). Of these novel genes, 23 out of 28 (82.1%) genes are supported by additional carriers affected by variants in similar classes and zygosity in ASD cohorts MSSNG, SSC, and SPARK. A further functional investigation is needed to determine the potential role of these Novel identified genes in ASD risk.
Notably, two families had multiple variants of the same type in known and novel genes, showing that finding a damaging variant in a known gene should not rule out searching for novel genes in the same family. First, the proband in (Family BRK-05) had a de novo Dmiss (p.Arg609His) in MYO5A (known gene, Table 2) and a de novo Dmiss (p.Asp428Gly) in DCAF17 (Novel gene, Table S4). Second, proband in (Family BRK-48) had a de novo Dmiss (p.Tyr126Phe) in WDR37 (known gene, Table 2) and a de novo Dmiss (p.Cys218Arg) in COPS5 (Novel gene, Table S4). The high level of genotype/phenotype heterogeneity in individuals with ASD may explain the multiple variants/genes that could collectively contribute to the genetic risk of ASD. Comprehensive searches of known and novel genes contributing to ASD in each family help to determine the total burden of the disorder.
The use of WGS at point of care for families with ASD is relatively new in Qatar, where the public understanding of research as opposed to clinical testing still in its early stages. Genetic consultation is offered to individuals with significant genetic findings (i.e., variants classified as pathogenic or likely-pathogenic) to explain basic aspects such as recurrent risk based on mode of inheritance (de novo versus inherited) and interpretation of results. While study begins to set the scene for the integration of research findings into clinical practice, it nevertheless has important limitations which must be considered. First, our study sample size of 100 families limits generalizations at present about the relationship between consanguinity and ASD. While we observe an enrichment in recessive inheritance in such families, larger numbers will be needed to confirm if this trend will hold. Indeed, the BARAKA study has recently surpassed 250 families enrolled, with an eventual aim of 1000 families by end of 2024. As the cohort size increases, in particular from the local population where consanguinity exceeds 50%, we shall have valuable additional data to investigate this. Moreover, larger cohort sizes will allow us to move away from a per-family pathogenic variant approach to a cohort-level approach, using tools such as rare variant burden analysis [78] and/or gene-set enrichment analysis, which may aid novel gene discovery and uncover new ASD-implicated biological pathways. Similarly, larger datasets could be valuable in case–control studies that produce GWAS-like summary statistics, which can then support explorations of polygenic risk in ASD; such an effort is currently undermined in the absence of summary statistics from individuals with similar ancestry. Finally, combining our growing data with MSSNG in coming releases will make data from this unique ancestry available to global research endeavors which can then investigate more fully the genetic architecture in this part of the world compared to largely outbred populations.
Conclusions
Taken as a whole, our study provides several important takeaways related to ASD research, especially in under-studied global populations. First, comprehensive characterization by WGS is a viable approach to identify genetic etiology in a substantial fraction of affected individuals. Second, we demonstrate the critical role played by de novo variants even in settings of high consanguinity, and thus the importance of enrolling parents where available to identify DNs with high specificity. Third, we observe a fourfold enrichment of homozygous causes in consanguineous families compared to non-consanguineous families; however, even in consanguineous and multiplex settings, the causative variant may be dominant/de novo, highlighting the necessity of comprehensively examining all variant classes before concluding a case study. Fourth, despite our cohort’s relatively high diagnostic yield, over 73% of families remain unresolved. The missing genetic risk could be due to common variants, rare variants in novel genes, variants in non-coding and regulatory regions, variants that could have been overlooked by subsequent prioritization and definition of damaging variants, or compound heterozygotes resulting from a combination of different variant classes (e.g., CNV on one allele and SNVs/indels on another). Accounting of these types of variants in the next release of the study may lead to genetic diagnosis in unresolved families. In all, we believe the BARAKA-Qatar study’s plans to continue growing cohorts with higher representation from the Middle East, North Africa and South Asia will help advance global understanding of ASD etiology in this region of the world.
Availability of data and materials
All data used and generated is available within the main manuscript and supporting files. The WGS data of families in this study have been submitted to MSSNG project and should be included in the coming DB7 release. Access to the data can be requested by completing data access agreement at https://research.mss.ng.
Abbreviations
- ASD:
-
Autism spectrum disorder
- BARAKA:
-
Building a Resource for the Advancement of Knowledge of Autism
- WGS:
-
Whole genome sequencing
- SNVs:
-
Single nucleotide variants
- CNVs:
-
Copy number variants
- SVs:
-
Structural variants
- TREs:
-
Tandem repeats expansions
- mtDNA:
-
Mitochondrial DNA
- ID:
-
Intellectual disability
- ADHD:
-
Attention-deficit hyperactivity disorder
- DD:
-
Developmental delay
- GI:
-
Gastrointestinal
- TADA:
-
Transmission and de novo association
- DN:
-
De novo
- LoF:
-
Loss of function
- Dmiss:
-
Damaging missense
- ACMG:
-
American College of Medical Genetics
- P:
-
Pathogenic
- LP:
-
Likely-pathogenic
- VUS:
-
Variant of uncertain significance
- QGP:
-
Qatar-genome project
- OMIM:
-
Online Mendelian Inheritance in Man
- SFARI:
-
Simons Foundation Autism Research Initiative
- MAF:
-
Minor allele frequencies
References
American Psychiatric Association. DSM-5 Autism Spectrum Disorder Fact Sheet. Am Psychiatr Assoc. 2014;(October):233–55.
Iakoucheva LM, Muotri AR, Sebat J. Getting to the cores of autism. Cell. 2019;178(6):1287–98. https://doi.org/10.1016/j.cell.2019.07.037.
Tick B, Bolton P, Happé F, Rutter M, Rijsdijk F. Heritability of autism spectrum disorders: a meta-analysis of twin studies. J Child Psychol Psychiatry Allied Discip. 2016;57(5):585–95.
Dias CM, Walsh CA. Recent advances in understanding the genetic architecture of autism. Annu Rev Genomics Hum Genet. 2020;21:289–304.
Zhao M, Havrilla J, Peng J, Drye M, Fecher M, Guthrie W, et al. Development of a phenotype ontology for autism spectrum disorder by natural language processing on electronic health records. J Neurodev Disord. 2022;14(1):1–12. https://doi.org/10.1186/s11689-022-09442-0.
Peter B, Dinu V, Liu L, Huentelman M, Naymik M, Lancaster H, et al. Exome sequencing of two siblings with sporadic autism spectrum disorder and severe speech sound disorder suggests pleiotropic and complex effects. Behav Genet. 2019;49(4):399–414. https://doi.org/10.1007/s10519-019-09957-8.
Wang T, Guo H, Xiong B, Stessman HAF, Wu H, Coe BP, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun. 2016;7(13):1–10. https://doi.org/10.1038/ncomms13316.
Trost B, Engchuan W, Nguyen CM, Thiruvahindrapuram B, Dolzhenko E, Backstrom I, et al. Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature. 2020;586(7827):80–6. https://doi.org/10.1038/s41586-020-2579-z.
Mitra I, Huang B, Mousavi N, Ma N, Lamkin M, Yanicky R, et al. Patterns of de novo tandem repeat mutations and their role in autism. Nature. 2021;589(7841):246–50.
Trost B, Thiruvahindrapuram B, Chan A, Engchuan W, Higginbotham E, Howe J, et al. 3. Genomic architecture of autism spectrum disorder from comprehensive whole-genome sequence annotation. Eur Neuropsychopharmacol. 2022;63:e45–6.
Fu JM, Satterstrom FK, Peng M, Brand H, Collins RL, Dong S, et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat Genet. 2022;54(9):1320–31.
Trost B, Thiruvahindrapuram B, Chan AJS, Engchuan W, Higginbotham EJ, Howe JL, et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell. 2022;185(23):4409-4427.e18.
Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46(8):881–5.
Lim ET, Raychaudhuri S, Sanders SJ, Stevens C, Sabo A, Macarthur DG, et al. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron. 2014;77(2):235–42.
Doan RN, Lim ET, De Rubeis S, Betancur C, Cutler DJ, Chiocchetti AG, et al. Recessive gene disruptions in autism spectrum disorder. Nat Genet. 2019;51(7):1092–8. https://doi.org/10.1038/s41588-019-0433-8.
Choi L, An JY. Genetic architecture of autism spectrum disorder: lessons from large-scale genomic studies. Neurosci Biobehav Rev. 2021;128:244–57. https://doi.org/10.1016/j.neubiorev.2021.06.028.
Tuncay IO, Parmalee NL, Khalil R, Kaur K, Kumar A, Jimale M, et al. Analysis of recent shared ancestry in a familial cohort identifies coding and noncoding autism spectrum disorder variants. NPJ Genom Med. 2022;7(1):13.
Alshaban F, Aldosari M, Al-Shammari H, El-Hag S, Ghazal I, Tolefat M, et al. Prevalence and correlates of autism spectrum disorder in Qatar: a national study. J Child Psychol Psychiatry Allied Discip. 2019;60(12):1254–68.
Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, et al. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 Sites, United States, 2014. MMWR Surveill Summ. 2018;67(6):1.
Xu G, Strathearn L, Liu B, Bao W. Corrected prevalence of autism spectrum disorder among US children and adolescents. JAMA. 2018;319(5):505.
Da’as SI, Aamer W, Hasan W, Al-Maraghi A, Al-Kurbi A, Kilani H, et al. PGAP3 Associated with hyperphosphatasia with mental retardation plays a novel role in brain morphogenesis and neuronal wiring at early development. Cells. 2020;9(8):1–25.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Depristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–501.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92.
Auton A, Abecasis GR, Altshuler DM, Durbin RM, Bentley DR, Chakravarti A, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–76.
Farrell JA. Expanding the horizons of next generation sequencing with RUFUS. Doctoral dissertation, Boston College; 2014.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. 2012;1–9. Available from: http://arxiv.org/abs/1207.3907.
Kohailan M, Aamer W, Syed N, Padmajeya S, Hussein S, Sayed A, et al. Patterns and distribution of de novo mutations in multiplex Middle Eastern families. J Hum Genet. 2022;67(10):579–88.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The Human Genome Browser at UCSC. Genome Res. 2002;12(6):996–1006.
Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26(22):2867–73.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–94.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–82.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19(9):1553–61.
Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):37–43.
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–6.
Samocha KE, Kosmicki JA, Karczewski KJ, O’Donnell-Luria AH, Pierce-Hoffman E, MacArthur DG, et al. Regional missense constraint improves variant deleteriousness prediction. bioRxiv. 2017;148353. Available from: https://www.biorxiv.org/content/10.1101/148353v1%0A; https://www.biorxiv.org/content/10.1101/148353v1.abstract.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745–7.
Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick’s Online Mendelian Inheritance in Man (OMIM®). Nucleic Acids Res. 2009;37(SUPPL. 1):793–6.
Ahmad N. Abou Tayoun1, 2, 3,*, Tina Pesaran4, Marina T. DiStefano5, Andrea Oza5, Heidi L. Rehm5, 6, 7, Leslie G. Biesecker8 and SMHCSVIWG (ClinGen S. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Physiol Behav. 2019;176(3):139–48.
Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017;100(2):267–80. https://doi.org/10.1016/j.ajhg.2017.01.004.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21(6):974–84.
Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28(18):333–9.
Zhu M, Need AC, Han Y, Ge D, Maia JM, Zhu Q, et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am J Hum Genet. 2012;91(3):408–21. https://doi.org/10.1016/j.ajhg.2012.07.004.
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32(8):1220–2.
Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, Marth GT, Quinlan AR, Hall IM. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods. 2015;12(10):966–8.
Wala JA, Bandopadhayay P, Greenwald NF, O’Rourke R, Sharpe T, Stewart C, et al. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res. 2018;28(4):581–91.
Jeffares DC, Jolly C, Hoti M, Speed D, Shaw L, Rallis C, et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun. 2016;2017(8):1–11.
Geoffroy V, Guignard T, Kress A, Gaillard JB, Solli-Nowlan T, Schalk A, et al. AnnotSV and knotAnnotSV: a web server for human structural variations annotations, ranking and analysis. Nucleic Acids Res. 2021;49(W1):W21–8.
Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, et al. A structural variation reference for medical and population genetics. Nature. 2020;581(7809):444–51.
Abel HJ, Larson DE, Regier AA, Chiang C, Das I, Kanchi KL, et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature. 2020;583(7814):83–9.
Devuyst O. The 1000 genomes project: welcome to a new world. Perit Dial Int. 2015;35(7):676–7.
Belyeu JR, Chowdhury M, Brown J, Pedersen BS, Cormier MJ, Quinlan AR, et al. Samplot: a platform for structural variant visual validation and automated filtering. Genome Biol. 2021;22(1):1–13.
Kohailan M, Al-Saei O, Padmajeya S, Aamer W, Elbashir N, Akil AAS, et al. A de novo start-loss in EFTUD2 associated with mandibulofacial dysostosis with microcephaly: case report. Cold Spring Harb Mol Case Stud. 2022;8(4):1–11.
Rossi N, Aliyev E, Visconti A, Akil ASA, Syed N, Aamer W, et al. Ethnic-specific association of amylase gene copy number with adiposity traits in a large Middle Eastern biobank. NPJ Genomic Med. 2021;6(1):1–9. https://doi.org/10.1038/s41525-021-00170-3.
Dolzhenko E, Bennett MF, Richmond PA, Trost B, Chen S, Van Vugt JJFA, et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol. 2020;21(1):1–14.
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180(3):568-584.e23.
Ruzzo EK, Pérez-Cano L, Jung JY, Wang L kai, Kashef-Haghighi D, Hartl C, et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell. 2019;178(4):850–866.e26.
Yuen RKC, Merico D, Bookman M, Howe JL, Thiruvahindrapuram B, Patel RV, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20(4):602–11.
Liang L, Li X, Moutton S, Schrier Vergano SA, Cogné B, Saint-Martin A, et al. De novo loss-of-function KCNMA1 variants are associated with a new multiple malformation syndrome and a broad spectrum of developmental and neurological phenotypes. Hum Mol Genet. 2019;28(17):2937–51.
Mbarek H, Devadoss Gandhi G, Selvaraj S, Al-Muftah W, Badji R, Al-Sarraj Y, et al. Qatar genome: insights on genomics from the Middle East. Hum Mutat. 2022;43(4):499–510.
Schmitz-Abe K, Sanchez-Schmitz G, Doan RN, Hill RS, Chahrour MH, Mehta BK, et al. Homozygous deletions implicate non-coding epigenetic marks in Autism spectrum disorder. Sci Rep. 2020;10(1):1–15. https://doi.org/10.1038/s41598-020-70656-0.
Wu J, Yu P, Jin X, Xu X, Li J, Li Z, et al. Genomic landscapes of Chinese sporadic autism spectrum disorders revealed by whole-genome sequencing. J Genet Genomics. 2018;45(10):527–38. https://doi.org/10.1016/j.jgg.2018.09.002.
Romeu A, Arola L. Classical dynamin DNM1 and DNM3 genes attain maximum expression in the normal human central nervous system. BMC Res Notes. 2014;7(1):1–4.
Nakashima M, Kouga T, Lourenço CM, Shiina M, Goto T, Tsurusaki Y, et al. De novo DNM1 mutations in two cases of epileptic encephalopathy. Epilepsia. 2016;57(1):e18-23.
Allen NM, Conroy J, Shahwan A, Lynch B, Correa RG, Pena SDJ, et al. Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia. 2016;57(1):e12–7.
Küry S, Zhang J, Besnard T, Caro-Llopis A, Zeng X, Robert SM, et al. Rare pathogenic variants in WNK3 cause X-linked intellectual disability. Genet Med. 2022;24(9):1941–51.
Prieto F, Badía L, Mulas F, Monfort A, Mora F. X-linked dysmorphic syndrome with mental retardation. Clin Genet. 1987;32(5):326–34.
Sheng M, Kim E. The Shank family of scaffold proteins. J Cell Sci. 2000;113(11):1851–6.
Yoo YE, Yoo T, Kang H, Kim E. Brain region and gene dosage-differential transcriptomic changes in Shank2-mutant mice. Front Mol Neurosci. 2022;15(October):1–15.
Worley G, Crissman BG, Cadogan E, Milleson C, Adkins DW, Kishnani PS. Down syndrome disintegrative disorder: new-onset autistic regression, dementia, and insomnia in older children and adolescents with down syndrome. J Child Neurol. 2015;30(9):1147–52.
Mottis A, Mouchiroud L, Auwerx J, Emmett MJ, Lazar MA, Zhou WW, et al. Integrative regulation of physiology by histone deacetylase 3. Nat Rev Mol Cell Biol. 2019;151(2):102–15.
Zhou W, He Y, Rehman AU, Kong Y, Hong S, Yalamanchili HK, et al. NCOR1 /2 loss of function impairs memory through a novel GABAergic hypothalamus–CA3 projection. 2019;22(2):205–17.
Chan AJS, Engchuan W, Reuter MS, Wang Z, Thiruvahindrapuram B, Trost B, et al. Genome-wide rare variant score associates with morphological subtypes of autism spectrum disorder. Nat Commun. 2022;13(1):6463.
Acknowledgements
We are grateful to Qatar National Research Fund (QNRF) for funding this research (BARAKA-Qatar project: NPRP10-0202-170320). We thank all individuals with ASD and their families that enrolled in this cohort. We also thank and acknowledge the assistance of Drs. Tasneim Abdalla, Amani Abdalla, Israa Salah Eldin Khidir, and Abdelrahman A. E'mar in the collection of phenotypic data from electronic health records. We thank the families participating in MSSNG as well as the donors who support this program. We also acknowledge all of the families participated in SSC and SPARK as well as the principal investigators (A.Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R.Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D.Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C.Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, and E. Wijsman). We appreciate obtaining access to genetic and phenotypic data on SFARI Base. Approved researchers can obtain the SSC and SPARK datasets by applying at https://base.sfari.org. S.W.S hold the Northbridge Chair in Pediatric Research at the Hospital for Sick Children and University of Toronto.
Funding
This research project (BARAKA-Qatar Study) was generously supported by the Qatar National Research Fund (QNRF) (NPRP10-0202–170320), the Qatar International Islamic Bank (QIIB) and Mohammed AlSaad (MAS) Holding.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.A and K.A.F. Methodology: M.A, E.A, B.Trost, M. Kohailan, W.A, N.S, R.S, W.E, and B. Thiruvahindrapuram. Software: E.A, M. Kohailan, B.Trost, and W.E. Formal analysis: M.A. Data curation: M.A and E.A. Investigation: M.A, E.A, B.Trost, W.A, N.S, R.S, G.GD, W.E, B. Thiruvahindrapuram, M.G. Project administration: A.A.A, S.P, J.H, J.W, J.L. Resources: A.A.A, A.S, J.L, S.H, N.Albashir, S.E, M.J, M.E, D.S, A.N, M.W.A, N.AbdelAati and M.Kamal. Validation: M. Kambouris, I.P and A.H. Visualization: M.A and E.A. Writing—original draft: MA. Writing—review and editing: B.Trost, R.S, M. Kohailan, E.A, W.A, W.E, MWA, A.N, M.W.A, Y.M, A.A.A, S.W.S, and K.A.F. Funding acquisition: A.A.A, Y.M, and K.A.F. Supervision: K.A.F, M.Kamal, S.W.S, A.A.A. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the institutional review board (IRB) of Sidra Medicine (IRB No. 1500767), and written informed consent was obtained from all participants. Our research conforms to the principles of the Helsinki Declaration.
Consent for publication
Not applicable.
Competing interests
S.W.S. is a scientific advisor to Population Bio and a Highly Cited Academic Advisor to the King Abdullaziz University. Intellectual property from his work held at the University of Toronto has been licensed to Athena Diagnostics. None of these relationships have influence the research described in this paper. The remaining authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Detail phenotypes of individuals with ASD, Table S2. Known ASD/NDD gene panels, Table S3. Known mtDNA pathogenic variants associated with hearing loss/Deafness at greater than 2% heteroplasmy, Table S4. Candidate dominant variants in Novel genes/regions associated with ASD.
Additional file 2:
Figure S1. Co-morbidities of Autism. Frequency of co-occurrence of phenotypes in individuals with ASD, Figure S2. Pairwise relationship of individuals. Related parents are in blue and above the threshold of kinship (>0.044) (dashed red line), Figure S3. Inbreeding coefficient (F). Per sample estimate of inbreeding of all individuals included. Individuals from inbred families are in blue, Figure S4. Sanger Sequencing. Validation of de novo variants.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Abdi, M., Aliyev, E., Trost, B. et al. Genomic architecture of autism spectrum disorder in Qatar: The BARAKA-Qatar Study. Genome Med 15, 81 (2023). https://doi.org/10.1186/s13073-023-01228-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-023-01228-w