
Abstract
Background
Mutations in the p110α catalytic subunit of phosphatidylinositol 3-kinase (PI3K), encoded by the PIK3CA gene, cause dysregulation of the PI3K pathway in 35–40% of patients with HR+/HER2– breast cancer. Preclinically, cancer cells harboring double or multiple PIK3CA mutations (mut) elicit hyperactivation of the PI3K pathway leading to enhanced sensitivity to p110α inhibitors.
Methods
To understand the role of multiple PIK3CAmut in predicting response to p110α inhibition, we estimated the clonality of multiple PIK3CAmut in circulating tumor DNA (ctDNA) from patients with HR+/HER2– metastatic breast cancer enrolled to a prospectively registered clinical trial of fulvestrant ± taselisib, and analyzed the subgroups against co-altered genes, pathways, and outcomes.
Results
ctDNA samples with clonal multiple PIK3CAmut had fewer co-alterations in receptor tyrosine kinase (RTK) or non-PIK3CA PI3K pathway genes compared to samples with subclonal multiple PIK3CAmut indicating a strong reliance on the PI3K pathway. This was validated in an independent cohort of breast cancer tumor specimens that underwent comprehensive genomic profiling. Furthermore, patients whose ctDNA harbored clonal multiple PIK3CAmut exhibited a significantly higher response rate and longer progression-free survival vs subclonal multiple PIK3CAmut.
Conclusions
Our study establishes clonal multiple PIK3CAmut as an important molecular determinant of response to p110α inhibition and provides rationale for further clinical investigation of p110α inhibitors alone or with rationally-selected therapies in breast cancer and potentially other solid tumor types.
Similar content being viewed by others

Background
The phosphatidylinositol 3-kinase (PI3K) pathway regulates pro-survival and pro-growth cellular signaling, but is frequently dysregulated in solid tumors [1,2,3,4]. In particular, approximately 40% of hormone-receptor positive, human epidermal growth factor receptor 2-negative (HR+/HER2–) breast cancers (BCs) exhibit aberrant activation of the PI3K pathway through gain-of-function mutations in the α isoform of PI3K. PI3Kα is an obligate complex of a regulatory p85 domain and the catalytic α isoform phosphatidylinositol-4,5-bisphosphate 3-kinase (p110α), which is encoded by the PIK3CA gene [2, 5, 6]. Although PIK3CA mutations (PIK3CAmut) can occur throughout the gene, the majority of PIK3CAmut occur in two “hotspot” regions of p110α: the helical domain (primarily amino acids E542 and E545); and the kinase domain (primarily amino acid H1047) [2, 7, 8].
PIK3CAmut have long been a target of clinical investigation as a likely predictive biomarker of response to PI3K pathway inhibitors. Early pan-PI3K isoform inhibitors and dual inhibitors of PI3K and mTOR such as buparlisib, pictilisib, and apitolisib, were unable to fully validate PIK3CAmut as a predictive biomarker due to dose-limiting toxicities [9,10,11,12]. However, more recent inhibitors, such as the p110β-sparing inhibitor, taselisib, and the p110α-selective inhibitor, alpelisib, have demonstrated statistically significant improvements in progression free survival in phase III studies [13, 14]. Alpelisib was approved by the Food and Drug Administration (FDA) in 2019 for the treatment of patients with HR+/HER2– PIK3CAmut advanced or metastatic breast cancer in combination with fulvestrant, an estrogen receptor degrader, thus validating PIK3CAmut as a predictive biomarker of response to PI3K pathway inhibition.
Despite these successes, there remains an unmet need to develop PI3K inhibitors with improved isoform specificity and anti-cancer efficacy. To guide this development, further refinement of our understanding of PIK3CAmut and subcategories of PIK3CAmut as predictive biomarkers for patients with cancer is necessary. Building from early observations of exceptional responders in an alpelisib phase I trial [15] Vasan, et al., determined that co-occurring PIK3CAmut within the same tumor (i.e., “double PIK3CAmut”), especially those occurring together on the same allele (in cis), were the likely source of these exceptional responses [16]. Through in silico and preclinical modeling efforts, the authors demonstrated that double PIK3CAmut in cis elicit hyperactivation of the PI3K pathway through enhanced membrane binding that resulted in both increased cellular proliferation and sensitivity to p110α-selective inhibitors, such as alpelisib and GDC-0077 (inavolisib) [16].
Taselisib was investigated in combination with fulvestrant in the phase III clinical trial, SANDPIPER, for patients with estrogen-receptor positive (ER+), HER2– locally advanced or metastatic breast cancer (NCT02340221) [14]. The study met its primary endpoint of improved progression-free survival (PFS) with taselisib + fulvestrant over placebo + fulvestrant albeit with modest clinical activity (7.4 vs 5.4 months, respectively; hazard ratio = 0.70) [14]. SANDPIPER is the largest randomized clinical trial of a PI3K inhibitor in patients with PIK3CAmut tumors. Of note, when categorizing patients’ tumors as PIK3CAmut by baseline circulating tumor DNA (ctDNA), a larger treatment effect was observed compared to outcomes based on tissue PIK3CAmut positivity (irrespective of ctDNA-based status), especially in patients with more than one PIK3CAmut in their ctDNA compared to those with a single PIK3CAmut in their ctDNA [14, 16].
To further investigate this observation, we employed an in silico methodology to infer the clonality of single and multiple PIK3CAmut identified in plasma-derived ctDNA from participants enrolled to SANDPIPER, and analyzed the resultant subgroups against co-altered genes and pathways, as well as against clinical outcomes. To validate our findings, we similarly interrogated a large genomic database of patient breast cancer tumors that underwent comprehensive genomic profiling [17, 18]. Herein, we establish the clonal aspects of single and multiple PIK3CAmut, enhance our understanding of how multiple PIK3CAmut impact ability of other cellular signaling pathways to influence anti-tumor responses of patients with HR+/HER2– metastatic BC to PI3K inhibition, and provide justification for new rational therapeutic combination approaches.
Methods
SANDPIPER clinical trial
Between 9 April 2015 and 4 September 2017, 631 postmenopausal patients aged 18 years or older with ER+, HER2– locally advanced or metastatic breast cancer and who experienced disease recurrence/progression during or after aromatase inhibitor therapy were enrolled to the prospectively registered phase III clinical trial, SANDPIPER (NCT02340221) in a 2:1 fashion for treatment with taselisib (GDC-0032) + fulvestrant (n = 417) versus placebo + fulvestrant (n = 214) [14]. Based on centralized testing of tumor tissue with the cobas® PIK3CA Mutation Test, patients with PIK3CAmut tumors were randomized separately from those in which no PIK3CAmut was detected (NMD). Stratification factors included visceral disease, endocrine sensitivity, and geographic region. Notable exclusion criteria include HER2-positive (HER2+) disease, prior treatment with fulvestrant, and prior treatment with a PI3K, mTOR, or AKT inhibitor. For details regarding ethical consent to participate, please refer to the Declarations section of this manuscript. In brief, the SANDPIPER study met its primary endpoint of improved progression-free survival (PFS) with taselisib + fulvestrant over placebo + fulvestrant in patients with PIK3CAmut tumors (7.4 vs 5.4 months, respectively; hazard ratio = 0.70). The secondary endpoints, including objective response rate (ORR), clinical benefit rate (CBR), duration of objective response (DoR), and PFS per blinded independent central review (BICR-PFS), showed consistent improvement with taselisib + fulvestrant over placebo + fulvestrant in the PIK3CAmut population. Overall survival (OS) data were immature at the time of the original report [14].
Plasma ctDNA collection and comprehensive genomic profiling (CGP)
ctDNA was isolated as previously described [19] from plasma collected immediately prior to treatment (i.e., at baseline) from 631 participants enrolled to the SANDPIPER clinical trial [14]. CGP of baseline plasma samples from 598 participants was performed with the FoundationOne®Liquid (F1L) assay in a Clinical Laboratory Improvement Amendments (CLIA)-certified, College of American Pathologists (CAP)-accredited reference laboratory (Foundation Medicine, Inc., Cambridge, MA, USA). F1L uses hybrid-capture, adapter ligation-based libraries to identify genomic alterations (base substitutions, small insertions and deletions, copy number alterations, and select rearrangements/fusion events) in 70 cancer-related genes. Comprehensive details on the F1L platform, sequencing, and mutation calling methodologies were previously described [19]. Of the 598 plasma samples that underwent CGP, 508 of these samples were successfully sequenced, having passed quality control parameters for DNA concentrations, library preparation, tumor purity, and target region coverage.
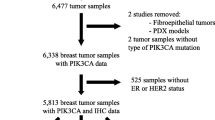
Three hundred thirty-nine of these baseline plasma ctDNA samples harbored one or more pathogenic PIK3CA single-nucleotide variant(s) (PIK3CAmut), defined as a variant of known or likely oncogenic significance, as described by Clark, et al. [19]. Two samples were excluded from subsequent analysis due to the inability to estimate clonality (see Methods subsection “PIK3CAmut clonality estimation” below for more details). The resultant 337 PIK3CAmut patient samples represent the final study population for the downstream analyses described herein, sub-categorized based on the number of PIK3CAmut detected in baseline ctDNA and the estimated clonality of those PIK3CAmut (see again, Methods subsection “PIK3CAmut clonality estimation” below). Samples with one detectable PIK3CAmut were classified as ‘single PIK3CAmut’ (n = 271) and those with ≥ 2 detectable PIK3CAmut were classified as ‘multiple PIK3CAmut’ (n = 66). See Fig. 1 for details.

Flow diagram of baseline ctDNA samples analyzed and estimated clonality collected from SANDPIPER participants. PIK3CAmut is defined as a single nucleotide variant in the PIK3CA gene that is predicted to be of known or likely oncogenic significance
Independent breast cancer dataset
Comprehensive genomic profiling (CGP) was performed on formalin-fixed paraffin-embedded (FFPE) breast tumor samples submitted to Foundation Medicine, Inc. (Cambridge, MA, USA) during the course of routine clinical care through March 31, 2021. Profiling was done for all classes of alterations in at least 324 genes using the FoundationOne® (F1) or FoundationOne®CDx (F1CDx) assay in a CLIA-certified, CAP-accredited laboratory (Foundation Medicine, Inc., Cambridge, MA, USA) [20]. For the analyses herein, samples were filtered from adult patients (≥ 18 years old) and for samples that harbored ≥ 2 pathogenic single-nucleotide variants in the PIK3CA gene. Processing of sequence data and identification of different classes of genomic alterations were performed as previously described [17, 18]. The variant post processing and harmonization process is maintained across the ctDNA and tissue-based assays.
PIK3CAmut clonality estimation
For the SANDPIPER cohort, the clonality of each PIK3CAmut was estimated based on the alteration allele fraction and the estimated ctDNA fraction. The ctDNA fraction was estimated based on tumor aneuploidy or maximum somatic allele fraction, as described previously [21,22,23]. Briefly, when there is clear evidence of aneuploidy (~ 10% tumor shed), an aneuploidy-based estimate of tumor fraction is used. In cases without clear aneuploidy, a maximum somatic allele fraction (MSAF), the highest variant allele fraction among all non-germline variants and after excluding specific clonal hematopoiesis-associated alterations, was used. Clonal fraction (i.e., estimated clonality) of a variant was calculated as the ratio of the variant allele fraction (VAF) to the sample estimated ctDNA fraction. Variants with a clonal fraction of ≥ 0.25 were classified as clonal; variants with a clonal fraction < 0.25 were classified as subclonal. This cutoff was chosen as it represents the clonal fraction where a heterozygous single copy alteration would be identified in a majority of diploid tumor cells. These clonality estimates do not take into account the zygosity and loci copy number estimates. In cases of variants on an amplified allele, this can result in overestimation of tumor fraction or variant clonal fraction. Herein, for samples with a single PIK3CAmut, a 'clonal single PIK3CAmut' is defined as a mutation for which the clonal fraction is ≥ 0.25; a 'subclonal single PIK3CAmut' is defined as a mutation for which the clonal fraction is < 0.25. For samples with multiple PIK3CAmut detected, the PIK3CA clonality category is defined as follows: ‘clonal multiple PIK3CAmut’: samples for which at least two (≥ 2) clonal PIK3CAmut were detected; ‘subclonal multiple PIK3CAmut’: samples for which at most one (≤ 1) clonal PIK3CAmut was detected. These classifications and nomenclature are also succinctly summarized in Additional file 1: Table S1. Two samples for which the clonality of the PIK3CAmut could not be estimated and therefore could not be classified, were excluded from the analysis (Fig. 1).
In the independent dataset of multiple PIK3CAmut breast cancer tissue samples, we utilized estimates of total and mutated copy number from the somatic-germline-zygosity (SGZ) algorithm [24] to calculate the clonality [25]. For each predicted somatic alteration observed in a sample, a tumor fraction was estimated using the following formula: 2AF / (mc – AF(wc + mc-2)), where AF indicates the variant allele fraction, and mc and wc indicate the mutated copies and wild-type copies, respectively. The tumor fraction of the sample was then calculated as the maximum estimated tumor fraction from all the somatic mutations in a sample. Clonal fraction of each mutation was then obtained as the ratio of the AF and sample estimated tumor fraction, with ≥ 50% considered clonal. PIK3CA clonality categories were derived similarly to the SANDPIPER cohort.
Signaling pathway cluster analysis
Signaling pathway alteration status was defined by the detection of at least one pathogenic alteration (i.e., base substitution, insertion and deletion, copy number alteration, or rearrangement of known or likely oncogenic significance) in any gene from the associated pathway gene list in Additional file 1: Table S4. Of note, PIK3CA was removed from the PI3K pathway gene list for the indicated pathway-level analyses; all participants in the analysis cohort harbored a pathogenic PIK3CA mutation. The gene list used for the signaling pathway level analysis for both the SANDPIPER dataset (F1L sequencing of baseline ctDNA) and the independent dataset from Foundation Medicine, Inc. (F1 or F1CDx sequencing of tumor tissue) was guided by and restricted to the genes included in the F1L gene panel.
Analysis of patient response to PI3K inhibition
Patients enrolled in SANDPIPER were stratified into PIK3CAmut subgroups (see Results) against which objective response rate (ORR), represented by the number of patients who responded to treatment (defined by best objective response per investigator assessment, including CR and PR) divided by the total number of patients in each subgroup, and progression-free survival (PFS), defined as time from randomization to first evidence of disease progression as determined by the investigator using Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1) or death from any cause, were assessed. Differences in the ORR among patients in the PIK3CAmut subgroups were compared (p-value) using a stratified exact conditional test. Differences in the PFS among patients in the PIK3CAmut subgroups were compared using the Kaplan–Meier method wherein associated hazard ratios and p-values were obtained from Cox proportional hazards regression models and log-rank test, respectively.
Statistical analyses and software
Statistics, computation, and plotting were performed using Python 2.7 and R 3.6.1. Because analyses were exploratory, p-values were not adjusted for multiple testing. A p-value ≤ 0.05 was considered significant for the analyses herein.
Results
Clonality estimation of multiple PIK3CAmut in ctDNA
Of the 631 patients enrolled to SANDPIPER, 339 baseline ctDNA samples were successfully sequenced and found to harbor at least one detectable PIK3CAmut (Fig. 1). After excluding two samples for the inability to estimate clonality, of these, one PIK3CAmut was detected in 271 samples and are herein termed single PIK3CAmut; ≥ 2 PIK3CAmut were detected in 66 samples and are herein termed multiple PIK3CAmut. Samples were further assigned to a PIK3CAmut clonality category of clonal or subclonal based on a clonality estimation algorithm. Refer to the Methods and Additional file 1: Table S1 for specific details.
Reflective of the 2:1 randomization schema for SANDPIPER, of the 66 samples in which multiple PIK3CAmut were identified, 23 (34.8%) were from patients treated with placebo + fulvestrant (pbo + fulv) and 43 (65.2%) were from patients treated with taselisib + fulvestrant (tas + fulv). Upon clonality estimation, the majority of the multiple PIK3CAmut were categorized as clonal [47 of 66 (71.2%); 18 of 23 (78.3%) in the pbo + fulv arm; 29 of 43 (67.4%) in the tas + fulv arm] and a lower number were categorized as subclonal [19 of 66 (28.8%); 5 of 23 (21.7%) in the pbo + fulv arm; 14 of 43 (32.6%) in the tas + fulv arm] (Fig. 1). Within the subclonal multiple PIK3CAmut cohort, the majority (78.9%; n = 15 of 19) harbored one PIK3CAmut with a clonality estimate of clonal and at least one PIK3CAmut with a clonality estimate of subclonal, and a minority (21.1%; n = 4 of 19) harbored only PIK3CAmut with a clonality estimate of subclonal. Similarly, of the 271 samples in which a single PIK3CAmut was identified, the majority were classified as clonal [247 of 271 (91.1%); 74 of 80 (92.5%) in the pbo + fulv arm; 173 of 191 (90.6%) in the tas + fulv arm] (Fig. 1). Twenty-four samples with a single PIK3CAmut were classified as subclonal [24 of 271 (8.9%); 6 of 80 (7.5%) in the pbo + fulv arm; 18 of 191 (9.4%) in the tas + fulv arm] (Fig. 1). The specific PIK3CAmut and combinations of PIK3CAmut (e.g., H1047R + E726K, E545K + P539R, etc.) identified, clonality categorizations, and distribution between study treatment arms can be found in Additional file 1: Tables S2 & S3.
Clonal multiple PIK3CAmut exhibit fewer RTK co-alterations
Using clonality estimation, we sought to determine whether the mutational landscape differs between breast cancers harboring clonal multiple PIK3CAmut compared to BCs harboring subclonal multiple PIK3CAmut regardless of treatment arm. As illustrated by tile plots, a comparison of the alteration rates of individual genes between clonal (Fig. 2A) vs subclonal (Fig. 2B) multiple PIK3CAmut samples revealed no statistically significant differences. Among the samples in which only a single PIK3CAmut was detected, a statistically lower prevalence of MDM2 amplifications and BRCA2 alterations was observed in those categorized as clonal single PIK3CAmut compared to those categorized as subclonal single PIK3CAmut [MDM2: 1.2% (3 of 247 samples) vs 12.5% (3 of 24 samples), p = 0.0103; BRCA2: 2.4% (6 of 247 samples) vs 12.5% (3 of 24 samples), p = 0.0361] (Additional file 1: Fig. S1).

Gene alteration rates in baseline ctDNA are not different between clonal vs subclonal multiple PIK3CAmut. Tile plots from FoundationOne®Liquid (F1L) sequencing of baseline ctDNA from SANDPIPER participants exhibit the somatic alterations co-occurrent with samples categorized as (A) clonal multiple PIK3CAmut or (B) subclonal multiple PIK3CAmut. Samples are represented in the columns, whereas genes are represented in the rows. No statistically significant difference was observed in the individual gene alteration rates between these PIK3CAmut clonality subgroups (p-value > 0.05; Fisher’s Exact Test). Orange boxes represent samples with multiple alteration types identified in one gene
Analysis of these data at the signaling pathway level (see Additional file 1: Table S4 for pathway gene lists), revealed that patients with ctDNA harboring clonal multiple PIK3CAmut have a lower prevalence of alterations in genes associated with receptor tyrosine kinase (RTK) signaling in baseline ctDNA samples [9 of 47 samples (19.1%)] compared to subclonal multiple PIK3CAmut [9 of 19 samples (47.4%), p = 0.032] (Fig. 3A). Cumulative alterations in other analyzed signaling pathways [i.e., the p53, MAPK, or PI3K (excluding PIK3CA) pathways] were not statistically different between multiple PIK3CAmut clonality groups, despite showing numerical differences. Between treatment groups, no differences were observed in the prevalence of RTK, MAPK, PI3K, or p53 pathway genes altered (Additional file 1: Table S5 and S6; p-value > 0.05, Fisher’s Exact Test). Additionally, the prevalences of RTK, MAPK, PI3K, and p53 pathway genes altered within the subclonal multiple PIK3CAmut subgroup, wherein one of the PIK3CA mutations has a clonality estimate of clonal (n = 15), were similar to the prevalences observed in the entire subclonal multiple PIK3CAmut cohort (data not shown). Furthermore, pathway-grouped (i.e., RTK, p53, MAPK, or PI3K) alterations were neither co-occurrent nor mutually exclusive of one another in samples categorized to either multiple PIK3CAmut clonality group (Fig. 3B-C). Interestingly, the same pathway-level analysis of samples in which only a single PIK3CAmut was identified revealed no statistically significant differences in pathway-grouped gene alterations between clonal and subclonal groups (Additional file 1: Fig. S2).

Clonal multiple PIK3CAmut samples exhibit fewer RTK and non-PIK3CA PI3K pathway gene alterations. A Summary diagram of pathway-level alteration rates in baseline ctDNA from SANDPIPER participants shows a significantly lower prevalence of alterations in genes associated with receptor tyrosine kinase (RTK) signaling in those samples with clonal vs subclonal multiple PIK3CAmut (p = 0.0317); (B & C), tile plot of sample-level pathway co-alteration analysis. Summary diagrams of pathway-level alteration rates in a larger dataset of (D) ERBB2 non-amplified metastatic breast cancer (BC) tumor tissue samples and (E) any BC tumor tissue samples from the Foundation Medicine database harboring clonal multiple PIK3CAmut exhibit a lower prevalence of alterations in RTK-related genes (p = 0.0233 and p = 4.01 × 10–5, respectively) and in non-PIK3CA PI3K-pathway genes (p = 0.0119 and p = 5.09 × 10–4, respectively) compared to samples harboring subclonal multiple PIK3CAmut. A higher proportion of alterations in MAPK pathway genes was observed in breast cancer samples with clonal multiple PIK3CAmut vs those with subclonal multiple PIK3CAmut (p = 9.34 × 10–3) (E). p-values were obtained from a Fisher’s Exact Test. KEYS: For the summary pathway co-alteration analyses, pink and lavender bars represent samples with clonal or subclonal multiple PIK3CAmut, respectively. For alteration type specifications, distinct from “multiple PIK3CAmut”, orange boxes represent samples with multiple alteration types identified in one gene
Independent dataset analysis of clonal PIK3CAmut findings
To determine whether our pathway-level findings in the SANDPIPER ctDNA dataset are supported by a second dataset, we interrogated all available breast tumor tissue samples in an independent database for which PIK3CAmut and clonality calls could be estimated (see Methods and Additional file 1: Fig. S3). The database consists of real-world genomic data from patients whose tumor tissue was submitted for comprehensive genomic profiling as part of routine clinical care. Within this dataset, 1,029 breast cancer tumor samples harbored multiple PIK3CAmut, with a majority estimated as clonal [738 of 1,029 (71.7%)] (Additional file 1: Fig. S3). Consistent with the SANDPIPER cohort, a similar proportion of clonal multiple PIK3CAmut [405 of 520 (77.9%)] was observed in the subgroup of breast cancer tissue samples that were ERBB2 non-amplified and biopsied from a metastatic site of disease (Additional file 1: Fig. S3), which more closely resembles the trial population.
Similar to our observations in the SANDPIPER ctDNA dataset, clonal multiple PIK3CAmut samples from the metastatic breast tumor tissue dataset exhibited significantly fewer alterations in genes associated with RTK signaling [82 of 405 samples (20.2%)] compared to samples with subclonal multiple PIK3CAmut [35 of 115 samples (30.4%), p = 0.0233] (Fig. 3D). In the SANDPIPER ctDNA dataset, we observed a trend of fewer alterations in PI3K (excluding PIK3CA) pathway genes in samples with clonal vs subclonal multiple PIK3CAmut, however this did not reach statistical significance. In the larger dataset of metastatic tumor tissue from patients with ERBB2 non-amplified breast cancer, however, this finding was statistically significant [22 of 405 samples (5.4%) vs 15 of 115 samples (13.0%), p = 0.0119] (Fig. 3D). Taking a broader look across all breast cancer samples (inclusive of primary and metastatic tumor tissue) in the independent dataset with multiple PIK3CAmut samples, a significantly lower proportion of alterations in RTK pathway genes and non-PIK3CA PI3K pathway genes was observed in samples categorized as clonal multiple PIK3CAmut compared to those categorized as subclonal multiple PIK3CAmut [Fig. 3E, RTK: 156 of 738 clonal multiple PIK3CAmut samples (21.1%) vs 98 of 291 subclonal multiple PIK3CAmut samples (33.7%), p = 4.01 × 10–5; PI3K: 45 of 738 clonal multiple PIK3CAmut samples (6.1%) vs 38 of 291 subclonal multiple PIK3CAmut samples (13.1%), p = 5.09 × 10–4]. Conversely, a significantly higher proportion of alterations in MAPK pathway genes (Additional file 1: Table S4) was observed in breast cancer samples categorized as clonal multiple PIK3CAmut compared to those categorized as subclonal multiple PIK3CAmut [Fig. 3E, 177 of 738 samples (24.0%) vs 48 of 291 samples (16.5%), p = 9.34 × 10–3]. Notwithstanding the similarities in the findings from the SANDPIPER baseline ctDNA and the independent breast cancer tumor tissue, differences exist in the timing of the specimen collection and the NGS-based methodologies (e.g., ctDNA- versus tissue-based) that may contribute to the datasets not being contemporary. For example, the baseline plasma samples from the SANDPIPER study were freshly collected during the study screening period prior to initiation of 2L treatment, whereas collection of the metastatic tumor tissue samples was not restricted to the early line metastatic setting.
Clonal multiple PIK3CAmut associate with improved clinical outcomes
To determine whether mutation clonality influences clinical outcomes in patients with ER+/HER2– mBC, we analyzed objective response rates (ORR) and progression-free survival (PFS) of the SANDPIPER participants against treatment regimen (i.e., pbo + fulv or tas + fulv) and against clonal or subclonal status in both the multiple and single PIK3CAmut cohorts. In the multiple PIK3CAmut cohort (n = 66), a higher ORR was observed in patients whose samples were categorized as clonal multiple PIK3CAmut [30% (95% CI, 17–45)] vs those whose samples were categorized as subclonal multiple PIK3CAmut [5.3% (95% CI, 0.13–26)], regardless of treatment regimen (p = 0.021) (Fig. 4A). Within the treatment arms, this finding was not significant for patients treated specifically with pbo + fulv [11% and 0.00% in the clonal vs subclonal subgroups, respectively (95% CI, 1.4–35 vs 0.00–52), p = 1.0] (Fig. 4B). However, this finding was significant for patients treated with tas + fulv [41% and 7.1% in the clonal vs subclonal subgroups, respectively (95% CI, 24–61 vs 0.18–34), p = 0.033] (Fig. 4C).

Clonal multiple PIK3CAmut status correlates with higher ORR and longer PFS. A – C Bar plots of overall objective response rate (ORR) for SANDPIPER study participants show that ORR trends higher or is significantly higher in those whose baseline ctDNA harbored clonal vs subclonal multiple PIK3CAmut. A Study participants who received either study treatment regimen: clonal [14 responses of 47 participants = 30% ORR (95% CI, 17–45)] vs subclonal [1 response of 19 participants = 5.3% ORR (95% CI, 0.13–26)]; p = 0.021. B Study participants who received placebo + fulvestrant: clonal [2 responses of 18 participants = 11% ORR (95% CI, 1.4–35)] vs subclonal [0 responses of 5 participants = 0.00% ORR (95% CI, 0.0–52)]; p = 1.0. C Study participants who received taselisib + fulvestrant: clonal [12 responses of 29 participants = 41% ORR (95% CI, 243.5–61)] vs subclonal [1 response of 14 participants = 7.1% ORR (95% CI, 0.18–34)]; p = 0.033. D Shown via Kaplan–Meier curves, median progression-free survival (PFS) was longer for those whose corresponding baseline ctDNA harbored clonal multiple PIK3CAmut vs subclonal multiple PIK3CAmut and were treated with either placebo + fulvestrant [median PFS (mPFS) = 3.9 vs 2.0 months, respectively; Hazard Ratio (HR) = 0.19 (95% CI, 0.051–0.73), p = 0.025] or with taselisib + fulvestrant [mPFS = 7.6 vs 5.1 months, respectively; HR = 0.37 (95% CI, 0.16–0.87), p = 0.027]. See Methods on details for calculations and statistics of ORR and PFS. mut, mutation(s); n, number of study participants or samples in the indicated subgroup
In comparing patient ORR for samples of single PIK3CAmut clonality status, ORR did not significantly differ in the total analyzed population [either treatment regimen, clonal vs subclonal single PIK3CAmut status: 15% vs 25% (95% CI, 11–20 vs 9.8–47), p = 0.24] or under either SANDPIPER treatment regimen [pbo + fulv, clonal vs subclonal status: 9.5% vs 17% (95% CI, 3.9–19 vs 0.42–64), p = 0.48; tas + fulv, clonal vs subclonal status: 17% vs 28% (95% CI 12–24 vs 9.7–53), p = 0.33]. Numerically higher ORR was seen in patients with ctDNA categorized as subclonal single PIK3CAmut vs clonal single PIK3CAmut, but did not reach significance (Additional File 1: Fig. S4A-SC). This may suggest that subclonal single PIK3CAmut tumors are less aggressive than clonal single PIK3CAmut tumors, however, further investigation and a larger sample size are warranted to confirm this observation and to better elucidate the potential associated biological underpinnings.
When we examined PFS within each SANDPIPER treatment arm, the results were complementary to the ORR analysis. Specifically, median PFS (mPFS) was longer for patients whose baseline ctDNA samples were categorized as clonal multiple PIK3CAmut vs subclonal multiple PIK3CAmut for both the pbo + fulv arm [mPFS = 3.9 vs 2.0 months, respectively; Hazard Ratio (HR) = 0.19 (95% CI, 0.051–0.73), p = 0.025] and the tas + fulv arm [mPFS = 7.6 vs 5.1 months, respectively; HR = 0.37 (95% CI, 0.16–0.87), p = 0.027] (Fig. 4D). mPFS was longest overall for patients who received tas + fulv and whose baseline ctDNA harbored clonal multiple PIK3CAmut (Fig. 4D). For patients whose baseline ctDNA harbored only a single PIK3CAmut, we observed no significant difference in mPFS between clonality subgroups under either treatment arm (Additional file 1: Fig. S4D). Lastly, the ORR and mPFS of the subclonal multiple PIK3CAmut subgroup wherein one of the PIK3CA mutations has a clonality estimate of clonal (n = 15) were similar to the clinical outcomes observed in the entire subclonal multiple PIK3CAmut cohort (data not shown).
Discussion
As previously described, tumor cells harboring multiple PIK3CAmut occurring in cis confer increased sensitivity to p110α inhibition compared to tumor cells harboring only a single PIK3CAmut or multiple PIK3CAmut occurring in trans [16]. Therein, the authors performed both long-range single-molecule real-time sequencing (SMRTseq) [26] of circular DNA templates from multiple PIK3CAmut breast cancer cell lines and a limited number of fresh tumor samples, as well as an in silico clonality estimation analysis [27] on a previously-published breast cancer sequencing dataset [6] to infer in cis vs in trans status of multiple PIK3CAmut. For the latter, co-occurring clonal PIK3CAmut were representative of multiple PIK3CAmut in cis, whereas a mix of co-occurring clonal and subclonal, or subclonal only, PIK3CAmut were representative of multiple PIK3CAmut in trans. Together, these efforts revealed that multiple PIK3CAmut identified in breast cancers more commonly occur in cis [16].
We have recently shown that clonal multiple PIK3CAmut predominantly occur in cis while subclonal multiple PIK3CAmut frequently occur in trans [25]. Based on this, we were able to confirm the preclinical findings from Vasan, et al. [16] in a phase III clinico-genomics dataset. Although some of the analysis subgroups were relatively small, we nevertheless observed prolonged PFS and increased ORR for patients with ctDNA harboring clonal vs subclonal multiple PIK3CAmut, which was enhanced for those patients randomized to treatment with taselisib + fulvestrant. This is analogous to the preclinical findings that multiple PIK3CAmut breast epithelial cells are more sensitive to alpelisib treatment when the PIK3CA mutations are in cis compared to when they are in trans [16]. These trends were also observed in a phase I/Ib population of patients with HR+/HER2– mBC treated with inavolisib (GDC-0077) alone or with endocrine therapy and with or without palbociclib (NCT03006172). Specifically, the fraction of patients who experienced partial response or stable disease in this study was higher in those patients from whom multiple PIK3CAmut were detected in baseline ctDNA compared to those in whom only a single PIK3CAmut was detected [28]. Furthermore, from a case report of a patient with ER+/HER2– mBC treated with alpelisib plus fulvestrant who achieved a partial response, molecular profiling revealed that both the primary tumor and metastatic lesions harbored multiple PIK3CAmut [29].
Supportive of the correlations we observed with clinical response, our analysis of multiple PIK3CAmut clonality against the broader genomic landscape of breast cancer ctDNA reveals an updated model for dependence on the PI3K pathway. To illustrate the interpretation of our findings in conjunction with previously published work, we have generated the model shown in Fig. 5. We hypothesize clonal multiple PIK3CAmut alone may be sufficient to drive tumor growth and proliferation through hyperactivation of the PI3K/AKT pathway (Fig. 5A) and as a result, are highly sensitive to PI3K inhibition (Fig. 5C). Conversely, our data also suggest subclonal multiple PIK3CAmut may require additional input from co-altered signaling pathway genes, including alterations in RTK pathway genes, to fully drive tumor growth and proliferation through the PI3K and parallel pathways (Fig. 5B). As such, tumor growth and proliferation may not be fully abrogated by PI3K inhibition alone due to potential bypass mechanisms. Ultimately, our observations suggest that rational therapeutic combinations should be considered – particularly for those patients with tumors harboring subclonal multiple PIK3CAmut, wherein co-targeting of concomitantly dysregulated RTK or non-PIK3CA PI3K pathway genes in addition to p110α may be beneficial (Fig. 5D-E).

Models for interpreting predicted reliance on PI3K pathway signaling and sensitivity to p110α inhibition. A Tumors with clonal multiple PIK3CAmut are sufficient to drive tumor growth and proliferation through hyperactivation of the PI3K pathway alone and (B) are highly sensitive to PI3K inhibition. C Tumors with subclonal multiple PIK3CAmut may be insufficient to adequately drive growth and proliferation alone and may have co-occurring alterations in RTK and/or non-PIK3CA PI3K pathway genes (C’, C’’, C’’’). In these instances, tumor growth and proliferation may not be fully abrogated by PI3K inhibition alone due to (D) activation of additional parallel signaling pathways, and/or (E) further enhanced PI3K/AKT pathway signaling
An open question, however, is whether the clonality of PIK3CAmut – especially those of subclonal status – is representative of co-occurrence or mutual exclusivity of alterations in RTK and PI3K pathway genes at the single-cell level. If PIK3CAmut and RTK/PI3K pathway alterations occur in separate cells, a treatment combination may provide more benefit than if these alterations were to occur in the same cell. Although we are likely a distant future away from integrating single-cell technologies into clinical practice, which may afford an opportunity to better characterize and refine our understanding of the clonality of alterations in key signaling pathways.
Our study also raises questions about the importance of multiple PIK3CAmut across cancer types. First and foremost, do our findings in HR+/HER2– metastatic breast cancer translate to other cancer types harboring multiple PIK3CAmut? One could surmise that other cancer types exhibit different levels of dependence on the PI3K/AKT pathway compared to their reliance on other cancer-dysregulated signaling pathways, and depending on the genomic background of those cancers, they may be differentially influenced by the presence of multiple PIK3CAmut. According to the analysis by Vasan, et al., colorectal (CRC) and uterine/endometrial (UC/EC) cancers harbor the next highest proportion of multiple PIK3CAmut after breast cancers [16]. Through the independent tumor tissue database, we analyzed 635 CRC and 681 UC/EC tumor tissue samples with clonal vs subclonal multiple PIK3CAmut against the potential for co-occurring RTK and non-PIK3CA PI3K pathway alterations and observed no differences (data not shown); as alluded to, this may be due to inherent differences in the biologies of these tumor types compared to breast cancer and/or due to an underpowered dataset. To address exactly those questions about the importance of multiple PIK3CAmut beyond breast cancer, new clinical studies are underway. One such study is the ongoing TAPISTRY (Tumor-Agnostic Precision Immuno-Oncology and Somatic Targeting Rational for You) platform study (NCT04589845). Leveraging site-based NGS results to augment enrollment, TAPISTRY will assess the efficacy of the p110α inhibitor, inavolisib, in adolescent and adult patients with a range of advanced or metastatic tumor types harboring multiple PIK3CAmut in patients that have failed conventional therapeutic options.
Conclusions
In summary, our study establishes the prospect that multiple clonal PIK3CAmut are prognostic of response to PI3K inhibition. These data provide additional rationale for the clinical investigation of the clonality status of PIK3CAmut with PI3K-inhibitors like the p110α-isoform selective inhibitor, inavolisib, alone or in combination with other rationally selected therapies.
Availability of data and materials
All relevant data are provided as supplementary information. Information on the availability of data from the SANDPIPER clinical trial can be found in the published primary clinical manuscript [14]. In brief, qualified researchers can request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). SANDPIPER trial participants did not give consent to publicly share their individualized comprehensive genomic profiling data. However, the clinical study data (https://vivli.org/) and requests for genomic profiling data and analyses can be released to qualified requestors along with necessary agreements to enforce terms such as security, patient privacy, and consent of specified data use, consistent with evolving and applicable data protection laws. For up-to-date details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here: https://go.roche.com/data_sharing.
Due to HIPAA requirements, Foundation Medicine, Inc. (FMI) is not consented to share individualized patient genomic data, which contains potentially identifying or sensitive information. Foundation Medicine is committed to collaborative data analysis and has well established and widely used mechanisms by which qualified researchers can query our genomic database of > 600,000 de-identified sequenced cancers. More information and mechanisms for data access can be obtained by contacting the corresponding author or the Foundation Medicine Data Governance Council at data.governance.council@foundationmedicine.com. This study made use of publicly available packages with R version 3.6.1 and Python 2.7. Code used to generate figures are available upon request.
Abbreviations
- 2L:
-
Second-line therapy
- AF:
-
Allele fraction or allele frequency
- AKT:
-
Protein kinase B
- BICR-PFS:
-
Progression-free survival (PFS) per blinded independent central review
- BC(s):
-
Breast cancer(s)
- BOR:
-
Best objective response
- BRCA2 :
-
BReast CAncer gene 2
- CAP:
-
College of American Pathologists
- CBR:
-
Clinical benefit rate
- CGP:
-
Comprehensive genomic profiling
- CI:
-
Confidence interval
- CLIA:
-
Clinical Laboratory Improvement Amendments
- CR:
-
Complete response
- CRC:
-
Colorectal
- ctDNA:
-
Circulating tumor DNA
- DNA:
-
Deoxyribonucleic acid
- DoR:
-
Duration of objective response
- EC:
-
Endometrial cancer
- ER+:
-
Estrogen receptor-positive
- ERBB2 :
-
Erb-b2 receptor tyrosine kinase 2
- F1:
-
FoundationOne®
- F1CDx:
-
FoundationOne®CDx
- F1L:
-
FoundationOne®Liquid assay
- FDA:
-
Food and Drug Administration
- FFPE:
-
Formalin-fixed, paraffin-embedded
- FMI:
-
Foundation Medicine, Inc.
- fulv:
-
Fulvestrant
- HER2–:
-
Human epidermal growth factor receptor 2-negative
- HER2+:
-
Human epidermal growth factor receptor 2-positive
- HR+:
-
Hormone receptor-positive
- IRB:
-
Institutional review board
- MAPK:
-
Mitogen activated protein kinase (pathway)
- mc:
-
Mutated copies
- MDM2 :
-
Murine double minute 2 gene
- MND:
-
Mutation not detected
- mPFS:
-
Median progression-free survival
- MSAF:
-
Maximum somatic allele fraction
- mTOR:
-
Mammalian target of rapamycin
- mut:
-
Mutated, mutation(s), or mutant
- NGS:
-
Next-generation sequencing
- NMD:
-
No mutation detected
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- p110α:
-
Phosphatidylinositol-4,5-bisphosphtate 3-kinase catalytic subunit alpha protein, 110 kilodaltons
- p110β:
-
Phosphatidylinositol-4,5-bisphosphtate 3-kinase catalytic subunit beta protein, 110 kilodaltons
- p53:
-
Transformation-related protein 53
- PFS:
-
Progression-free survival
- PI3K:
-
Phosphatidylinositol 3-kinase complex
- PI3Kα:
-
Phosphoinositol 3-kinase alpha
- PIK3CA :
-
Phosphatidylinositol-4,5-bisphosphtate 3-kinase catalytic subunit alpha gene
- PIK3CAmut:
-
PIK3CA mutated, mutation(s), or mutant
- pbo:
-
Placebo
- PR:
-
Partial response
- RECIST:
-
Response Evaluation Criteria in Solid Tumors
- RTK:
-
Receptor tyrosine kinase
- SGZ:
-
Somatic-germline-zygosity
- SMRTseq:
-
Single-molecule real-time sequencing
- SNV:
-
Single nucleotide variant
- TAPISTRY:
-
Tumor-Agnostic Precision Immuno-Oncology and Somatic Targeting Rational for You platform study
- tas:
-
Taselisib
- UC:
-
Uterine cancer
- VAF:
-
Variant allele fraction
- wc:
-
Wild-type copies
References
Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170(4):605–35.
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–13.
Zhang Y, Kwok-Shing Ng P, Kucherlapati M, Chen F, Liu Y, Tsang YH, de Velasco G, Jeong KJ, Akbani R, Hadjipanayis A, et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell. 2017;31(6):820-832 e823.
Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, Szabo S, Konishi H, Karakas B, Blair BG, Lin C, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3(8):772–5.
Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, Cai Y, Bielski CM, Donoghue MTA, Jonsson P, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. 2018;34(3):427-438 e426.
Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65(23):10992–1000.
Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102(3):802–7.
Baselga J, Im SA, Iwata H, Cortes J, De Laurentiis M, Jiang Z, Arteaga CL, Jonat W, Clemons M, Ito Y, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(7):904–16.
Di Leo A, Johnston S, Lee KS, Ciruelos E, Lonning PE, Janni W, O’Regan R, Mouret-Reynier MA, Kalev D, Egle D, et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018;19(1):87–100.
Powles T, Lackner MR, Oudard S, Escudier B, Ralph C, Brown JE, Hawkins RE, Castellano D, Rini BI, Staehler MD, et al. Randomized open-label phase II trial of Apitolisib (GDC-0980), a novel inhibitor of the PI3K/Mammalian target of rapamycin pathway, versus Everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. 2016;34(14):1660–8.
Vuylsteke P, Huizing M, Petrakova K, Roylance R, Laing R, Chan S, Abell F, Gendreau S, Rooney I, Apt D, et al. Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3-kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor-positive, HER2-negative, locally recurrent, or metastatic breast cancer: interim analysis of the multicentre, placebo-controlled, phase II randomised PEGGY study. Ann Oncol. 2016;27(11):2059–66.
Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, Iwata H, Conte P, Mayer IA, Kaufman B, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380(20):1929–40.
Dent S, Cortes J, Im YH, Dieras V, Harbeck N, Krop IE, Wilson TR, Cui N, Schimmoller F, Hsu JY, et al. Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor-positive, PIK3CA-mutant, HER2-negative, advanced breast cancer: the SANDPIPER trial. Ann Oncol. 2021;32(2):197–207.
Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, Middleton MR, Berlin J, Schuler M, Gil-Martin M, et al. Phosphatidylinositol 3-Kinase alpha-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J Clin Oncol. 2018;36(13):1291–9.
Vasan N, Razavi P, Johnson JL, Shao H, Shah H, Antoine A, Ladewig E, Gorelick A, Lin TY, Toska E, et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kalpha inhibitors. Science. 2019;366(6466):714–23.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–31.
Milbury CA, Creeden J, Yip WK, Smith DL, Pattani V, Maxwell K, Sawchyn B, Gjoerup O, Meng W, Skoletsky J, et al. Clinical and analytical validation of FoundationOne(R)CDx, a comprehensive genomic profiling assay for solid tumors. PLoS ONE. 2022;17(3):e0264138.
Clark TA, Chung JH, Kennedy M, Hughes JD, Chennagiri N, Lieber DS, Fendler B, Young L, Zhao M, Coyne M, et al. Analytical validation of a hybrid capture-based next-generation sequencing clinical assay for genomic profiling of cell-free circulating tumor DNA. J Mol Diagn. 2018;20(5):686–702.
Westphalen CB, Krebs MG, Le Tourneau C, Sokol ES, Maund SL, Wilson TR, Jin DX, Newberg JY, Fabrizio D, Veronese L, et al. Genomic context of NTRK1/2/3 fusion-positive tumours from a large real-world population. NPJ Precis Oncol. 2021;5(1):69.
Husain H, Pavlick DC, Fendler BJ, Madison RW, Decker B, Gjoerup O, Parachoniak CA, McLaughlin-Drubin M, Erlich RL, Schrock AB, et al. Tumor fraction correlates with detection of actionable variants across > 23,000 circulating tumor DNA samples. JCO Precis Oncol. 2022;6:e2200261.
Tukachinsky H, Madison RW, Chung JH, Gjoerup OV, Severson EA, Dennis L, Fendler BJ, Morley S, Zhong L, Graf RP, et al. Genomic analysis of circulating tumor DNA in 3,334 patients with advanced prostate cancer identifies targetable BRCA alterations and AR resistance mechanisms. Clin Cancer Res. 2021;27(11):3094–105.
Woodhouse R, Li M, Hughes J, Delfosse D, Skoletsky J, Ma P, Meng W, Dewal N, Milbury C, Clark T, et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS ONE. 2020;15(9):e023780.
Sun JX, He Y, Sanford E, Montesion M, Frampton GM, Vignot S, Soria JC, Ross JS, Miller VA, Stephens PJ, et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched norma. PLoS Comput Biol. 2018;14(2):e1005965.
Sivakumar S, Jin DX, Rathod R, Ross J, Cantley LC, Scaltriti M, et al. Genetic heterogeneity and tissue-specific patterns of tumors with multiple PIK3CA mutations. Clin Cancer Res. 2023;29(6):1125–36.
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323(5910):133–8.
Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44(16):e131.
Jhaveri K, Juric D, Varga A, Turner N, Schmid P, Saura C, et al. Preliminary correlative analysis of clinical outcomes with PIK3CA mutation (mut) status from a phase I/Ib study of GDC-0077 in patients (pts) with hormone receptor-positive/HER2-negative metastatic breast cancer (HR+/HER2- mBC). San Antonio Breast Cancer Symposium 2020. Poster, abstract published in Cancer Res. 2021;81(4_Supplement):PS5–12.
Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, Ebbesen SH, Ainscough BJ, Ramu A, Iyer G, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature. 2015;518(7538):240–4.
Acknowledgements
We thank the participants, their families, the nurses, the investigators, and the other site staff who participated in the SANDPIPER study as well as the SANDPIPER study team. In addition, we thank Steven Gendreau for helpful discussions and Sophia Maund for general coordination and support with the team at Foundation Medicine, Inc.
Funding
This work was supported by F. Hoffmann-La Roche Ltd, Basel, Switzerland.
Author information
Authors and Affiliations
Contributions
Conception & Design: K.E. Hutchinson, T.R. Wilson, J.W. Chen. Acquisition of Data: H.M. Savage, T.R. Wilson, S.E. Trabucco, S. Sivakumar, E.S. Sokol. Analysis (e.g. statistical analyses, biostatistics, computational analyses) and interpretation of data: K.E. Hutchinson, J. W. Chen, H.M. Savage, T. R. Wilson, S. Sivakumar, E.S. Sokol. Writing, review, and/or revision of the manuscript: All authors. Administrative, technical, or material support (i.e. reporting or organizing data): H.M. Savage, T.R. Wilson. Study supervision: K.E. Hutchinson, T.R. Wilson, E.S. Sokol. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Written informed consent to participate was obtained from all participants of the GO29058 (SANDPIPER) study. Approval for the analysis of samples from the Foundation Medicine database, including a waiver of informed consent and Health Insurance Portability and Accountability Act waiver of authorization, was obtained from the Western Institutional Review Board (protocol #20152817). The Institutional Review Board granted a waiver of informed consent under 45 CFR § 45.116 based on review and determination that this research meets the following requirements: (i) the research involves no more than minimal risk to the subjects; (ii) the research could not practicably be carried out without the requested waiver; and (iii) the waiver will not adversely affect the rights and welfare of the subjects.
Details of the prospectively registered phase III SANDPIPER trial, as well as the presentation of the primary and secondary endpoints of the trial (clinicaltrials.gov: NCT02340221) were reported previously [14]. In brief, SANDPIPER studied the clinical efficacy of taselisib (GDC-0032) plus fulvestrant vs placebo plus fulvestrant in patients with ER+/HER2– PIK3CAmut locally advanced or metastatic BC. See Methods for additional details. SANDPIPER was approved by the relevant ethics bodies or institutional review boards (IRBs) at each participating site and conducted per the principles of the Declaration of Helsinki, International Council for Harmonisation Guidelines, and the laws and regulations of the countries in which it was conducted. All patients provided written informed consent for participation in the study, including tumor tissue and blood sampling and subsequent protocol-defined and exploratory biomarker analyses. SANDPIPER enrolled participants on the basis of a valid cobas® PIK3CA Mutation Test result from freshly collected or archival tumor tissue; both PIK3CAmut and PIK3CA mutation-not-detected (MND) were eligible.
Consent for publication
Not applicable.
Competing interests
J.W. Chen, K.E. Hutchinson, H.M. Savage, T.J. Stout, F. Schimmoller, and T.R. Wilson are all employees of Roche/Genentech and own stock in Roche.
J. Cortés: Consulting/Advisor: Roche, Celgene, Cellestia, AstraZeneca, Seattle Genetics, Daiichi Sankyo, Erytech, Athenex, Polyphor, Lilly, Merck Sharp&Dohme, GSK, Leuko, Bioasis, Clovis Oncology, Boehringer Ingelheim, Ellipses, Hibercell, BioInvent, Gemoab, Gilead, Menarini, Zymeworks, Reveal Genomics; Honoraria: Roche , Novartis , Celgene, Eisai, Pfizer, Samsung Bioepis, Lilly, Merck Sharp&Dohme, Daiichi Sankyo; Research funding to the Institution: Roche, Ariad pharmaceuticals, AstraZeneca, Baxalta GMBH/Servier Affaires, Bayer healthcare, Eisai, F.Hoffman-La Roche, Guardanth health, Merck Sharp&Dohme, Pfizer, Piqur Therapeutics, Puma C, Queen Mary University of London; Stock: MedSIR, Nektar Pharmaceuticals, Leuko (relative); Travel, accommodation, expenses: Roche, Novartis, Eisai, pfizer, Daiichi Sankyo, Astrazeneca, Gilead. Patents: “Pharmaceutical Combinations of A Pi3k Inhibitor And A Microtubule Destabilizing Agent. Javier Cortés Castán, Alejandro Piris Giménez, Violeta Serra Elizalde. WO 2014/199294 A” & “Her2 as a predictor of response to dual HER2 blockade in the absence of cytotoxic therapy. Aleix Prat, Antonio Llombart, Javier Cortés.US 2019/ 0338368 A1”.
S. Dent: Honararia for consulting: Novartis, Astra Zeneca.
N. Harbeck: Honoraria for lectures and/or consulting: Amgen, AstraZeneca, Daiichi-Sankyo, Exact Sciences, Gilead, Lilly, MSD, Novartis, Pierre-Fabre, Pfizer, Roche, Sandoz, Seagen.
W. Jacot: Dr. Jacot reports grants, personal fees and non-financial support from Astra Zeneca; personal fees and non-financial support from Eisai, Novartis, Roche, Pfizer, Eli Lilly, and Chugai; personal fees from MSD, Bristol Myers Squibb, and Seagen; and grants and personal fees from Daiichi Sankyo, outside the submitted work.
I. Krop: Honoraria from Genentech, AstraZeneca, and Celltrion; fees from a consulting or advisory role from Genentech/Roche, Seattle Genetics, Daiichi Sankyo, MacroGenics, Taiho Pharmaceutical, Context Therapeutics, Novartis, Merck, and Ionis; research funding from Genentech and Pfizer; employment/leadership/stock and other ownership interests (for an immediate family member) from AMAG Pharmaceuticals.
S.E. Trabucco, S. Sivakumar, and E.S. Sokol are all employees of Foundation Medicine, a wholly owned subsidiary of Roche and are shareholders in Roche.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
PDF file, containing the article-associated supplementary tables and figures. Table S1. Clonality subgroup nomenclature and associated definitions for single and multiple PIK3CAPIK3CAmut; Table S2. Clonality estimation and tabulation of multiple PIK3CAPIK3CAmut identified in baseline ctDNA from SANDPIPER participants; Table S3. Clonality estimation and tabulation of single PIK3CAPIK3CAmut identified in baseline ctDNA from SANDPIPER participants; Table S4. Signaling pathway gene lists utilized in pathway-level analyses; Table S5. Fraction of samples with altered signaling pathway genes between SANDPIPER treatment groups; Table S6. Fraction of samples with altered signaling pathway genes between SANDPIPER treatment groups stratified by clonality status; Fig. S1. Gene alteration rates between SANDPIPER baseline ctDNA samples categorized as clonal vs subclonal single PIK3CAPIK3CAmut; Fig. S2. Pathway-level co-alteration analyses between ctDNA samples harboring clonal vs subclonal single PIK3CAPIK3CAmut; Fig. S3. Consort diagram for the clonality analysis of an independent breast tumor tissue dataset; Fig. S4. ORR and PFS of SANDPIPER participants whose baseline ctDNA samples harbored single PIK3CAPIK3CAmut.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hutchinson, K.E., Chen, J.W., Savage, H.M. et al. Multiple PIK3CA mutation clonality correlates with outcomes in taselisib + fulvestrant-treated ER+/HER2–, PIK3CA-mutated breast cancers. Genome Med 15, 28 (2023). https://doi.org/10.1186/s13073-023-01181-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-023-01181-8