
Abstract
Background
We previously reported that impaired type I IFN activity, due to inborn errors of TLR3- and TLR7-dependent type I interferon (IFN) immunity or to autoantibodies against type I IFN, account for 15–20% of cases of life-threatening COVID-19 in unvaccinated patients. Therefore, the determinants of life-threatening COVID-19 remain to be identified in ~ 80% of cases.
Methods
We report here a genome-wide rare variant burden association analysis in 3269 unvaccinated patients with life-threatening COVID-19, and 1373 unvaccinated SARS-CoV-2-infected individuals without pneumonia. Among the 928 patients tested for autoantibodies against type I IFN, a quarter (234) were positive and were excluded.
Results
No gene reached genome-wide significance. Under a recessive model, the most significant gene with at-risk variants was TLR7, with an OR of 27.68 (95%CI 1.5–528.7, P = 1.1 × 10−4) for biochemically loss-of-function (bLOF) variants. We replicated the enrichment in rare predicted LOF (pLOF) variants at 13 influenza susceptibility loci involved in TLR3-dependent type I IFN immunity (OR = 3.70[95%CI 1.3–8.2], P = 2.1 × 10−4). This enrichment was further strengthened by (1) adding the recently reported TYK2 and TLR7 COVID-19 loci, particularly under a recessive model (OR = 19.65[95%CI 2.1–2635.4], P = 3.4 × 10−3), and (2) considering as pLOF branchpoint variants with potentially strong impacts on splicing among the 15 loci (OR = 4.40[9%CI 2.3–8.4], P = 7.7 × 10−8). Finally, the patients with pLOF/bLOF variants at these 15 loci were significantly younger (mean age [SD] = 43.3 [20.3] years) than the other patients (56.0 [17.3] years; P = 1.68 × 10−5).
Conclusions
Rare variants of TLR3- and TLR7-dependent type I IFN immunity genes can underlie life-threatening COVID-19, particularly with recessive inheritance, in patients under 60 years old.
Similar content being viewed by others

Background
Clinical variability is high in unvaccinated individuals infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), ranging from silent infection to lethal disease. In ~ 3% of cases, infection leads to critical COVID-19 pneumonia, requiring high-flow oxygen (O2 > 6 L/min), mechanical ventilation (non-invasive or by intubation), or extracorporeal membrane oxygenation (ECMO) [1]. Advanced age is by far the strongest predictor of COVID-19 severity, with the risk of death doubling every 5 years of age from childhood onward [2, 3]. Men are also at greater risk of death than women [3,4,5]. Genome-wide (GW) association studies have identified several common loci associated with COVID-19 severity, the most significant being a region on chromosome 3p21.31 that was introduced by archaic introgression from Neanderthals [6,7,8,9,10]. The risk haplotype encompasses six genes (SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6, and XCR1) and confers an estimated OR per copy of between 1.6 and 2.1, with higher values for individuals under 60 years old [7, 11]. Twenty-four GW regions have been shown to be significantly associated with critical COVID-19 [10,11,12]. Four of these regions encompass genes involved in type I IFN immunity. The first, on chr12q24.13, containing protective variants, is also a Neanderthal haplotype [13] and includes the OAS1, OAS2, and OAS3 cluster, these interferon-stimulated genes (ISGs) being required for the activation of antiviral RNaseL. The second, a region on chr21q22.1, includes IFNAR2. The third, a region on chr19p13.2, includes TYK2. The fourth, a region on chr9p21, includes IFNA10. However, common variants have a modest effect size and explain only a very small fraction of the clinical variability [6, 8]. This prompted us to search for rare variants conferring a stronger predisposition to life-threatening COVID-19.
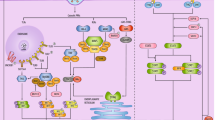
Through a candidate approach focusing on influenza susceptibility genes, the COVID Human Genetics Effort (CHGE [14]) provided proof-of-concept that autosomal inborn errors of TLR3-dependent and -independent type I interferon (IFN) immunity, including autosomal recessive (AR) deficiencies of IFNAR1 or IRF7, can underlie critical COVID-19 [15]. Other children with AR IFNAR1, IFNAR2, TBK1, or STAT2 deficiency were subsequently reported, as well as children with AR TYK2 deficiency [16,17,18,19,20] (Fig. 1). Some other groups were unable to replicate these findings, but the variants were not tested biochemically and it is unclear whether recessive defects were considered [11, 21,22,23]. There may also be other reasons for their findings [1, 24], the most important being the age distribution of the case cohorts. The other case cohorts were much older than ours (mean age of 66 vs. 52 years) and we found that inborn errors of immunity (IEI) were more frequent in patients under 60 years old [25]. Consistently, we recently reported that ~ 10% of children with moderate, severe, or critical COVID-19 pneumonia had recessive inborn errors of type I IFN immunity [19]. Moreover, older patients are more likely to carry pre-existing autoantibodies (auto-Abs) neutralizing type I IFN, which are found in about 15% of critical cases and up to 21% of patients over the age of 80 years [26, 27]. The presence of such auto-Abs has been replicated by at least 26 studies worldwide [28, 29], and we also recently showed that autoimmunity to type I IFNs is a strong common predictor of COVID-19 death in unvaccinated individuals, providing further evidence for the role of type I IFN immunity in life-threatening COVID-19 [29].

Type I IFN immunity genes associated with life-threatening COVID-19. Inborn errors of type I IFN immunity and autoantibodies neutralizing type I IFNs (α, β, ω) underlie life-threatening COVID-19 pneumonia by interfering with type I IFN immunity in respiratory epithelial cells (RECs) and blood plasmacytoid dendritic cells (pDCs). SARS-CoV-2 infection can induce type I IFN production in a TLR3-dependent manner in tissue-resident RECs (which express TLR3 but not TLR7) and in a TLR7-dependent manner in circulating pDCs (which express TLR7 but not TLR3). IRF7 is constitutively expressed in pDCs, at higher levels than in other cell types, whereas it is mostly induced by viral infection in RECs. Reported in red are the 13 genes (IFNAR1, IFNAR2, IRF3, IRF7, IRF9, IKBKG, STAT1, STAT2, TBK1, TICAM1, TLR3, TRAF3, and UNC93B1) investigated in a previous study [15]; TYK2 and TLR7 were subsequently shown to underlie severe COVID-19 [19, 30]
Using an unbiased X-wide gene burden test, we also identified X-linked recessive (XR) TLR7 deficiency in 17 male patients aged 7–71 years with critical COVID-19 pneumonia, accounting for ~ 1% of cases in men (Fig. 1) [30]. Moreover, six of the 11 TLR7 variants previously reported in patients from other studies were deleterious (carried by nine of 16 patients) [31,32,33,34,35,36], whereas the TLR7 variants in other studies were not disclosed [21, 22]. TLR3 senses viral dsRNA in respiratory epithelial cells, whereas TLR7 senses ssRNA in plasmacytoid dendritic cells [25, 28]. Both pathways induce the production of type I IFNs. TLR7 gain-of-function variants were recently shown to be associated with human systemic lupus erythematosus [37], providing an example of mirror genetic effects between infectious and inflammatory/autoimmune diseases [38]. Collectively, these findings suggest that type I IFNs are essential for protective immunity to SARS-CoV-2 in the respiratory tract, with insufficient type I IFN activity accounting for up to 15–20% of cases of life-threatening COVID-19. Despite this high proportion, the determinants of critical COVID-19 pneumonia remain to be identified in ~ 80% of cases. Here, we tested the hypotheses that other IEI may underlie critical COVID-19 pneumonia in at least some patients and that our initial findings could be replicated in a new cohort. With the CHGE, we performed a GW gene-based rare variant association analysis. This analysis was performed in both previously investigated patients who had not been screened at the GW level [15, 19, 30], and in newly recruited patients. We also tested the hypothesis that we could replicate our initial finding of an enrichment in pLOF variants of candidate type I IFN-related genes in newly recruited patients, given the controversy from other groups. We extended the analysis to two other type I IFN-related genes, TLR7 and TYK2, that we had recently found to be associated with critical COVID-19 [19, 30], and to branchpoint (BP) variants with a potentially strong impact on the splicing of the 15 type I IFN-related genes [39]. Finally, we refined the analysis of the type I IFN-related genes by taking age, sex, and zygosity into account.
Methods
Cohort
Since the beginning of the pandemic, we have enrolled more than 9000 individuals with SARS-CoV-2 infection and broad clinical manifestations from all over the world through the COVID Human Genetic Effort (CHGE). In this study, we focused on 3503 patients with life-threatening COVID-19 and 1373 individuals with asymptomatic/mild infection. Life-threatening COVID-19 cases were defined as patients with pneumonia who developed critical disease, whether pulmonary with high-flow oxygen (> 6 L/min) or mechanical ventilation [continuous positive airway pressure (CPAP), bilevel positive airway pressure (BIPAP), and intubation], septic shock, or any other type of organ damage requiring intensive care unit admission. We screened for the presence of autoantibodies (auto-Abs) against type I IFNs in all patients for whom plasma was available (N = 928), as previously described [26, 27], and we excluded 234 patients who tested positive for auto-Abs as they already have a major risk factor for developing critical COVID-19 [29]. In total, 3269 patients with life-threatening COVID-19 were included in the analysis. Among those 3269 patients, 1301 had been included in previous studies restricted to a short list of 18 candidate genes [15, 19] or to the X chromosome [30], and 1968 had not been studied before. Controls were defined as individuals infected with SARS-CoV-2 who remained asymptomatic or pauci-symptomatic, with the presence of mild, self-healing, ambulatory disease (N = 1373). The presence of infection was assessed on the basis of a positive PCR test and/or serological test and/or the presence of typical symptoms such as anosmia or agueusia after exposure to a confirmed COVID-19 case. Whole-exome (N = 2003 cases and 866 controls) or whole-genome (N = 1266 cases and 507 controls) sequencing was performed for the cases and controls, and high-quality variants were obtained from the sequencing data as detailed in the Additional file 1: Supplementary Methods.
Population stratification
Principal component analysis (PCA) was performed with PLINK v1.9 software [40] on a pruned subset of ~ 14,600 SNPs not in linkage disequilibrium (maximum r2 value for linkage disequilibrium 0.4 between pairs of SNPs) with a minor allele frequency (MAF) > 1%, call rate > 99%, and P value for departure from Hardy–Weinberg equilibrium > 10−5, as previously described [41]. Ethnic origin was inferred from the PCA as previously described [41].
Variant selection
For each gene, we considered several sets of candidate coding variants, defined according to (i) functional annotation: predicted loss-of-function (pLOF) variants only (including stop gain/lost, start lost, frameshift, or splice variants), or pLOF with missense and in-frame variants (MISSLOF); (ii) the gnomAD v2.1 allele frequency (AF): variants with a gnomAD allele frequency below 1%, 0.1%, or 0.01%; and (iii) Combined Annotation Dependent Depletion (CADD) score [42] for missense and in-frame variants: CADD score ≥ mutation significance cut-off (MSC) for the corresponding gene [43] or all variants regardless of the CADD score. We considered nine sets of variants in total: (1) pLOF variants with gnomAD AF < 1%; (2) pLOF variants with gnomAD AF < 0.1%; (3) pLOF variants with gnomAD AF < 0.01%; (4) MISSLOF with CADD > MSC and gnomAD AF < 1%; (5) MISSLOF with CADD > MSC and gnomAD AF < 0.1%; (6) MISSLOF with CADD > MSC and gnomAD AF < 0.01%; (7) MISSLOF with gnomAD AF < 1%; (8) MISSLOF with gnomAD AF < 0.1%; (9) MISSLOF with gnomAD AF < 0.01%.
Rare variant burden analysis
We performed a genome-wide gene-based rare variants burden analysis. For each gene, the genotypic information for candidate rare variants was summarized into a genetic score defined according to three genetic models: (1) co-dominant: samples were coded 2 if they carried at least one biallelic variant, 1 if they carried at least one monoallelic variant, and 0 otherwise; (2) heterozygous: samples were coded 1 if they carried at least one monoallelic variant and 0 otherwise; and (3) recessive: samples were coded 1 if they carried at least one biallelic variant and 0 otherwise. For the X chromosome, hemizygous males are considered to be equivalent to homozygous females. The association between the genetic score for each gene and the disease status was assessed with a logistic regression-based likelihood ratio test (LRT) from EPACTS (Efficient and Parallelizable Association Container Toolbox) [44] for the genome-wide burden analysis or R 3.6.0 [45] for the candidate type I IFN-related pathway. Firth’s bias correction, with the fast.logistf.fit function of EPACTS or the logistf function of the R logistf package [46], was applied if the P value of the LRT was below 0.05. Analyses were adjusted for sex, age (in years), and the first five PCs of the PCA In Firth’s regression, a penalty term is assigned to the standard maximum likelihood function used to estimate the parameters of a logistic regression model when there are rare events or when complete separation exists [47]. With no covariates, this corresponds to adding 0.5 to every cell of a 2 by 2 table of allele counts versus case–control status. For a given gene and variant set, the burden test was not performed if the number of carriers across all samples was below 3.
We used three analysis strategies: (1) joint analysis of all samples; (2) trans-ethnic meta-analysis: the analysis was stratified according to 7 inferred ancestry subgroups (African, North African, European, admixed American, Middle Eastern, South Asian, East Asian). For each subgroup, an ethnicity specific PCA was performed and used in the logistic regression model; and (3) trans-pipeline meta-analysis to account for heterogeneity due to the type of sequencing data: the analysis was stratified according to the type of data shared (FASTQ vs. VCF). Subgroup P values were subjected to further meta-analysis, accounting for the direction of the effect and sample size, with METAL [48].
Correction for multiple testing
For each gene, up to 9 burden tests were performed per genetic model. These tests were not independent; we therefore assessed the effective number of burden tests Meff with a method adapted from that described by Patin et al. [49], based on the approach of Li and Ji [50]. This approach makes use of the variance of the eigenvalues of the observed statistics correlation matrix to estimate Meff. The Bonferroni-corrected threshold was then defined as 0.05/Meff.
Odds ratio (OR) equality for homozygous/hemizygous versus heterozygous carriers of pLOF variants at type I IFN genes
We investigated whether the odds of critical COVID-19 differed for carriers and non-carriers of pLOF variants at the type I IFN immunity loci as a function of zygosity (homozygous/hemizygous vs heterozygous). In the full sample, we used LRT to compare a full Firth bias-corrected logistic regression model including two different parameters for carriers of pLOF as a function of zygosity (alternative hypothesis) with a Firth bias-corrected logistic regression model including only one parameter for carriers of pLOF, not taking zygosity into account (null hypothesis). The analysis was performed with the R logistf package.
Biochemical characterization of TLR7 variants with a luciferase reporter assay
We tested the TLR7 variants as previously described [30]. Briefly, TLR7 variants were generated by site-directed mutagenesis. The WT or variant alleles were re-introduced into a Myc-DDK-pCMV6 vector (Origene). HEK293T cells, which have no endogenous TLR7 expression, were transfected with 50 ng of Myc-DDK-pCMV6 vector, empty or containing the WT or a variant allele the reporter construct pGL4.32 (100 ng), and an expression vector for Renilla luciferase (10 ng), with the X-tremeGENE™ 9 DNA Transfection Reagent kit (Sigma-Aldrich). The pGL4.32 (luc2P/NF-κB–RE/Hygo) (Promega) reporter vector contains five copies of the NF-κB–responsive element (NF-κB–RE) linked to the luc2P luciferase reporter gene. After 24 h, the transfected cells were left unstimulated or were stimulated with R848 (1 μg/ml; resquimod), for activation via TLR7/8 (Invivogen), or R837 (5 μg/ml; imiquimod) (Invivogen), or CL264 (5 μg/ml; Invivogen), human TLR7-specific agonists, for 24 h. Relative luciferase activity was then determined by normalizing the values obtained against the firefly:Renilla luciferase signal ratio.
Results
Cohort description
Through the CHGE, we collected whole-exome sequencing (WES) or whole-genome sequencing (WGS) data for 3503 patients with life-threatening COVID-19 pneumonia (hereafter referred to as “patients”; see Supplemental Methods) and 1373 individuals with mild or asymptomatic infection, i.e., without pneumonia (hereafter referred to as “controls”). In total, 928 of the 3503 patients were screened for the presence of auto-Abs against type I IFN [26, 27] (Supplemental Methods) and the 234 patients who tested positive were excluded from this analysis as they already have a major risk factor for the development of critical COVID-19 [29]. In total, 1301 of the 3269 remaining patients had been included in previous studies restricted to a short list of 18 candidate genes [15, 19] or to the X chromosome [30], and 1968 had not been studied before. The mean age (SD) of the patients was 55.7 (17.4) years, with a male-to-female ratio of 2.4 (Table 1). The controls were significantly younger than the patients (P < 0.0001), with a mean age (SD) of 43.8 years (20.1 years) and were more likely to be female (P < 0.0001; male-to-female ratio = 0.7). The patients and controls were of various ethnic origins, mostly of European and Middle Eastern ancestry, according to principal component analysis (PCA) (Fig. 2). Raw sequencing data were either centralized in the HGID laboratory and processed with the HGID pipeline (2492 cases and 870 controls) or processed separately by each sequencing hub (777 cases and 503 controls; See Supplemental Methods). A joint analysis was performed first on the combined sample of 3269 patients and 1373 controls. Given the heterogeneity of the cohort due to different ancestries and processing pipelines, we also performed a trans-ethnic and a trans-pipeline meta-analysis; only results consistent across the three analyses are reported here (See Supplemental Methods).

Principal component analysis of patients with life-threatening COVID-19 (red) and SARS-CoV-2-infected controls (green). Principal component analysis (PCA) was performed with PLINK v1.9 software [40] on a pruned subset of ~ 14,600 exonic SNPs in linkage equilibrium (maximum r2 value for linkage disequilibrium of 0.4 between pairs of SNPs) with a minor allele frequency (MAF) > 1%, call rate > 99% and P value for departure from Hardy–Weinberg equilibrium > 10.−5. Samples were of diverse ethnic origins, including European (EUR), admixed American (AMR), North African (NAFR), sub-Saharan African (AFR), Middle Eastern (ME), South Asian (SAS), and East Asian (EAS)
Genome-wide analysis under a co-dominant model
We first performed a GW rare variant burden analysis on the 3269 patients with life-threatening COVID-19 and 1373 controls with asymptomatic/mild COVID-19 under a co-dominant model, using nine sets of variants (See Supplemental Methods). The QQ plots for the joint analysis of the samples revealed no systematic deviations from the null hypothesis, and the genomic inflation factors (λ) were close to 1 (Additional file 2: Table S1). In total, 18,064 genes were analyzed with at least one of the nine variant sets, resulting in an effective number of independent tests (Meff) for the joint analysis of 108,384, giving a Bonferroni-corrected significance threshold of 4.61 × 10−7. No gene was found to be of GW significance (see the Manhattan plot in Fig. 3A, Additional file 2: Table S2). The gene with the strongest association was TREH, encoding the trehalase enzyme, which hydrolyses trehalose, with rare (gnomAD allele frequency [AF] < 10−4) nonsynonymous variants associated with a lower risk of life-threatening COVID-19 (OR = 0.12[95% CI 0.05–0.28], P = 1.9 × 10−6; Additional file 2: Table S3). In analyses of genes for which rare predicted loss-of-function (pLOF) variants were associated with an increase in the risk of life-threatening COVID-19 (Table 2), the strongest association was that for NPC2, for rare (gnomAD AF < 0.01) pLOF variants, with 28 heterozygous carriers among patients (0.9%), and four heterozygous carriers (0.3%) among controls (OR = 5.41 [95% CI 1.8–16.4], P = 5.8 × 10−4). NPC2 encodes the Niemann-Pick disease type C2 protein and homozygous LOF mutations of this gene cause Niemann-Pick disease [51]. NPC2 interacts with NPC1, which is also an essential endosomal receptor for the Ebola virus [52, 53]. Both NPC1 and NPC2 were implicated in the regulation of SARS-CoV-2 entry in a CRISPR screen [54]. The GW burden analysis under a dominant model yielded similar conclusions (Additional file 2: Table S3).

Manhattan plot for genome-wide burden analysis under the co-dominant (top) and recessive (bottom) models. For each gene, the negative log-transformed p value of the joint analysis for the most significant variant set under a co-dominant (top) or recessive (bottom) model is plotted. For each gene, variant sets providing inconsistent results across the joint analysis, the trans-ethnic meta-analysis, and the trans-pipeline meta-analysis (i.e., P < 0.001 in the joint analysis and P > 0.05 in the trans-ethnic or trans-pipeline meta-analysis) were discarded. The red lines represent the significance threshold after Bonferroni correction to account for the total number of independent tests (P = 4.61 × 10−7 under a co-dominant model and 1.85 × 10−6 under a recessive model). The names of the top-ranked genes with a joint P < 10−4 are shown in red for rare variants associated with an increase in the risk of critical COVID-19 and in blue otherwise
Genome-wide analysis under a recessive model
We then performed a GW screen under a recessive model (autosomal and X-linked). In total, 4511 genes were analyzed with at least one of the nine variant sets, resulting in 27,066 independent tests, giving a Bonferroni-corrected significance threshold of 1.85 × 10−6. No gene reached GW significance (Fig. 3B). In analyses of genes with rare variants increasing the risk of life-threatening COVID-19, TLR7 was, by two orders of magnitude, the most significant gene, with 51 carriers (1.6%) of at least one rare (gnomAD AF < 0.01) missense or pLOF variant in patients versus two carriers (0.1%) in controls (OR = 8.41[95% CI 1.9–35.5], P = 8.95 × 10−5) (Table 3). Most of the carriers were male, with only one carrier among the patients and one among the controls being female. The variants carried by the two controls were previously shown to be biochemically neutral [19, 30] (Additional file 2: Table S4). The 51 cases carried 33 different variants, 13 of which had been shown to be neutral; 16 were previously shown to be hypomorphic or amorphic [19, 30], and four were previously unknown. The four new variants were tested: one was found to be neutral and the other three were deleterious (Additional file 1: Fig S1). Restricting the analysis to biochemically proven LOF variants (bLOF) decreased the number of carriers (20 cases vs. 0 controls), but the association signal remained highly significant, with a much higher odds ratio (OR = 27.68 [95% CI 1.5–528.7], P = 1.08 × 10−4) (Table 3). These findings confirm that TLR7 is a critical COVID-19 susceptibility locus, responsible for 0.9% of critical cases in male patients.
Genome-wide gene-based analysis including common variants
Published GWAS identified a number of common variants associated with severe COVID-19 pneumonia [8, 10, 12, 55]. We then assessed the combined effect of common and rare candidate coding variants at the gene level, in a weighted burden approach [56], as detailed in the Supplemental Methods. Briefly, for each individual, we calculated a genetic score by summing the number of minor alleles for each variant and weighting this sum by the frequency of the variant [57]. We then tested the association between this genetic score and case–control status in a logistic regression framework. As described above, we focused on pLOF only, pLOF and in-frame variants with CADD > MSC, or pLOF and in-frame variants without filtering on CADD score (Additional file 2: Table S5). As in the analysis focusing on rare variants only, no gene reached genome-wide significance after correction for multiple testing in this analysis considering both rare and common variants. The top-ranked gene, with consistent results across the joint analysis and the trans-ethnic and trans-pipeline meta-analyses, was TREH, with a protective effect against life-threatening COVID-19 of pLOF or nonsynonymous variants with a CADD score greater than the MSC (OR = 0.85 [95%CI 0.78–0.91], P = 3.6 × 10−6). Finally, we analyzed 20 candidate genes identified by GWAS for critical pneumonia in more detail [8, 10, 12, 55]. No significant association was detected for any of these genes (Additional file 2: Table S6), even with a relaxed Bonferroni threshold of 2.5 × 10−3, accounting for the number of GWAS genes.
Enrichment in rare pLOF variants at 13 type I IFN-related influenza susceptibility loci
Following on from our initial analysis [15], we also performed a candidate pathway enrichment analysis focusing on the 13 genes involved in Toll-like receptor 3 (TLR3)– and interferon regulatory factor 7 (IRF7)–dependent type I IFN immunity to influenza virus (IFNAR1, IFNAR2, IRF3, IRF7, IRF9, IKBKG, STAT1, STAT2, TBK1, TICAM1, TLR3, TRAF3, and UNC93B1) (Fig. 1). We confirmed the significant enrichment in rare (gnomAD AF < 10−3) pLOF variants at the 13 loci in patients with critical COVID-19, with 34 carriers among patients versus six among controls (OR = 3.70 [95% CI 1.7–9.5], P = 2.1 × 10−4 under a co-dominant model; Table 4). We also estimated this p value in a simulation study taking 13 loci randomly selected from a set of genes with similar pLI and CoNeS values (see Additional file 1: Supplemental Methods); we obtained an empirical p value of 3.7 × 10−4. Our cohort included 551 patients and 314 controls already screened for pLOF variants of the 13 genes included in our previous study [15] (Additional file 2: Table S7). The exclusion of these 551 cases and 314 controls resulted in a similar conclusion of enrichment in rare pLOF at the 13 loci (OR = 3.21 [95% CI 1.3–8.2], P = 5.97 × 10−3) formally replicating our initial association. Significant replication was also observed in the trans-ethnic (P = 0.01) and the trans-pipeline (P = 0.009) analyses. We found that 31 of the 34 carriers of pLOF variants were heterozygous, and three were homozygous: one for a frameshift variant of IRF7 described in a previous study [15], one for a previously reported deletion spanning 4394 base pairs in IFNAR1 [16, 19], and one for a previously unknown deletion spanning 6624 base pairs of IFNAR1 (Additional file 2: Table S8). All the biallelic pLOF variants were found in patients. Consequently, the OR for homozygous carriers (OR = 15.79 [95%CI 1.4–2170.4], P = 0.02) was higher than that for heterozygous carriers (OR = 3.11 [95%CI 1.4–8.6], P = 5.2 × 10−3), but both were significant.
Inclusion of TYK2 and TLR7 genes and branchpoint variants
Since the publication of the aforementioned study [15], AR TYK2 deficiency has been reported in children with COVID-19 pneumonia [19]. We identified two patients homozygous for a rare pLOF variant of TYK2 already described in a previous study [19] and one patient and one control heterozygous for a rare pLOF variant (Additional file 2: Table S8). Adding these patients to the analysis gave very similar results under a co-dominant model (OR = 3.30[95% CI 1.6–7.8], P = 1.4 × 10−4) and strengthened the evidence for association under a recessive model (OR = 19.65[95% CI 2.1–2635.4], P = 3.4 × 10−3) (Table 4). An analysis of the rare pLOF variants at these 14 loci plus the bLOF variants of TLR7 revealed highly significant enrichment (OR = 3.82 [95%CI 2.0–7.2], P = 1.3 × 10−7 under a co-dominant model). The effect was stronger for homozygous/hemizygous carriers (OR = 39.19 [95%CI 5.2–5037.01], P = 4.7 × 10−7) than for heterozygous carriers (OR = 2.27 [95%CI 1.0–5.2], P = 0.04), and these two ORs were significantly different (P = 0.008). We further screened the full cohort of cases and controls for intronic branchpoint (BP) variants, which might potentially have a strong impact on splicing and be considered pLOF variants, in the 15 type I IFN-related genes, with our new tool BPHunter [39]. We identified six branchpoint (BP) variants (Additional file 2: Table S9) carried in heterozygous state by 10 additional cases and no controls. Adding these BP variants to the analysis of the 15 type I IFN-related loci under a co-dominant model further strengthened the association signal (OR = 4.40 [2.3–8.4], P = 7.7 × 10−8) (Table 4).
Age and sex stratified analysis of the 15 type I IFN-related loci
Advanced age is the strongest risk factor for life-threatening COVID-19. Male individuals are also at higher risk than female individuals. As for the main GWAS hits [58, 59], we performed an analysis stratified for age and sex for the 15 type I IFN-related loci. The analysis stratified for sex revealed a much stronger association signal in male than in female individuals, as expected given the X-linked recessive mode of inheritance of TLR7 deficiency (Additional file 2: Table S10). Nevertheless, the enrichment in rare pLOF variants at the 15 loci in female subjects remained significant under a co-dominant model (P = 0.02) and a recessive model (P = 0.05). The addition of the BP variants strengthened the association signal in female subjects under a co-dominant model (P = 3.7 × 10−3). In the analysis stratified for age, we assigned the cases to two age groups (under 60 years of age vs. 60 years and over), which we compared with all controls. We used an age cut-off of 60 years, which was close to the median age of the cases, in accordance with the analyses performed in [7, 59]. The age stratified analysis revealed a strong impact of age, the genetic effect being restricted to younger cases (OR = 4.65 [2.4–9.0], P = 2.2 × 10−9, Additional file 2: Table S10). Accordingly, the 67 patients with critical COVID-19 carrying a rare pLOF or bLOF variant of one of the 15 genes were significantly younger than the remaining 3202 patients in the cohort (mean age [SD] in years: 43.68 [19.4] vs. 56.0 [17.3] years; P = 2.3 × 10−6), consistent with our previous reports that IEIs conferring a predisposition to life-threatening COVID-19 are more frequent in young patients [1, 15, 30]. Moreover, the homozygous/hemizygous carriers were significantly younger than the heterozygous carriers (35.2 [20.3] vs. 48.7 [17.1] years, P = 0.008, Additional file 1: Fig S2). Overall, these results clearly demonstrate that the search for additional rare variants conferring a strong predisposition to life-threatening COVID-19 benefits from focus on younger patients.
In-frame nonsynonymous variants at the 15 loci
We further screened our cohort for rare in-frame nonsynonymous variants with a gnomAD AF < 10−3 at these type I IFN-related susceptibility loci. For the 13 initial loci, the enrichment disappeared when in-frame nonsynonymous variants were added to pLOF variants under a co-dominant model (OR = 1.08 [95%CI 0.9–1.3], P = 0.42) (Additional file 2: Table S11), whereas a non-significant trend persisted under the recessive model (OR = 5.02 [95% CI 0.7–52.7], P = 0.06). Focusing exclusively on in-frame variants decreased the strength of this trend considerably, with only eight homozygous carriers among patients and one among controls (OR = 1.14 [0.2–912.5], P = 0.68). Adding TYK2 variants led to similar conclusions (Additional file 2: Table S11). We then added TLR7 variants and considered the 15 loci together. Under a co-dominant model, the enrichment became non-significant when in-frame nonsynonymous variants were added (OR = 1.15 [1.0–1.4], P = 0.09), but enrichment remained significant under a recessive model (OR = 6.54 [2.4–24.8], P = 5.3 × 10−6; Additional file 2: Table S11). In analyses considering only rare in-frame homozygous/hemizygous nonsynonymous variants, the effect size was smaller, but the enrichment remained significant (OR = 3.52 [1.3–13.3], P = 2.8 × 10−3). In total, 41 patients carried a rare homozygous/hemizygous in-frame nonsynonymous variant at one of the 15 loci, and 16 of these variants (carried by 16 patients) were TLR7 in-frame variants already shown to be bLOF. After excluding the TLR7 bLOF variants, there was no residual significant enrichment in rare in-frame nonsynonymous variants in patients relative to controls, whatever the genetic model considered.
Discussion
In this exome-wide gene burden analysis for rare variants underlying critical COVID-19, no gene reached GW statistical significance after accounting for multiple testing. We used simulations to determine the power of our sample to detect an association at the 2.5 × 10−6 exome-wide significance threshold (Additional file 1: Fig S3); our sample had a power of more than 80% for detecting alleles with a carrier frequency of 5 × 10−3 in the general population and a relative risk of critical COVID-19 of at least 6. These results are consistent with those of two previous large exome-wide studies including more than 1000 critical cases and thousands of population-based controls that found no statistically significant autosomal gene burden associations at stringent significance thresholds accounting for the number of phenotypes and variant sets analyzed [11, 21]. However, under a recessive model, the strongest association—albeit not statistically significant at GW level—was obtained with the X-linked TLR7 gene, for which association has consistently been reported across studies [21, 22, 30, 32], reaching the less conservative exome-wide significance threshold of 2.5 × 10−6 in some of these previous studies [21, 22]. It should be stressed that stringent correction for multiple testing, while necessary to avoid false positives, is a conservative strategy, and that the lack of formal statistical significance at a GW level does not preclude biological causality and medical significance. The burden of proof can be provided experimentally via biochemical, virological, and immunological experiments, as our previous studies of TLR7 in which we showed that biochemically deleterious TLR7 variants blunted the pDC-dependent sensing of SARS-CoV-2 and induction of type I IFN, thereby accounting for ~ 1% of critical pneumonia cases in men [30]. Additional genes may be found by restricting the association analysis to variants experimentally proven to be deleterious.
This analysis also confirms our previous findings of an enrichment in rare pLOF variants of 13 genes involved in TLR3- and IRF7-dependent type I IFN immunity to seasonal influenza virus in critical cases relative to controls with mild/asymptomatic infection [15]. These results were strengthened by the addition of TYK2, which was recently shown to underlie severe COVID-19 [19, 20], and TLR7, especially under a recessive model. We found that homozygous/hemizygous carriers of rare pLOF or bLOF variants at the 15 loci had a significantly higher risk of life-threatening COVID-19 than heterozygotes. This is consistent with the generally higher clinical penetrance of recessive than dominant IEI [1]. Overall, 1.7% of the patients with life-threatening COVID-19 carried a rare pLOF or bLOF variant at one of the 15 loci, these variants being homozygous/hemizygous in 0.8% of cases. Adding the BP variants at the 15 loci increased the proportion of carriers among patients with life-threatening COVID-19 to 2.1%. One of the STAT2 BP variants identified (2:56749159:T > A) has already been validated experimentally [39], but the effects of the five other BP variants identified require confirmation. The study of in-frame nonsynonymous variants might also increase this proportion, but would require the experimental characterization of all these variants. Indeed, in analyses restricted to rare in-frame nonsynonymous variants, we detected no significant enrichment in patients relative to controls. This result is not surprising, as we showed in a previous study [15] that less than 15% of the rare in-frame nonsynonymous variants at the 13 loci carried by cases initially studied were bLOF variants, whereas all the pLOF variants were found to be bLOF. Similar results were obtained for TLR7, with only 10 of 108 (9.2%) in-frame nonsynonymous variants observed in gnomAD being bLOF [30]. This high proportion of neutral variants strongly affects the power of burden tests and highlights the need for the experimental characterization of variants.
We also showed that patients carrying rare pLOF or bLOF variants of these 15 type I IFN-related genes were significantly younger than the remaining patients (mean age [SD] in years: 43.3 [20.3] vs. 56.0 [17.3] years). This was particularly true for homozygous/hemizygous carriers of rare pLOF or bLOF variants (35.2 [20.3] years), potentially accounting for the lack of replication of this finding by other studies including older patients [11, 21,22,23]. Consistent with this result, we recently found that ~ 10% of children hospitalized for COVID-19 pneumonia carry recessive inborn errors of type I IFN immunity [19]. In addition, older patients are more likely to carry auto-Abs against type I IFN, and unlike previous studies, we excluded patients carrying such antibodies from this analysis. None of the 234 patients with critical COVID-19 excluded from this study due to the presence of auto-Abs against type I IFN carried a rare pLOF variant of the 15 genes. Hence, samples in which the vast majority of patients are over the age of 60 years and of unknown status for auto-Abs against type I IFNs would have a much lower power to identify these rare inborn errors of type I IFN immunity.
Conclusions
Rare autosomal inborn errors of type I IFN-dependent immunity to influenza viruses can underlie critical forms of COVID-19, especially in subjects below 60 years of age, in addition to X-linked TLR7 deficiency. The search for additional rare mutations conferring a strong predisposition to life-threatening COVID-19 should focus on young patients with critical COVID-19 without auto-Abs against type I IFNs.
Availability of data and materials
Data supporting the findings of this study are available within the manuscript and supplemental files. The whole-genome sequencing data of anonymized patients recruited through the National Institutes of Health (NIH) and sequenced at the National Institute of Allergy and Infectious Diseases (NIAID) through the Uniformed Services University of the Health Sciences (USUHS)/the American Genome Center (TAGC) are available under dbGaP submission phs002245.v1. Other patients were not consented to share the raw WES/WGS data files beyond the research and clinical teams.
Change history
06 January 2024
A Correction to this paper has been published: https://doi.org/10.1186/s13073-023-01278-0
References
Zhang Q, Bastard P, Covid Human Genetic Effort, Cobat A, Casanova JL. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature. 2022;603:587–98.
Covid Forecasting Team. Variation in the COVID-19 infection-fatality ratio by age, time, and geography during the pre-vaccine era: a systematic analysis. Lancet. 2022;399:1469–88.
O’Driscoll M, Ribeiro Dos Santos G, Wang L, Cummings DAT, Azman AS, Paireau J, et al. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature. 2021;590:140–5.
Bennett TD, Moffitt RA, Hajagos JG, Amor B, Anand A, Bissell MM, et al. Clinical characterization and prediction of clinical severity of SARS-CoV-2 infection among US adults using data from the US National COVID Cohort Collaborative. JAMA Netw Open. 2021;4:e2116901.
Takahashi T, Ellingson MK, Wong P, Israelow B, Lucas C, Klein J, et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature. 2020;588:315–20.
Initiative CHG. Mapping the human genetic architecture of COVID-19. Nature. 2021;600:472–7.
Nakanishi T, Pigazzini S, Degenhardt F, Cordioli M, Butler-Laporte G, Maya-Miles D, et al. Age-dependent impact of the major common genetic risk factor for COVID-19 on severity and mortality. J Clin Invest. 2021;131. Available from: https://www.ncbi.nlm.nih.gov/pubmed/34597274. [Cited 2022 Apr 29].
Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591:92–8.
Zeberg H, Paabo S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature. 2020;587:610–2.
Initiative CHG. A first update on mapping the human genetic architecture of COVID-19. Nature. 2022;608:E1-10.
Kousathanas A, Pairo-Castineira E, Rawlik K, Stuckey A, Odhams CA, Walker S, et al. Whole-genome sequencing reveals host factors underlying critical COVID-19. Nature. 2022;607:97–103.
Namkoong H, Edahiro R, Takano T, Nishihara H, Shirai Y, Sonehara K, et al. DOCK2 is involved in the host genetics and biology of severe COVID-19. Nature. 2022;609:754–60.
Zeberg H, Paabo S. A genomic region associated with protection against severe COVID-19 is inherited from Neandertals. Proc Natl Acad Sci U A. 2021;118:e2026309118.
Casanova JL, Su HC, Covid Human Genetic Effort. A Global Effort to Define the Human Genetics of Protective Immunity to SARS-CoV-2 Infection. Cell. 2020;181:1194–9.
Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370:eabd4570.
Abolhassani H, Landegren N, Bastard P, Materna M, Modaresi M, Du L, et al. Inherited IFNAR1 deficiency in a child with both critical COVID-19 pneumonia and multisystem inflammatory syndrome. J Clin Immunol. 2022;42:471–83.
Khanmohammadi S, Rezaei N, Khazaei M, Shirkani A. A case of autosomal recessive interferon alpha/beta receptor alpha chain (IFNAR1) deficiency with severe COVID-19. J Clin Immunol. 2022;42:19–24.
Schmidt A, Peters S, Knaus A, Sabir H, Hamsen F, Maj C, et al. TBK1 and TNFRSF13B mutations and an autoinflammatory disease in a child with lethal COVID-19. NPJ Genom Med. 2021;6:55.
Zhang Q, Matuozzo D, Le Pen J, Lee D, Moens L, Asano T, et al. Recessive inborn errors of type I IFN immunity in children with COVID-19 pneumonia. J Exp Med. 2022;219:e20220131.
Ogishi M, Arias A, Yang R, Han JE, Zhang P, Rinchai D, et al. Impaired IL-23–dependent induction of IFN-γ underlies mycobacterial disease in patients with inherited TYK2 deficiency. J Exp Med. 2022;219(10):e20220094. https://doi.org/10.1084/jem.20220094.
Butler-Laporte G, Povysil G, Kosmicki JA, Cirulli ET, Drivas T, Furini S, et al. Exome-wide association study to identify rare variants influencing COVID-19 outcomes: Results from the Host Genetics Initiative. PLoS Genet. 2022;18:e1010367.
Kosmicki JA, Horowitz JE, Banerjee N, Lanche R, Marcketta A, Maxwell E, et al. Pan-ancestry exome-wide association analyses of COVID-19 outcomes in 586,157 individuals. Am J Hum Genet. 2021;108:1350–5.
Povysil G, Butler-Laporte G, Shang N, Wang C, Khan A, Alaamery M, et al. Rare loss-of-function variants in type I IFN immunity genes are not associated with severe COVID-19. J Clin Invest. 2021;131. Available from: https://www.ncbi.nlm.nih.gov/pubmed/34043590. [Cited 2022 Apr 29].
Zhang Q, Cobat A, Bastard P, Notarangelo LD, Su HC, Abel L, et al. Association of rare predicted loss-of-function variants of influenza-related type I IFN genes with critical COVID-19 pneumonia. J Clin Invest. 2021;131. Available from: https://www.ncbi.nlm.nih.gov/pubmed/34166232. [Cited 2022 Apr 29].
Casanova JL, Abel L. Mechanisms of viral inflammation and disease in humans. Science. 2021;374(6571):1080–6. https://doi.org/10.1126/science.abj7965
Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:eabd4585.
Bastard P, Gervais A, Le Voyer T, Rosain J, Philippot Q, Manry J, et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. Sci Immunol. 2021;6:eabl4340.
Casanova JL, Abel L. From rare disorders of immunity to common determinants of infection: following the mechanistic thread. Cell. 2022;185:3086–103.
Manry J, Bastard P, Gervais A, Le Voyer T, Rosain J, Philippot Q, et al. The risk of COVID-19 death is much greater and age dependent with type I IFN autoantibodies. Proc Natl Acad Sci U A. 2022;119:e2200413119.
Asano T, Boisson B, Onodi F, Matuozzo D, Moncada-Velez M, Maglorius Renkilaraj MRL, et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci Immunol. 2021;6:eabl4348.
van der Made CI, Netea MG, van der Veerdonk FL, Hoischen A. Clinical implications of host genetic variation and susceptibility to severe or critical COVID-19. Genome Med. 2022;14:96.
Fallerini C, Daga S, Mantovani S, Benetti E, Picchiotti N, Francisci D, et al. Association of Toll-like receptor 7 variants with life-threatening COVID-19 disease in males: findings from a nested case-control study. Elife. 2021;10:e67569.
Mantovani S, Daga S, Fallerini C, Baldassarri M, Benetti E, Picchiotti N, et al. Rare variants in Toll-like receptor 7 results in functional impairment and downregulation of cytokine-mediated signaling in COVID-19 patients. Genes Immun. 2022;23:51–6.
Pessoa NL, Bentes AA, de Carvalho AL, de Souza Silva TB, Alves PA, de Sousa Reis EV, et al. Case report: hepatitis in a child infected with SARS-CoV-2 presenting toll-like receptor 7 Gln11Leu single nucleotide polymorphism. Virol J. 2021;18:180.
Solanich X, Vargas-Parra G, van der Made CI, Simons A, Schuurs-Hoeijmakers J, Antoli A, et al. Genetic screening for TLR7 variants in young and previously healthy men with severe COVID-19. Front Immunol. 2021;12:719115.
van der Made CI, Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, et al. Presence of genetic variants among young men with severe COVID-19. JAMA. 2020;324:663–73.
Brown GJ, Canete PF, Wang H, Medhavy A, Bones J, Roco JA, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. 2022;605:349–56.
Barreiro LB, Quintana-Murci L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet. 2010;11:17–30.
Zhang P, Philippot Q, Ren W, Lei WT, Li J, Stenson PD, et al. Genome-wide detection of human variants that disrupt intronic branchpoints. Proc Natl Acad Sci. 2022;119:e2211194119.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Belkadi A, Pedergnana V, Cobat A, Itan Y, Vincent QB, Abhyankar A, et al. Whole-exome sequencing to analyze population structure, parental inbreeding, and familial linkage. Proc Natl Acad Sci U A. 2016;113:6713–8.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–94.
Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods. 2016;13:109–10.
Efficient and Parallelizable Association Container Toolbox (EPACTS). Available from: https://genome.sph.umich.edu/wiki/EPACTS.
R Core Team. R: A language and environment for statistical computing. Vienna, Austria; 2022. Available from: https://www.R-project.org/.
Heinze G, Ploner M, Jiricka L. logistf: Firth’s Bias-Reduced Logistic Regression. R package version 1.24.1.; Available from: https://CRAN.R-project.org/package=logistf.
Firth D. Bias reduction of maximum likelihood estimates. Biometrika. 1993;80:27.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
Patin E, Kutalik Z, Guergnon J, Bibert S, Nalpas B, Jouanguy E, et al. Genome-wide association study identifies variants associated with progression of liver fibrosis from HCV infection. Gastroenterology. 2012;143:1244-1252 e12.
Li J, Ji L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Hered Edinb. 2005;95:221–7.
Verot L, Chikh K, Freydiere E, Honore R, Vanier MT, Millat G. Niemann-Pick C disease: functional characterization of three NPC2 mutations and clinical and molecular update on patients with NPC2. Clin Genet. 2007;71:320–30.
Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–3.
Zhao Y, Ren J, Harlos K, Stuart DI. Structure of glycosylated NPC1 luminal domain C reveals insights into NPC2 and Ebola virus interactions. FEBS Lett. 2016;590:605–12.
Zhu Y, Feng F, Hu G, Wang Y, Yu Y, Zhu Y, et al. A genome-wide CRISPR screen identifies host factors that regulate SARS-CoV-2 entry. Nat Commun. 2021;12:961.
Ostendorf BN, Patel MA, Bilanovic J, Hoffmann H-H, Carrasco SE, Rice CM, et al. Common human genetic variants of APOE impact murine COVID-19 mortality. Nature. 2022;611:346–51.
Curtis D. A weighted burden test using logistic regression for integrated analysis of sequence variants, copy number variants and polygenic risk score. Eur J Hum Genet. 2019;27:114–24.
Curtis D. A rapid method for combined analysis of common and rare variants at the level of a region, gene, or pathway. Adv Appl Bioinforma Chem. 2012;5:1–9. https://doi.org/10.2147/AABC.S33049.
Karlsen TH. Understanding COVID-19 through genome-wide association studies. Nat Genet. 2022;54:368–9.
Degenhardt F, Ellinghaus D, Juzenas S, Lerga-Jaso J, Wendorff M, Maya-Miles D, et al. Detailed stratified GWAS analysis for severe COVID-19 in four European populations. Hum Mol Genet. 2022;31:3945–66.
Acknowledgements
We thank the patients and their families for agreeing to participate in our research. We thank all members of the consortia listed below:
Members of COVID Human Genetic Effort: Laurent Abel1, Alessandro Aiuti2, Saleh Al-Muhsen3, Fahd Al-Mulla4, Mark S. Anderson5, Evangelos Andreakos6, Andrés A. Arias7, Hagit Baris Feldman8, Alexandre Belot9, Catherine M. Biggs10, Dusan Bogunovic11, Alexandre Bolze12, Anastasiia Bondarenko13, Ahmed A. Bousfiha14, Petter Brodin15, Yenan Bryceson16, Carlos D. Bustamante17, Manish J. Butte18, Giorgio Casari19, Samya Chakravorty20, John Christodoulou21, Antonio Condino-Neto22, Stefan N. Constantinescu23, Megan A. Cooper24, Clifton L. Dalgard25, Murkesh Desai26, Beth A. Drolet27, Jamila El Baghdadi28, Sara Espinosa-Padilla29, Jacques Fellay30, Carlos Flores31, José Luis Franco7, Antoine Froidure32, Peter K. Gregersen33, Filomeen Haerynck34, David Hagin35, Rabih Halwani36, Lennart Hammarström37, James R. Heath38, Sarah E. Henrickson39, Elena W. Y. Hsieh40, Eystein Husebye41, Kohsuke Imai42, Yuval Itan43, Erich D. Jarvis44, Timokratis Karamitros45, Kai Kisand46, Cheng-Lung Ku47, Yu-Lung Lau48, Yun Ling49, Carrie L. Lucas50, Tom Maniatis51, Davood Mansouri52, László Maródi53, Isabelle Meyts54, Joshua D. Milner55, Kristina Mironska56, Trine H. Mogensen57, Tomohiro Morio58, Lisa F. P. Ng59, Luigi D. Notarangelo60, Antonio Novelli61, Giuseppe Novelli62, Cliona O’Farrelly63, Satoshi Okada64, Tayfun Ozcelik65, Qiang Pan-Hammarström37, Rebeca Perez de Diego66, Anna M. Planas67, Carolina Prando68, Aurora Pujol69, Lluis Quintana-Murci70, Laurent Renia59, Igor Resnick71, Carlos Rodríguez-Gallego72, Vanessa Sancho-Shimizu73, Anna Sediva74, Mikko R. J. Seppänen75, Mohammed Shahrooei76, Anna Shcherbina77, Ondrej Slaby78, Andrew L. Snow79, Pere Soler-Palacín80, András N. Spaan81, Ivan Tancevski82, Stuart G. Tangye83, Ahmad Abou Tayoun84, Sathishkumar Ramaswamy84, Stuart E. Turvey85, Furkan Uddin86, Mohammed J. Uddin87, Diederik van de Beek88, Donald C. Vinh89, Horst von Bernuth90, Mayana Zatz91, Pawel Zawadzki92, Helen C. Su60, Jean-Laurent Casanova93
1INSERM U1163, University of Paris, Imagine Institute, Paris, France. 2San Raffaele Telethon Institute for Gene Therapy, IRCCS Ospedale San Raffaele, and Vita Salute San Raffaele University, Milan, Italy. 3Immunology Research Laboratory, Department of Pediatrics, College of Medicine and King Saud University Medical City, King Saud University, Riyadh, Saudi Arabia. 4Dasman Diabetes Institute, Department of Genetics and Bioinformatics, Dasman, Kuwait. 5Diabetes Center, University of California, San Francisco, San Francisco, CA, USA. 6Laboratory of Immunobiology, Center for Clinical, Experimental Surgery and Translational Research, Biomedical Research Foundation of the Academy of Athens, Athens, Greece. 7Group of Primary Immunodeficiencies, Universidad de Antioquia UdeA, Medellín, Colombia. 8Genetics Institute, Tel Aviv Sourasky Medical Center and Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel. 9Pediatric Nephrology, Rheumatology, Dermatology, HFME, Hospices Civils de Lyon, National Referee Centre RAISE, and INSERM U1111, Université de Lyon, Lyon, France. 10Department of Pediatrics, British Columbia Children’s Hospital, University of British Columbia, Vancouver, BC, Canada. 11Icahn School of Medicine at Mount Sinai, New York, NY, USA. 12Helix, San Mateo, CA, USA. 13Shupyk National Medical Academy for Postgraduate Education, Kiev, Ukraine. 14Clinical Immunology Unit, Department of Pediatric Infectious Disease, CHU Ibn Rushd and LICIA, Laboratoire d’Immunologie Clinique, Inflammation et Allergie, Faculty of Medicine and Pharmacy, Hassan II University, Casablanca, Morocco. 15SciLifeLab, Department Of Women’s and Children’s Health, Karolinska Institutet, Stockholm, Sweden. 16Department of Medicine, Center for Hematology and Regenerative Medicine, Karolinska Institutet, Stockholm, Sweden. 17Stanford University, Stanford, CA, USA. 18Division of Immunology, Allergy, and Rheumatology, Department of Pediatrics and the Department of Microbiology, Immunology, and Molecular Genetics, University of California, Los Angeles, Los Angeles, CA, USA. 19Clinical Genomics, IRCCS San Raffaele Scientific Institute and Vita-Salute San Raffaele University, Milan, Italy. 20Department of Pediatrics and Children’s Healthcare of Atlanta, Emory University, Atlanta, GA, USA. 21Murdoch Children’s Research Institute and Department of Paediatrics, University of Melbourne, Melbourne, VIC, Australia. 22Department of Immunology, Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brazil. 23de Duve Institute and Ludwig Cancer Research, Brussels, Belgium. 24Washington University School of Medicine, St. Louis, MO, USA. 25Department of Anatomy, Physiology and Genetics, Uniformed Services University of the Health Sciences, Bethesda, MD, USA. 26Bai Jerbai Wadia Hospital for Children, Mumbai, India. 27School of Medicine and Public Health, University of Wisconsin, Madison, WI, USA. 28Genetics Unit, Military Hospital Mohamed V, Rabat, Morocco. 29Instituto Nacional de Pediatria (National Institute of Pediatrics), Mexico City, Mexico. 30School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland; Precision Medicine Unit, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland. 31Genomics Division, Instituto Tecnológico y de Energías Renovables (ITER), Santa Cruz de Tenerife, Spain; Research Unit, Hospital Universitario N.S. de Candelaria, Santa Cruz de Tenerife, Spain; Faculty of Health Sciences, University of Fernando Pessoa Canarias, Las Palmas de Gran Canaria, Spain; CIBER de Enfermedades Respiratorias, Instituto de Salud Carlos III, Madrid, Spain. 32Pulmonology Department, Cliniques Universitaires Saint-Luc; Institut de Recherche Expérimentale et Clinique (IREC), Université Catholique de Louvain, Brussels, Belgium. 33Feinstein Institute for Medical Research, Northwell Health USA, Manhasset, NY, USA. 34Department of Paediatric Immunology and Pulmonology, Centre for Primary Immunodeficiency Ghent (CPIG), PID Research Laboratory, Jeffrey Modell Diagnosis and Research Centre, Ghent University Hospital, Ghent, Belgium. 35Genetics Institute Tel Aviv Sourasky Medical Center, Tel Aviv, Israel. 36Sharjah Institute of Medical Research, College of Medicine, University of Sharjah, Sharjah, United Arab Emirates. 37Department of Biosciences and Nutrition, Karolinska Institutet, Stockholm, Sweden. 38Institute for Systems Biology, Seattle, WA, USA. 39Department of Pediatrics, Division of Allergy Immunology, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Microbiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA. 40Departments of Pediatrics, Immunology and Microbiology, University of Colorado, School of Medicine, Aurora, CO, USA. 41Department of Medicine, Haukeland University Hospital, Bergen, Norway. 42Department of Community Pediatrics, Perinatal and Maternal Medicine, Tokyo Medical and Dental University (TMDU), Tokyo, Japan. 43Institute for Personalized Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, USA; Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA. 44Laboratory of Neurogenetics of Language and Howard Hughes Medical Institute, Rockefeller University, New York, NY, USA. 45Bioinformatics and Applied Genomics Unit, Hellenic Pasteur Institute, Athens, Greece. 46Molecular Pathology, Department of Biomedicine, Institute of Biomedicine and Translational Medicine, University of Tartu, Tartu, Estonia. 47Chang Gung University, Taoyuan County, Taiwan. 48Department of Paediatrics and Adolescent Medicine, University of Hong Kong, Hong Kong, China. 49Shanghai Public Health Clinical Center, Fudan University, Shanghai, China. 50Department of Immunobiology, Yale University School of Medicine, New Haven, CT, USA. 51Columbia University Zuckerman Institute, New York, NY, USA. 52Department of Clinical Immunology and Infectious Diseases, National Research Institute of Tuberculosis and Lung Diseases, Clinical Tuberculosis and Epidemiology Research Center, National Research Institute of Tuberculosis and Lung Diseases (NRITLD), Masih Daneshvari Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran. 53Primary Immunodeficiency Clinical Unit and Laboratory, Department of Dermatology, Venereology and Dermatooncology, Semmelweis University, Budapest, Hungary. 54Department of Pediatrics, University Hospitals Leuven; KU Leuven, Department of Microbiology, Immunology and Transplantation; Laboratory for Inborn Errors of Immunity, KU Leuven, Leuven, Belgium. 55Department of Pediatrics, Columbia University Irving Medical Center, New York, NY, USA. 56University Clinic for Children’s Diseases, Department of Pediatric Immunology, Medical Faculty, University “St.Cyril and Methodij,” Skopje, North Macedonia. 57Department of Biomedicine, Aarhus University, Aarhus, Denmark. 58Tokyo Medical and Dental University Hospital, Tokyo, Japan. 59A*STAR Infectious Disease Labs, Agency for Science, Technology and Research, Singapore, Singapore; Lee Kong Chian School of Medicine, Nanyang Technology University, Singapore, Singapore. 60National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA. 61Laboratory of Medical Genetics, IRCCS Bambino Gesù Children’s Hospital, Rome, Italy. 62Department of Biomedicine and Prevention, Tor Vergata University of Rome, Rome, Italy. 63Comparative Immunology Group, School of Biochemistry and Immunology, Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin, Ireland. 64Department of Pediatrics, Graduate School of Biomedical and Health Sciences, Hiroshima University, Hiroshima, Japan. 65Department of Molecular Biology and Genetics, Bilkent University, Bilkent-Ankara, Turkey. 66Laboratory of Immunogenetics of Human Diseases, Innate Immunity Group, IdiPAZ Institute for Health Research, La Paz Hospital, Madrid, Spain. 67IIBB-CSIC, IDIBAPS, Barcelona, Spain. 68Faculdades Pequeno Príncipe, Instituto de Pesquisa Pelé Pequeno Príncipe, Curitiba, Brazil. 69Neurometabolic Diseases Laboratory, Bellvitge Biomedical Research Institute (IDIBELL), L’Hospitalet de Llobregat, Barcelona, Spain; Catalan Institution of Research and Advanced Studies (ICREA), Barcelona, Spain; Center for Biomedical Research on Rare Diseases (CIBERER), ISCIII, Barcelona, Spain. 70Human Evolutionary Genetics Unit, CNRS U2000, Institut Pasteur, Paris, France; Human Genomics and Evolution, Collège de France, Paris, France. 71University Hospital St. Marina, Varna, Bulgaria. 72Department of Immunology, University Hospital of Gran Canaria Dr. Negrín, Canarian Health System, Las Palmas de Gran Canaria, Spain; Department of Clinical Sciences, University Fernando Pessoa Canarias, Las Palmas de Gran Canaria, Spain. 73Department of Paediatric Infectious Diseases and Virology, Imperial College London, London, UK; Centre for Paediatrics and Child Health, Faculty of Medicine, Imperial College London, London, UK. 74Department of Immunology, Second Faculty of Medicine Charles University, V Uvalu, University Hospital in Motol, Prague, Czech Republic. 75Adult Immunodeficiency Unit, Infectious Diseases, Inflammation Center, University of Helsinki and Helsinki University Hospital, Helsinki, Finland; Rare Diseases Center and Pediatric Research Center, Children’s Hospital, University of Helsinki and Helsinki University Hospital, Helsinki, Finland. 76Saeed Pathobiology and Genetics Lab, Tehran, Iran; Department of Microbiology and Immunology, Clinical and Diagnostic Immunology, KU Leuven, Leuven, Belgium. 77Department of Immunology, Dmitry Rogachev National Medical Research Center of Pediatric Hematology, Oncology and Immunology, Moscow, Russia. 78Central European Institute of Technology and Department of Biology, Faculty of Medicine, Masaryk University, Brno, Czech Republic. 79Department of Pharmacology and Molecular Therapeutics, Uniformed Services University of the Health Sciences, Bethesda, MD, USA. 80Pediatric Infectious Diseases and Immunodeficiencies Unit, Vall d’Hebron Barcelona Hospital Campus, Barcelona, Spain. 81St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, Rockefeller University, New York, NY, USA; Department of Medical Microbiology, University Medical Center Utrecht, Utrecht, Netherlands. 82Department of Internal Medicine II, Medical University of Innsbruck, Innsbruck, Austria. 83Garvan Institute of Medical Research, Darlinghurst, NSW, Australia; St Vincent’s Clinical School, Faculty of Medicine, UNSW Sydney, NSW, Australia. 84Al Jalila Children’s Hospital, Dubai, UAE. 85BC Children’s Hospital, University of British Columbia, Vancouver, BC, Canada. 86Centre for Precision Therapeutics, Genetic and Genomic Medicine Centre, NeuroGen Children Healthcare, Dhaka, Bangladesh; Holy Family Red Crescent Medical College, Dhaka, Bangladesh. 87College of Medicine, Mohammed Bin Rashid University of Medicine and Health Sciences, Dubai, UAE; Cellular Intelligence (Ci) Lab, GenomeArc Inc., Toronto, ON, Canada. 88Department of Neurology, Amsterdam Neuroscience, Amsterdam University Medical Center, University of Amsterdam, Amsterdam, Netherlands. 89Department of Medicine, Division of Infectious Diseases, McGill University Health Centre, Montréal, QC, Canada; Infectious Disease Susceptibility Program, Research Institute, McGill University Health Centre, Montréal, QC, Canada. 90Department of Pediatric Pneumology, Immunology and Intensive Care, Charité Universitätsmedizin, Berlin University Hospital Center, Berlin, Germany; Labor Berlin GmbH, Department of Immunology, Berlin, Germany; Berlin Institutes of Health (BIH), Berlin-Brandenburg Center for Regenerative Therapies, Berlin, Germany. 91Biosciences Institute, University of São Paulo, São Paulo, Brazil. 92Molecular Biophysics Division, Faculty of Physics, A. Mickiewicz University, Poznań, Poland. 93Rockefeller University and Howard Hughes Medical Institute, New York, NY, USA; Necker Hospital for Sick Children and INSERM, Paris, France.
Members of COVID-STORM Clinicians: Giuseppe Foti1, Giacomo Bellani1, Giuseppe Citerio1, Ernesto Contro1, Alberto Pesci2, Maria Grazia Valsecchi3, Marina Cazzaniga4
1Department of Emergency, Anesthesia and Intensive Care, School of Medicine and Surgery, University of Milano-Bicocca, San Gerardo Hospital, Monza, Italy. 2Department of Pneumology, School of Medicine and Surgery, University of Milano-Bicocca, San Gerardo Hospital, Monza, Italy. 3Center of Bioinformatics and Biostatistics, School of Medicine and Surgery, University of Milano-Bicocca, San Gerardo Hospital, Monza, Italy. 4Phase I Research Center, School of Medicine and Surgery, University of Milano-Bicocca, San Gerardo Hospital, Monza, Italy.
Members of COVID Clinicians: Jorge Abad1, Giulia Accordino2, Cristian Achille3, Sergio Aguilera-Albesa4, Aina Aguiló-Cucurull5, Alessandro Aiuti6, Esra Akyüz Özkan7, Ilad Alavi Darazam8, Jonathan Antonio Roblero Albisures9, Juan C. Aldave10, Miquel Alfonso Ramos11, Taj Ali Khan12, Anna Aliberti13, Seyed Alireza Nadji14, Gulsum Alkan15, Suzan A. AlKhater16, Jerome Allardet-Servent17, Luis M. Allende18, Rebeca Alonso-Arias19, Mohammed S. Alshahrani20, Laia Alsina21, Marie-Alexandra Alyanakian22, Blanca Amador Borrero23, Zahir Amoura24, Arnau Antolí25, Romain Arrestier26, Mélodie Aubart27, Teresa Auguet28, Iryna Avramenko29, Gökhan Aytekin30, Axelle Azot31, Seiamak Bahram32, Fanny Bajolle33, Fausto Baldanti34, Aurélie Baldolli35, Maite Ballester36, Hagit Baris Feldman37, Benoit Barrou38, Federica Barzaghi6, Sabrina Basso39, Gulsum Iclal Bayhan40, Alexandre Belot41, Liliana Bezrodnik42, Agurtzane Bilbao43, Geraldine Blanchard-Rohner44, Ignacio Blanco45, Adeline Blandinières46, Daniel Blázquez-Gamero47, Alexandre Bleibtreu48, Marketa Bloomfield49, Mireia Bolivar-Prados50, Anastasiia Bondarenko51, Alessandro Borghesi3, Raphael Borie52, Elisabeth Botdhlo-Nevers53, Ahmed A. Bousfiha54, Aurore Bousquet55, David Boutolleau56, Claire Bouvattier57, Oksana Boyarchuk58, Juliette Bravais59, M. Luisa Briones60, Marie-Eve Brunner61, Raffaele Bruno62, Maria Rita P. Bueno63, Huda Bukhari64, Jacinta Bustamante33, Juan José Cáceres Agra65, Ruggero Capra66, Raphael Carapito67, Maria Carrabba68, Giorgio Casari6, Carlos Casasnovas69, Marion Caseris70, Irene Cassaniti34, Martin Castelle71, Francesco Castelli72, Martín Castillo de Vera73, Mateus V. Castro63, Emilie Catherinot74, Jale Bengi Celik75, Alessandro Ceschi76, Martin Chalumeau77, Bruno Charbit78, Matthew P. Cheng79, Père Clavé50, Bonaventura Clotet80, Anna Codina81, Yves Cohen82, Roger Colobran83, Cloé Comarmond84, Alain Combes85, Patrizia Comoli39, Angelo G. Corsico2, Betul Sozeri86, Taner Coşkuner86, Aleksandar Cvetkovski87, Cyril Cyrus88, David Dalmau89, François Danion90, David Ross Darley91, Vincent Das92, Nicolas Dauby93, Stéphane Dauger94, Paul De Munter95, Loic de Pontual96, Amin Dehban97, Geoffroy Delplancq98, Alexandre Demoule99, Isabelle Desguerre100, Antonio Di Sabatino101, Jean-Luc Diehl102, Stephanie Dobbelaere103, Elena Domínguez-Garrido104, Clément Dubost105, Olov Ekwall106, Şefika Elmas Bozdemir107, Marwa H. Elnagdy108, Melike Emiroglu15, Akifumi Endo109, Emine Hafize Erdeniz110, Selma Erol Aytekin111, Maria Pilar Etxart Lasa112, Romain Euvrard113, Giovanna Fabio68, Laurence Faivre114, Antonin Falck115, Muriel Fartoukh116, Morgane Faure117, Miguel Fernandez Arquero118, Ricard Ferrer119, Jose Ferreres120, Carlos Flores121, Bruno Francois122, Victoria Fumadó123, Kitty S. C. Fung124, Francesca Fusco125, Alenka Gagro126, Blanca Garcia Solis127, Pierre Garçon345, Pascale Gaussem128, Zeynep Gayretli129, Juana Gil-Herrera130, Laurent Gilardin131, Audrey Giraud Gatineau132, Mònica Girona-Alarcón133, Karen Alejandra Cifuentes Godínez134, Jean-Christophe Goffard135, Nacho Gonzales136, Luis I. Gonzalez-Granado137, Rafaela González-Montelongo138, Antoine Guerder139, Belgin Gülhan140, Victor Daniel Gumucio141, Leif Gunnar Hanitsch142, Jan Gunst143, Marta Gut144, Jérôme Hadjadj145, Filomeen Haerynck146, Rabih Halwani147, Lennart Hammarström148, Selda Hancerli149, Tetyana Hariyan150, Nevin Hatipoglu151, Deniz Heppekcan152, Elisa Hernandez-Brito153, Po-ki Ho154, María Soledad Holanda-Peña155, Juan P. Horcajada156, Sami Hraiech157, Linda Humbert158, Ivan F. N. Hung159, Alejandro D. Iglesias160, Antonio Íñigo-Campos138, Matthieu Jamme161, María Jesús Arranz89, Marie-Thérèse Jimeno162, Iolanda Jordan133, Saliha Kanık Yüksek163, Yalcin Burak Kara164, Aydın Karahan165, Adem Karbuz166, Kadriye Kart Yasar167, Ozgur Kasapcopur168, Kenichi Kashimada169, Sevgi Keles111, Yasemin Kendir Demirkol170, Yasutoshi Kido171, Can Kizil172, Ahmet Osman Kılıç173, Adam Klocperk174, Antonia Koutsoukou175, Zbigniew J. Król176, Hatem Ksouri177, Paul Kuentz178, Arthur M. C. Kwan179, Yat Wah M. Kwan180, Janette S. Y. Kwok181, Jean-Christophe Lagier182, David S. Y. Lam183, Vicky Lampropoulou184, Fanny Lanternier185, Yu-Lung Lau186, Fleur Le Bourgeois94, Yee-Sin Leo187, Rafael Leon Lopez188, Daniel Leung186, Michael Levin189, Michael Levy94, Romain Lévy33, Zhi Li78, Daniele Lilleri34, Edson Jose Adrian Bolanos Lima190, Agnes Linglart191, Eduardo López-Collazo192, José M. Lorenzo-Salazar138, Céline Louapre193, Catherine Lubetzki193, Kwok-Cheung Lung194, Charles-Edouard Luyt195, David C. Lye196, Cinthia Magnone197, Davood Mansouri198, Enrico Marchioni199, Carola Marioli2, Majid Marjani200, Laura Marques201, Jesus Marquez Pereira202, Andrea Martín-Nalda203, David Martínez Pueyo204, Javier Martinez-Picado205, Iciar Marzana206, Carmen Mata-Martínez207, Alexis Mathian24, Larissa R. B. Matos63, Gail V. Matthews208, Julien Mayaux209, Raquel McLaughlin-Garcia210, Philippe Meersseman211, Jean-Louis Mège212, Armand Mekontso-Dessap213, Isabelle Melki115, Federica Meloni2, Jean-François Meritet214, Paolo Merlani215, Özge Metin Akcan216, Isabelle Meyts217, Mehdi Mezidi218, Isabelle Migeotte219, Maude Millereux220, Matthieu Million221, Tristan Mirault222, Clotilde Mircher223, Mehdi Mirsaeidi224, Yoko Mizoguchi225, Bhavi P. Modi226, Francesco Mojoli13, Elsa Moncomble227, Abián Montesdeoca Melián228, Antonio Morales Martinez229, Francisco Morandeira230, Pierre-Emmanuel Morange231, Cléemence Mordacq158, Guillaume Morelle232, Stéphane J. Mouly233, Adrián Muñoz-Barrera138, Cyril Nafati234, Shintaro Nagashima235, Yu Nakagama171, Bénédicte Neven236, João Farela Neves237, Lisa F. P. Ng238, Yuk-Yung Ng239, hubert Nielly105, Yeray Novoa Medina210, Esmeralda Nuñez Cuadros240, Semsi Nur Karabela167, J. Gonzalo Ocejo-Vinyals241, Keisuke Okamoto109, Mehdi Oualha33, Amani Ouedrani22, Tayfun Özçelik242, Aslinur Ozkaya-Parlakay140, Michele Pagani13, Qiang Pan-Hammarström148, Maria Papadaki243, Christophe Parizot209, Philippe Parola244, Tiffany Pascreau245, Stéphane Paul246, Estela Paz-Artal247, Sigifredo Pedraza248, Nancy Carolina González Pellecer134, Silvia Pellegrini249, Rebeca Pérez de Diego127, Xosé Luis Pérez-Fernández141, Aurélien Philippe250, Quentin Philippot116, Adrien Picod251, Marc Pineton de Chambrun85, Antonio Piralla34, Laura Planas-Serra252, Dominique Ploin253, Julien Poissy254, Géraldine Poncelet70, Garyphallia Poulakou175, Marie S. Pouletty255, Persia Pourshahnazari256, Jia Li Qiu-Chen257, Paul Quentric209, Thomas Rambaud258, Didier Raoult212, Violette Raoult259, Anne-Sophie Rebillat223, Claire Redin260, Léa Resmini261, Pilar Ricart262, Jean-Christophe Richard263, Raúl Rigo-Bonnin264, Nadia rivet46, Jacques G. Rivière265, Gemma Rocamora-Blanch25, Mathieu P. Rodero266, Carlos Rodrigo267, Luis Antonio Rodriguez190, Carlos Rodriguez-Gallego268, Agustí Rodriguez-Palmero269, Carolina Soledad Romero270, Anya Rothenbuhler271, Damien Roux272, Nikoletta Rovina175, Flore Rozenberg273, Yvon Ruch90, Montse Ruiz274, Maria Yolanda Ruiz del Prado275, Juan Carlos Ruiz-Rodriguez119, Joan Sabater-Riera141, Kai Saks276, Maria Salagianni184, Oliver Sanchez277, Adrián Sánchez-Montalvá278, Silvia Sánchez-Ramón279, Laire Schidlowski280, Agatha Schluter252, Julien Schmidt281, Matthieu Schmidt282, Catharina Schuetz283, Cyril E. Schweitzer284, Francesco Scolari285, Anna Sediva286, Luis Seijo287, Analia Gisela Seminario42, Damien Sene23, Piseth Seng221, Sevtap Senoglu167, Mikko Seppänen288, Alex Serra Llovich289, Mohammad Shahrooei97, Anna Shcherbina290, Virginie Siguret291, Eleni Siouti292, David M. Smadja293, Nikaia Smith78, Ali Sobh294, Xavier Solanich25, Jordi Solé-Violán295, Catherine Soler296, Pere Soler-Palacín297, Betül Sözeri86, Giulia Maria Stella2, Yuriy Stepanovskiy298, Annabelle Stoclin299, Fabio Taccone219, Yacine Tandjaoui-Lambiotte300, Jean-Luc Taupin301, Simon J. Tavernier302, Loreto Vidaur Tello112, Benjamin Terrier303, Guillaume Thiery304, Christian Thorball260, Karolina Thorn305, Caroline Thumerelle158, Imran Tipu306, Martin Tolstrup307, Gabriele Tomasoni308, Julie Toubiana77, Josep Trenado Alvarez309, Vasiliki Triantafyllia310, Sophie Trouillet-Assant311, Jesús Troya312, Owen T. Y. Tsang313, Liina Tserel314, Eugene Y. K. Tso315, Alessandra Tucci316, Şadiye Kübra Tüter Öz15, Matilde Valeria Ursini125, Takanori Utsumi225, Yurdagul Uzunhan317, Pierre Vabres318, Juan Valencia-Ramos319, Ana Maria Van Den Rym127, Isabelle Vandernoot320, Valentina Velez-Santamaria321, Silvia Patricia Zuniga Veliz134, Mateus C. Vidigal322, Sébastien Viel253, Cédric Villain323, Marie E. Vilaire-Meunier223, Judit Villar-García324, Audrey Vincent57, Guillaume Voiriot326, Alla Volokha327, Fanny Vuotto158, Els Wauters328, Joost Wauters329, Alan K. L. Wu330, Tak-Chiu Wu331, Aysun Yahşi332, Osman Yesilbas333, Mehmet Yildiz168, Barnaby E. Young187, Ufuk Yükselmiş334, Mayana Zatz63, Marco Zecca39, Valentina Zuccaro62, Jens Van Praet335, Bart N. Lambrecht336, Eva Van Braeckel336, Cédric Bosteels336, Levi Hoste337, Eric Hoste338, Fré Bauters336, Jozefien De Clercq336, Catherine Heijmans339, Hans Slabbynck340, Leslie Naesens341, Benoit Florkin342, Cécile Boulanger343, Dimitri Vanderlinden344
1Germans Trias i Pujol University Hospital and Research Institute, Badalona, Barcelona, Spain. 2Respiratory Diseases Division, IRCCS Policlinico San Matteo Foundation, University of Pavia, Pavia, Italy. 3Neonatal Intensive Care Unit, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy. 4Navarra Health Service Hospital, Pamplona, Spain. 5Jeffrey Modell Diagnostic and Research Center for Primary Immunodeficiencies, Barcelona, Catalonia, Spain; Immunology Division, Genetics Department, Vall d’Hebron University Hospital (HUVH), Vall d’Hebron Research Institute (VHIR), Vall d’Hebron Barcelona Hospital Campus, Universitat Autònoma de Barcelona (UAB), Barcelona, Catalonia, Spain. 6Immunohematology Unit, San Raffaele Hospital, Milan, Italy. 7Ondokuz Mayıs University Medical Faculty Pediatrics, Samsun, Turkey. 8Department of Infectious Diseases, Loghman Hakim Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran. 9Hospital Regional de Huehuetenango, “Dr. Jorge Vides de Molina,” Huehuetenango, Guatemala. 10Hospital Nacional Edgardo Rebagliati Martins, Lima, Peru. 11Parc Sanitari Sant Joan de Déu, Sant Boi de Llobregat Spain. 12Khyber Medical University, Khyber Pakhtunkhwa, Pakistan. 13Anesthesia and Intensive Care, Rianimazione I, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy. 14Virology Research Center, National Institutes of Tuberculosis and Lung Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran. 15Department of Pediatrics, Division of Pediatric Infectious Diseases, Selcuk University Faculty of Medicine, Konya, Turkey. 16College of Medicine, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia; Department of Pediatrics, King Fahad Hospital of the University, Al-Khobar, Saudi Arabia. 17Intensive Care Unit, Hôpital Européen, Marseille, France. 18Immunology Department, Hospital 12 de Octubre, Research Institute imas12, Complutense University, Madrid, Spain. 19Immunology Department, Asturias Central University Hospital, Biosanitary Research Institute of the Principality of Asturias (ISPA), Oviedo, Spain. 20Emergency and Critical Care Medicine Departments, College of Medicine, Imam AbdulRahman Ben Faisal University, Dammam, Saudi Arabia. 21Clinical Immunology and Primary Immunodeficiencias Unit, Hospital Sant Joan de Déu, Institut de Recerca Sant Joan de Déu, Barcelona, Spain; Universitat de Barcelona, Barcelona, Spain. 22Department of Biological Immunology, Necker Hospital for Sick Children, AP-HP and INEM, Paris, France. 23Internal Medicine Department, Hôpital Lariboisière, AP-HP, Paris, France; Université de Paris, Paris, France. 24Internal Medicine Department, Pitié-Salpétrière Hospital, Paris, France. 25Department of Internal Medicine, Hospital Universitari de Bellvitge, IDIBELL, Barcelona, Spain. 26Service de Médecine Intensive Réanimation, Hôpitaux Universitaires Henri Mondor, AP-HP, Créteil, France; Groupe de Recherche Clinique CARMAS, Faculté de Santé de Créteil, Université Paris Est Créteil, Créteil, France. 27INSERM U1163, University of Paris, Imagine Institute, Paris, France and Pediatric Neurology Department, Necker-Enfants malades Hospital, AP-HP, Paris, France. 28Hospital U. de Tarragona Joan XXIII. Universitat Rovira i Virgili (URV). IISPV, Tarragona, Spain. 29Department of Propedeutics of Pediatrics and Medical Genetics, Danylo Halytsky Lviv National Medical University, Lviv, Ukraine. 30Department of Immunology and Allergy, Konya City Hospital, Konya, Turkey. 31Private Practice, Paris, France. 32INSERM U1109, University of Strasbourg, Strasbourg, France. 33Necker Hospital for Sick Children, AP-HP, Paris, France. 34Molecular Virology Unit, Microbiology and Virology Department, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy. 35Department of Infectious Diseases, CHU de Caen, Caen, France. 36Consorcio Hospital General Universitario, Valencia, Spain. 37Genetics Institute, Tel Aviv Sourasky Medical Center and Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel. 38Department of Urology, Nephrology, Transplantation, APHP-SU, Sorbonne Université, INSERM U 1082, Paris, France. 39Cell Factory and Pediatric Hematology-Oncology, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy. 40Yildirim Beyazit University, Faculty of Medicine, Ankara City Hospital, Children’s Hospital, Ankara, Turkey. 41University of Lyon, CIRI, INSERM U1111, National Referee Centre RAISE, Pediatric Rheumatology, HFME, Hospices Civils de Lyon, Lyon, France. 42Center for Clinical Immunology, CABA, Buenos Aires, Argentina. 43Cruces University Hospital, Bizkaia, Spain. 44Paediatric Immunology and Vaccinology Unit, Geneva University Hospitals and Faculty of Medicine, Geneva, Switzerland. 45University Hospital and Research Institute “Germans Trias i Pujol,” Badalona, Spain. 46Hematology, Georges Pompidou Hospital, AP-HP, Paris, France. 47Pediatric Infectious Diseases Unit, Instituto de Investigación Hospital 12 de Octubre (imas12), Hospital Universitario 12 de Octubre, Universidad Complutense, Madrid, Spain. 48Infectious disease Unit, Pitié-Salpêtrière Hospital, AP-AP, Paris, France. 49Department of Pediatrics, Thomayer’s Hospital, first Faculty of Medicine, Charles University, Prague, Czech Republic; Department of Immunology, Motol University Hospital, Second Faculty of Medicine, Charles University, Prague, Czech Republic. 50Centro de Investigación Biomédica en Red de Enfermedades Hepàticas y Digestivas (Ciberehd), Hospital de Mataró, Consorci Sanitari del Maresme, Mataró, Spain. 51Shupyk National Healthcare University of Ukraine, Kyiv, Ukraine. 52Service de Pneumologie, Hopital Bichat, AP-HP, Paris, France. 53Department of Infectious Diseases, CIC1408, GIMAP CIRI INSERM U1111, University Hospital of Saint-Etienne, Saint-Etienne, France. 54Clinical Immunology Unit, Pediatric Infectious Disease Department, Faculty of Medicine and Pharmacy, Averroes University Hospital, LICIA Laboratoire d’immunologie clinique, d’inflammation et d’allergie, Hassann Ii University, Casablanca, Morocco. 55Bégin Military Hospital, St Mandé, France. 56Sorbonne Université, INSERM, Institut Pierre Louis d’Epidémiologie et de Santé Publique (iPLESP), AP-HP, Hôpital Pitié Salpêtrière, Service de Virologie, Paris, France. 57Endocrinology Unit, AP-HP Hôpitaux Universitaires Paris-Sud, Le Kremlin-Bicêtre, France. 58Department of Children’s Diseases and Pediatric Surgery, I. Horbachevsky Ternopil National Medical University, Ternopil, Ukraine. 59Pneumology Unit, Tenon Hospital, AP-HP, Paris, France. 60Department of Respiratory Diseases, Hospital Clínico y Universitario de Valencia, Valencia, Spain. 61Intensive Care Unit, Réseau Hospitalier Neuchâtelois, Neuchâtel, Switzerland. 62Infectious Diseases Unit, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy. 63Human Genome and Stem Cell Research Center, University of São Paulo, São Paulo, Brazil. 64Department of Internal Medicine, College of Medicine, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia. 65Hospital Insular, Las Palmas de Gran Canaria, Spain. 66MS Center, Spedali Civili, Brescia, Italy. 67Laboratoire d’ImmunoRhumatologie Moléculaire, plateforme GENOMAX, INSERM UMR_S 1109, Faculté de Médecine, ITI TRANSPLANTEX NG, Université de Strasbourg, Strasbourg, France. 68Fondazione IRCCS Ca′ Granda Ospedale Maggiore Policlinico, Milan, Italy. 69Neuromuscular Unit, Neurology Department, Hospital Universitari de Bellvitge–IDIBELL and CIBERER, Barcelona, Spain. 70Hopital Robert Debré, Paris, France. 71Pediatric Immunohematology Unit, Necker Enfants Malades Hospital, AP-HP, Paris, France. 72Department of Infectious and Tropical Diseases, University of Brescia, ASST Spedali Civili di Brescia, Brescia, Italy. 73Doctoral Health Care Center, Canarian Health System, Las Palmas de Gran Canaria, Spain. 74Hôpital Foch, Suresnes, France. 75Selcuk University Faculty of Medicine, Department of Anesthesiology and Reanimation, Intensive Care Medicine Unit, Konya, Turkey. 76Division of Clinical Pharmacology and Toxicology, Institute of Pharmacological Sciences of Southern Switzerland, Ente Ospedaliero Cantonale and Faculty of Biomedical Sciences, Università della Svizzera italiana, Lugano, Switzerland. 77Necker Hospital for Sick Children, Paris University, AP-HP, Paris, France. 78Pasteur Institute, Paris, France. 79McGill University Health Centre, Montreal, Canada. 80University Hospital and Research Institute “Germans Trias i Pujol,” IrsiCaixa AIDS Research Institute, UVic-UCC, Badalona, Spain. 81Clinical Biochemistry, Pathology, Paediatric Neurology and Molecular Medicine Departments and Biobank, Institut de Recerca Sant Joan de Déu and CIBERER-ISCIII, Esplugues, Spain. 82AP-HP, Avicenne Hospital, Intensive Care Unit, Bobigny, France; University Sorbonne Paris Nord, Bobigny, France; INSERM, U942, F-75010, Paris, France. 83Hospital Universitari Vall d’Hebron, Barcelona, Spain. 84Pitié-Salpêtrière Hospital, Paris, France. 85Service de médecine Intensive Réanimation, Groupe Hospitalier Pitié-Salpêtrière, Sorbonne Université, Paris, France. 86Umraniye Training and Research Hospital, Istanbul, Turkey. 87Faculty of Medical Sciences at University “Goce Delcev,” Shtip, North Macedonia. 88Department of Biochemistry, College of Medicine, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia. 89Fundació Docencia i Recerca Mutua Terrassa, Barcelona, Spain. 90Maladies Infectieuses et Tropicales, Nouvel Hôpital Civil, CHU Strasbourg, Strasbourg, France. 91UNSW Medicine, St Vincent’s Clinical School, Sydney, NSW, Australia; Department of Thoracic Medicine, St Vincent’s Hospital Darlinghurst, Sydney, NSW, Australia. 92Intensive Care Unit, Montreuil Hospital, Montreuil, France. 93CHU Saint-Pierre, Université Libre de Bruxelles (ULB), Brussels, Belgium. 94Pediatric Intensive Care Unit, Robert-Debré University Hospital, AP-HP, Paris, France. 95General Internal Medicine, University Hospitals Leuven, Leuven, Belgium. 96Hôpital Jean Verdier, AP-HP, Bondy, France. 97Specialized Immunology Laboratory of Dr. Shahrooei, Sina Medical Complex, Ahvaz, Iran. 98Centre de génétique humaine, CHU Besançon, Besançon, France. 99Sorbonne Université médecine and AP-HP Sorbonne université site Pitié-Salpêtrière, Paris, France. 100Pediatric Neurology Department, Necker-Enfants Malades Hospital, AP-HP, Paris, France. 101Department of Internal Medicine, Fondazione IRCCS Policlinico San Matteo, University of Pavia, Pavia, Italy. 102Intensive Care Unit, Georges Pompidou Hospital, AP-HP, Paris, France. 103Department of Pneumology, AZ Delta, Roeselare, Belgium. 104Molecular Diagnostic Unit, Fundación Rioja Salud, Logroño, La Rioja, Spain. 105Bégin Military Hospital, Saint Mandé, France. 106Department of Pediatrics, Institute of Clinical Sciences, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden; Department of Rheumatology and Inflammation Research, Institute of Medicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden. 107Bursa City Hospital, Bursa, Turkey. 108Department of Medical Biochemistry and Molecular Biology, Faculty of Medicine, Mansoura University, Mansoura, Egypt. 109Tokyo Medical and Dental University, Tokyo, Japan. 110Ondokuz Mayıs University Faculty of Medicine, Samsun, Turkey. 111Necmettin Erbakan University, Meram Medical Faculty, Division of Pediatric Allergy and Immunology, Konya, Turkey. 112Intensive Care Medicine, Donostia University Hospital, Biodonostia Institute of Donostia, CIBER Enfermedades Respiratorias ISCIII, Donostia, Spain. 113Internal Medicine, University Hospital Edouard Herriot, Hospices Civils de Lyon, Lyon, France. 114Centre de Génétique, CHU Dijon, Dijon, France. 115Robert Debré Hospital, Paris, France. 116AP-HP Tenon Hospital, Paris, France. 117Sorbonne Universités, UPMC University of Paris, Paris, France. 118Department of Clinical Immunology, Hospital Clínico San Carlos, Madrid, Spain. 119Intensive Care Department, Vall d’Hebron University Hospital (HUVH), Vall d’Hebron Barcelona Hospital Campus, Barcelona, Catalonia, Spain; Shock, Organ Dysfunction and Resuscitation Research Group, Vall d’Hebron Research Institute (VHIR), Vall d’Hebron Barcelona Hospital Campus, Barcelona, Catalonia, Spain. 120Intensive Care Unit, Hospital Clínico y Universitario de Valencia, Valencia, Spain. 121Genomics Division, Instituto Tecnológico y de Energías Renovables (ITER), Santa Cruz de Tenerife, Spain; CIBER de Enfermedades Respiratorias, Instituto de Salud Carlos III, Madrid, Spain; Research Unit, Hospital Universitario N.S. de Candelaria, Santa Cruz de Tenerife, Spain; Faculty of Health Sciences, University of Fernando Pessoa Canarias, Las Palmas de Gran Canaria, Spain. 122CHU Limoges and INSERM CIC 1435 and UMR 1092, Limoges, France. 123Infectious Diseases Unit, Department of Pediatrics, Hospital Sant Joan de Déu, Barcelona, Spain; Institut de Recerca Sant Joan de Déu, Spain; Universitat de Barcelona (UB), Barcelona, Spain. 124Department of Pathology, United Christian Hospital, Hong Kong, China. 125Institute of Genetics and Biophysics “Adriano Buzzati-Traverso,” IGB-CNR, Naples, Italy. 126Department of Pediatrics, Children’s Hospital Zagreb, University of Zagreb School of Medicine, Zagreb, Josip Juraj Strossmayer University of Osijek, Medical Faculty Osijek, Osijek, Croatia. 127Laboratory of Immunogenetics of Human Diseases, IdiPAZ Institute for Health Research, La Paz Hospital, Madrid, Spain. 128Hematology, AP-HP, Hopital Européen Georges Pompidou and INSERM UMR-S1140, Paris, France. 129Faculty of Medicine, Department of Pediatrics, Division of Pediatric Infectious Diseases, Karadeniz Technical University, Trabzon, Turkey. 130Division of Immunology, Hospital General Universitario and Instituto de Investigación Sanitaria “Gregorio Marañón,” Madrid, Spain. 131Bégin Military Hospital, Bégin, France. 132Aix Marseille Univ, IRD, AP-HM, SSA, VITROME, IHU Méditerranée Infection, Marseille, France, French Armed Forces Center for Epidemiology and Public Health (CESPA), Marseille, France. 133Pediatric Intensive Care Unit, Hospital Sant Joan de Déu, Barcelona, Spain. 134Gestion Integral en Salud, Guatemala. 135Department of Internal Medicine, Hôpital Erasme, Université Libre de Bruxelles, Brussels, Belgium. 136Immunodeficiencies Unit, Research Institute Hospital, Madrid, Spain. 137Primary Immunodeficiencies Unit, Pediatrics, University Hospital 12 de Octubre, Madrid, Spain; School of Medicine Complutense University of Madrid, Madrid, Spain. 138Genomics Division, Instituto Tecnológico y de Energías Renovables (ITER), Santa Cruz de Tenerife, Spain. 139Assistance Publique Hôpitaux de Paris, Paris, France. 140Ankara City Hospital, Ankara, Turkey. 141Department of Intensive Care, Hospital Universitari de Bellvitge, IDIBELL, Barcelona, Spain. 142Immunodeficiency Outpatient Clinic, Institute for Medical Immunology, FOCIS Center of Excellence, Charité Universitätsmedizin Berlin, Germany. 143Surgical Intensive Care Unit, University Hospitals Leuven, Leuven, Belgium. 144CNAG-CRG, Barcelona Institute of Science and Technology, Barcelona, Spain. 145Department of Internal Medicine, National Reference Center for Rare Systemic Autoimmune Diseases, AP-HP, APHP-CUP, Hôpital Cochin, Paris, France. 146Department of Paediatric Immunology and Pulmonology, Center for Primary Immunodeficiency Ghent, Jeffrey Modell Diagnosis and Research Center, PID Research Lab, Ghent University Hospital, Ghent, Belgium. 147Sharjah Institute of Medical Research, College of Medicine, University of Sharjah, Sharjah, UAE, Sharjah, UAE. 148Department of Biosciences and Nutrition, SE14183, Huddinge, Karolinska Institutet, Stockholm, Sweden. 149Department of Pediatrics (Infectious Diseases), Istanbul Faculty of Medicine, Istanbul University, Istanbul, Turkey. 150I. Horbachevsky Ternopil National Medical University, Ternopil, Ukraine. 151Pediatric Infectious Diseases Unit, Bakirkoy Dr. Sadi Konuk Training and Research Hospital, University of Health Sciences, Istanbul, Turkey. 152Health Sciences University, Darıca Farabi Education and Research Hospital, Kocaeli, Turkey. 153Department of Immunology, Hospital Universitario de Gran Canaria Dr. Negrín, Canarian Health System, Las Palmas de Gran Canaria, Spain. 154Department of Paediatrics, Queen Elizabeth Hospital, Hong Kong, China. 155Intensive Care Unit. Marqués de Valdecilla Hospital, Santander, Spain. 156Hospital del Mar, Institut Hospital del Mar d’Investigacions Mèdiques (IMIM), UAB, UPF, Barcelona, Spain. 157Intensive Care Unit, APHM, Marseille, France. 158CHU Lille, Lille, France. 159Department of Medicine, University of Hong Kong, Hong Kong, China. 160Department of Pediatrics, Columbia University, New York, NY, USA. 161Centre hospitalier intercommunal Poissy Saint Germain en Laye, Poissy, France. 162IHU Méditerranée Infection, Service de l’Information Médicale, Hôpital de la Timone, Marseille, France. 163Health Science University Ankara City Hospital, Ankara, Turkey. 164School of Medicine, General Surgery Department Fevzi Çakmak Mah, Marmara University, Istanbul, Turkey. 165Mersin City Education and Research Hospital, Mersin, Turkey. 166Division of Pediatric Infectious Diseases, Prof. Dr. Cemil Tascıoglu City Hospital, Istanbul, Turkey. 167Departments of Infectious Diseases and Clinical Microbiology, Bakirkoy Dr. Sadi Konuk Training and Research Hospital, University of Health Sciences, Istanbul, Turkey. 168Department of Pediatric Rheumatology, Istanbul University-Cerrahpasa, Istanbul, Turkey. 169Department of Pediatrics, Tokyo Medical and Dental University, Tokyo, Japan. 170Health Sciences University, Umraniye Education and Research Hospital, Istanbul, Turkey. 171Department of Parasitology and Research Center for Infectious Disease Sciences, Graduate School of Medicine, Osaka City University, Osaka, Japan. 172Pediatric Infectious Diseases Unit of Osman Gazi University Medical School in Eskişehir, Turkey. 173Meram Medical Faculty, Necmettin Erbakan University, Konya, Turkey. 174Department of Immunology, Second Faculty of Medicine, Charles University and University Hospital in Motol, Prague, Czech Republic. 175ICU, First Department of Respiratory Medicine, National and Kapodistrian University of Athens, Medical School, “Sotiria” General Hospital of Chest Diseases, Athens, Greece. 176Central Clinical Hospital of the Ministry of Interior and Administration, Warsaw, Poland. 177Clinique des soins intensifs, HFR Fribourg, Fribourg, Switzerland. 178Oncobiologie Génétique Bioinformatique, PC Bio, CHU Besançon, Besançon, France. 179Department of Intensive Care, Tuen Mun Hospital, Hong Kong, China. 180Paediatric Infectious Disease Unit, Hospital Authority Infectious Disease Center, Princess Margaret Hospital, Hong Kong (Special Administrative Region), China. 181Department of Pathology, Queen Mary Hospital, Hong Kong, China. 182Aix Marseille Univ, IRD, MEPHI, IHU Méditerranée Infection, Marseille, France. 183Department of Paediatrics, Tuen Mun Hospital, Hong Kong, China. 184Biomedical Research Foundation of the Academy of Athens, Athens, Greece. 185Necker Hospital, Paris, France. 186Department of Paediatrics and Adolescent Medicine, University of Hong Kong, Hong Kong, China. 187National Centre for Infectious Diseases, Singapore, Singapore. 188Hospital Universitario Reina Sofía, Cordoba, Spain. 189Imperial College, London, England. 190Hospital General San Juan de Dios, Ciudad de Guatemala, Guatemala. 191Endocrinology and Diabetes for Children, AP-HP, Bicêtre Paris-saclay hospital, Le Kremlin-Bicêtre, France. 192Innate Immunity Group, IdiPAZ Institute for Health Research, La Paz Hospital, Madrid, Spain. 193Neurology Unit, AP-HP Pitié-Salpêtrière Hospital, Paris University, Paris, France. 194Department of Medicine, Pamela Youde Nethersole Eastern Hospital, Hong Kong, China. 195Intensive Care Unit, AP-HP Pitié-Salpêtrière Hospital, Paris University, Paris, France. 196National Centre for Infectious Diseases, Singapore, Singapore; Tan Tock Seng Hospital, Singapore, Singapore; Yong Loo Lin School of Medicine, Singapore, Singapore; Lee Kong Chian School of Medicine, Singapore, Singapore. 197Hospital de Niños Dr. Ricardo Gutierrez, Buenos Aires, Argentina. 198Department of Clinical Immunology and Infectious Diseases, National Research Institute of Tuberculosis and Lung Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran. 199Neurooncology and Neuroinflammation Unit, IRCCS Mondino Foundation, Pavia, Italy. 200Clinical Tuberculosis and Epidemiology Research Center, National Research Institute of Tuberculosis and Lung Diseases (NRITLD), Shahid Beheshti University of Medical Sciences, Tehran, Iran. 201Coordenadora da Unidade de Infeciologia e Imunodeficiências do Serviço de Pediatria, Centro Materno-Infantil do Norte, Porto, Portugal. 202Hospital Sant Joan de Déu and University of Barcelona, Barcelona, Spain. 203Pediatric Infectious Diseases and Immunodeficiencies Unit, Hospital Universitari Vall d’Hebron, Vall d’Hebron Research Institute, Vall d’Hebron Barcelona Hospital Campus, Universitat Autònoma de Barcelona (UAB), Barcelona, Catalonia, Spain. 204Hospital Universitari Mutua de Terrassa, Universitat de Barcelona, Barcelona, Spain. 205IrsiCaixa AIDS Research Institute, ICREA, UVic-UCC, Research Institute “Germans Trias i Pujol,” Badalona, Spain. 206Department of Laboratory, Cruces University Hospital, Barakaldo, Bizkaia, Spain, Bizkaia, Spain. 207Intensive Care Unit, Hospital General Universitario “Gregorio Marañón,” Madrid, Spain. 208University of New South Wales, Sydney, NSW, Australia. 209AP-HP Pitié-Salpêtrière Hospital, Paris, France. 210Department of Pediatrics, Complejo Hospitalario Universitario Insular-Materno Infantil, Canarian Health System, Las Palmas de Gran Canaria, Spain. 211Medical Intensive Care Unit, University Hospitals Leuven, Leuven, Belgium. 212Aix-Marseille University, APHM, Marseille, France. 213Service de Médecine Intensive Réanimation, Hôpitaux Universitaires Henri Mondor, Assistance Publique–Hôpitaux de Paris (AP-HP), Groupe de Recherche Clinique CARMAS, Faculté de Santé de Créteil, Université Paris Est Créteil, France. 214AP-HP Cohin Hospital, Paris, France. 215Department of Critical Care Medicine, Ente Ospedaliero Cantonale, Bellinzona, Switzerland. 216Necmettin Erbakan University, Meram Medical Faculty, Division of Pediatric Infectious Diseases, Konya, Turke4y. 217Department of Pediatrics, University Hospitals Leuven, Leuven, Belgium; KU Leuven, Department of Microbiology, Immunology and Transplantation; Laboratory for Inborn Errors of Immunity, KU Leuven, Leuven, Belgium. 218Hospices Civils de Lyon, Hôpital de la Croix-Rousse, Lyon, France. 219Hôpital Erasme, Brussels, Belgium. 220Centre hospitalier de gonesse, Gonesse, France. 221Aix Marseille Univ, IRD, AP-HM, MEPHI, IHU Méditerranée Infection, Marseille, France. 222Vascular Medicine, Georges Pompidou Hospital, AP-HP, Paris, France. 223Institut Jérôme Lejeune, Paris, France. 224Division of Pulmonary and Critical Care, College of Medicine-Jacksonville, University of Florida, Jacksonville, FL, USA. 225Department of Pediatrics, Hiroshima University Graduate School of Biomedical and Health Sciences, Hiroshima, Japan. 226BC Children’s Hospital Research Institute, University of British Columbia, Vancouver, BC, Canada. 227Médecine Intensive Réanimation, Hôpitaux Universitaires Henri Mondor, Assistance Publique–Hôpitaux de Paris (AP-HP), Créteil, France. 228Guanarteme Health Care Center, Canarian Health System, Las Palmas de Gran Canaria, Spain. 229Regional University Hospital of Malaga, Malaga, Spain. 230Department of Immunology, Hospital Universitari de Bellvitge, IDIBELL, Barcelona, Spain. 231Aix Marseille Univ, INSERM, INRAE, C2VN, Marseille, France. 232Department of General Paediatrics, Hôpital Bicêtre, AP-HP, University of Paris Saclay, Le Kremlin-Bicêtre, France. 233INSERM U1144, Université de Paris, DMU INVICTUS, AP-HP.Nord, Département de Médecine Interne, Lariboisière Hospital, Paris, France. 234CHU de La Timone, Marseille, France. 235Department of Epidemiology, Infectious Disease Control and Prevention, Graduate School of Biomedical and Health Sciences, Hiroshima University, Hiroshima, Japan. 236Pediatric Immunology and Rheumatology Department, Necker Hospital, AP-HP, Paris, France. 237Centro Hospitalar Universitário de Lisboa Central, Lisbon, Portugal. 238Infectious Disease Horizontal Technology Centre, A*STAR, Singapore, Singapore; Singapore Immunology Network, A*STAR, Singapore. 239Department of Medicine and Geriatrics, Tuen Mun Hospital, Hong Kong, China. 240Regional Universitary Hospital of Malaga, Málaga, Spain. 241Department of Immunology, Hospital Universitario Marqués de Valdecilla, Santander, Spain. 242Bilkent University, Department of Molecular Biology and Genetics, Ankara, Turkey. 243BRFAA, Athens, Greece. 244IHU Méditerranée Infection, Aix Marseille Univ, IRD, AP-HM, SSA, VITROME, IHU Méditerranée Infection, Marseille, France. 245L’Hôpital Foch, Suresnes, France. 246Department of Immunology, CIC1408, GIMAP CIRI INSERM U1111, University Hospital of Saint-Etienne, Saint-Etienne, France. 247Department of Immunology, Hospital Universitario 12 de Octubre, Instituto de Investigación Sanitaria Hospital 12 de Octubre (imas12), Madrid, Spain. 248Unit of Biochemistry, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Mexico City, Mexico. 249Diabetes Research Institute, IRCCS San Raffaele Hospital, Milan, Italy. 250AP-HP Hôpitaux Universitaires Paris-Sud, Le Kremlin-Bicêtre, France. 251AP-HP, Avicenne Hospital, Intensive Care Unit, Bobigny, France; INSERM UMR-S 942, Cardiovascular Markers in Stress Conditions (MASCOT), University of Paris, Paris, France. 252Neurometabolic Diseases Laboratory, IDIBELL-Hospital Duran i Reynals, Barcelona; CIBERER U759, ISCiii Madrid, Spain. 253Hospices Civils de Lyon, Lyon, France. 254Univ. Lille, INSERM U1285, CHU Lille, Pôle de médecine intensive-réanimation, CNRS, UMR 8576–Unité de Glycobiologie Structurale et Fonctionnelle, Lille, France. 255Department of General pediatrics, Robert Debre Hospital, Paris, France. 256University of British Columbia, Vancouver, BC, Canada. 257Jeffrey Modell Diagnostic and Research Center for Primary Immunodeficiencies, Barcelona, Catalonia, Spain; Diagnostic Immunology Research Group, Vall d’Hebron Research Institute (VHIR), Vall d’Hebron University Hospital (HUVH), Vall d’Hebron Barcelona Hospital Campus, Barcelona, Catalonia, Spain. 258AP-HP, Avicenne Hospital, Intensive Care Unit, Bobigny, France; University Sorbonne Paris Nord, Bobigny, France. 259Centre Hospitalier de Saint-Denis, St Denis, France. 260Precision Medicine Unit, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland. 261Paris Cardiovascular Center, PARCC, INSERM, Université de Paris, Paris, France. 262Germans Trias i Pujol Hospital, Badalona, Spain. 263Medical Intensive Care Unit, Hopital de la Croix-Rousse, Hospices Civils de Lyon, Lyon, France. 264Department of Clinical Laboratory, Hospital Universitari de Bellvitge, IDIBELL, Barcelona, Spain. 265Pediatric Infectious Diseases and Immunodeficiencies Unit, Hospital Universitari Vall d’Hebron, Vall d’Hebron Research Institute, Vall d’Hebron Barcelona Hospital Campus., Barcelona, Spain. 266Université de Paris, CNRS UMR-8601, Paris, France; Team Chemistry and Biology, Modeling and Immunology for Therapy, CBMIT, Paris, France. 267Germans Trias i Pujol University Hospital and Research Institute, Badalona, Spain. 268Department of Immunology, University Hospital of Gran Canaria Dr. Negrín, Canarian Health System, Las Palmas de Gran Canaria, Spain; Department of Clinical Sciences, University Fernando Pessoa Canarias, Las Palmas de Gran Canaria, Spain. 269Neurometabolic Diseases Laboratory, Bellvitge Biomedical Research Institute (IDIBELL), 08908 L’Hospitalet de Llobregat, Barcelona, Spain; University Hospital Germans Trias i Pujol, Badalona, Barcelona, Catalonia, Spain. 270Consorcio Hospital General Universitario, Valencia, Spain. 271AP-HP Hôpitaux Universitaires Paris-Sud, Paris, France. 272Intensive Care Unit, Louis-Mourier Hospital, Colombes, France. 273Virology Unit, Université de Paris, Cohin Hospital, AP-HP, Paris, France. 274Neurometabolic Diseases Laboratory and CIBERER U759, Barcelona, Spain. 275Hospital San Pedro, Logroño, Spain. 276University of Tartu, Institute of Biomedicine and Translational Medicine, Tartu, Estonia. 277Respiratory Medicine, Georges Pompidou Hospital, AP-HP, Paris, France. 278Infectious Diseases Department, International Health Program of the Catalan Institute of Health (PROSICS), Vall d’Hebron University Hospital (HUVH), Vall d’Hebron Barcelona Hospital Campus, Universitat Autónoma de Barcelona, Barcelona, Spain. 279Hospital Clínico San Carlos and IdSSC, Madrid, Spain. 280Faculdades Pequeno Príncipe, Instituto de Pesquisa Pelé Pequeno Príncipe, Curitiba, Brazil. 281AP-HP, Avicenne Hospital, Intensive Care Unit, Bobigny, France. 282Service de Médecine Intensive Réanimation, Institut de Cardiologie, Hopital Pitié-Salpêtrière, Paris, France. 283Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany. 284CHRU de Nancy, Hôpital d’Enfants, Vandoeuvre, France. 285Chair of Nephrology, University of Brescia, Brescia, Italy. 286Department of Immunology, Second Faculty of Medicine, Charles University and Motol University Hospital, Prague, Czech Republic. 287Clínica Universidad de Navarra and Ciberes, Madrid, Spain. 288HUS Helsinki University Hospital, Children and Adolescents, Rare Disease Center, and Inflammation Center, Adult Immunodeficiency Unit, Majakka, Helsinki, Finland. 289Fundació Docencia i Recerca Mutua Terrassa, Terrassa, Spain. 290D. Rogachev National Medical and Research Center of Pediatric Hematology, Oncology, Immunoogy, Moscow, Russia. 291Haematology Laboratory, Lariboisière Hospital, University of Paris, Paris, France. 292Biomedical Research Foundation of the Academy of Athens, Athens, Greece. 293INSERM U1140, University of Paris, European Georges Pompidou Hospital, Paris, France. 294Department of Pediatrics, Faculty of Medicine, Mansoura University, Mansoura, Egypt. 295Intensive Care Medicine, Hospital Universitario de Gran Canaria Dr. Negrín, Canarian Health System, Las Palmas de Gran Canaria, Spain. 296CHU de Saint Etienne, Saint-Priest-en-Jarez, France. 297Pediatric Infectious Diseases and Immunodeficiencies Unit, Hospital Universitari Vall d’Hebron, Vall d’Hebron Research Institute, Vall d’Hebron Barcelona Hospital Campus. Universitat Autònoma de Barcelona (UAB), Barcelona, Catalonia, Spain; EU, Barcelona, Spain. 298Department of Pediatric Infectious Diseases and Pediatric Immunology, Shupyk National Healthcare University of Ukraine, Kyiv, Ukraine. 299Gustave Roussy Cancer Campus, Villejuif, France. 300Intensive Care Unit, Avicenne Hospital, AP-HP, Bobigny, France. 301Laboratory of Immunology and Histocompatibility, Saint-Louis Hospital, Paris University, Paris, France. 302Center for Inflammation Research, Laboratory of Molecular Signal Transduction in Inflammation, VIB, Ghent, Belgium. 303Department of Internal Medicine, Université de Paris, INSERM, U970, PARCC, F-75015, Paris, France. 304Service de médecine intensive réanimation, CHU de Saint-Etienne, France. 305Department of Rheumatology and Inflammation Research, Institute of Medicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden. 306University of Management and Technology, Lahore, Pakistan. 307Department of Infectious Diseases, Aarhus University Hospital, Aarhus, Denmark. 308First Division of Anesthesiology and Critical Care Medicine, University of Brescia, ASST Spedali Civili di Brescia, Brescia, Italy. 309Intensive Care Department, Hospital Universitari MutuaTerrassa, Universitat Barcelona, Terrassa, Spain. 310Laboratory of Immunobiology, Center for Clinical, Experimental Surgery and Translational Research, Biomedical Research Foundation of the Academy of Athens, Athens, Greece. 311International Center of Research in Infectiology, Lyon University, INSERM U1111, CNRS UMR 5308, ENS, UCBL, Lyon, France; Hospices Civils de Lyon, Lyon Sud Hospital, Pierre-Bénite, France. 312Infanta Leonor University Hospital, Madrid, Spain. 313Department of Medicine and Geriatrics, Princess Margaret Hospital, Hong Kong, China. 314University of Tartu, Institute of Clinical Medicine, Tartu, Estonia. 315Department of Medicine, United Christian Hospital, Hong Kong, China. 316Hematology Department, ASST Spedali Civili di Brescia, Brescia, Italy. 317Pneumologie, Hôpital Avicenne, AP-HP, INSERM U1272, Université Sorbonne Paris Nord, Bobigny, France. 318Dermatology Unit, Laboratoire GAD, INSERM UMR1231 LNC, Université de Bourgogne, Dijon, France. 319University Hospital of Burgos, Burgos, Spain. 320Center of Human Genetics, Hôpital Erasme, Université Libre de Bruxelles, Brussels, Belgium. 321Bellvitge University Hospital, L’Hospitalet de Llobregat, Barcelona, Spain. 322University of São Paulo, São Paulo, Brazil. 323CHU de Caen, Caen, France. 324Hospital del Mar–IMIM Biomedical Research Institute, Barcelona, Catalonia, Spain. 326Sorbonne Université, Service de Médecine Intensive Réanimation, Hôpital Tenon, Assistance Publique-Hôpitaux de Paris, Paris, France. 327Pediatric Infectious Disease and Pediatric Immunology Department, Shupyk National Healthcare University of Ukraine, Kyiv, Ukraine. 328Department of Pneumology, University Hospitals Leuven, Leuven, Belgium. 329Laboratory for Clinical Infectious and Inflammatory Disorders, Department of Microbiology, Immunology, and Transplantation, Leuven, Belgium. 330Department of Clinical Pathology, Pamela Youde Nethersole Eastern Hospital, Hong Kong, China. 331Department of Medicine, Queen Elizabeth Hospital, Hong Kong, China. 332Ankara City Hospital, Children’s Hospital, Ankara, Turkey. 333Division of Pediatric Infectious Disease, Department of Pediatrics, Faculty of Medicine, Karadeniz Technical University, Trabzon, Turkey. 334Health Sciences University, Lütfi Kırdar Kartal Education and Research Hospital, İstanbul, Turkey. 335Department of Nephrology and Infectiology, AZ Sint-Jan, Bruges, Belgium. 336Department of Pulmonology, Ghent University Hospital, Belgium. 337Department of Pediatric Pulmonology and Immunology, Ghent University Hospital, Ghent, Belgium. 338Department of Intensive Care Unit, Ghent University Hospital, Ghent, Belgium. 339Department of Pediatric Hemato-oncology, Jolimont Hospital, La louvière, Belgium. 340Department of Pulmonology, ZNA Middelheim, Antwerp, Belgium. 341Department of Internal Medicine, Ghent University Hospital, Ghent, Belgium. 342Department of Pediatric Immuno-hémato-rheumatology, CHR Citadelle, Liége, Belgium. 343Department of Pediatric Hemato-oncology, UCL Louvain, Brussels, Belgium. 344Department of Pediatrics, Saint Luc, UCL Louvain, Brussels Belgium. 345Intensive Care Unit, Grand Hôpital de l’Est Francilien Site de Marne-La-Vallée, Jossigny, France.
Members of Orchestra Working Group: Laurent Abel1, Matilda Berkell2, Valerio Carelli3, Alessia Fiorentino3, Surbi Malhotra2, Alessandro Mattiaccio3, Tommaso Pippucci3, Marco Seri3, Evelina Tacconelli4
1Inserm, University Paris cité, Imagine Institute, Paris, France, 2University of Antwerp, Antwerp, Belgium, 3University of Bologna, Bologna, 40138, Italy, 4University of Verona, 37129 Verona, Italy
Members of French COVID Cohort Study Group: Laurent Abel1, Claire Andrejak2, François Angoulvant3, Delphine Bachelet4, Marie Bartoli5, Romain Basmaci6, Sylvie Behilill7, Marine Beluze8, Dehbia Benkerrou9, Krishna Bhavsar4, Lila Bouadma4, Sabelline Bouchez10, Maude Bouscambert11, Minerva Cervantes-Gonzalez4, Anissa Chair4, Catherine Chirouze12, Alexandra Coelho13, Camille Couffignal4, Sandrine Couffin-Cadiergues14, Eric d’Ortenzio5, Marie-Pierre Debray4, Lauren Deconinck4, Dominique Deplanque15, Diane Descamps4, Mathilde Desvallée16, Alpha Diallo5, Alphonsine Diouf13, Céline Dorival9, François Dubos17, Xavier Duval4, Brigitte Elharrar18, Philippine Eloy4, Vincent Enouf7, Hélène Esperou14, Marina Esposito-Farese4, Manuel Etienne19, Eglantine Ferrand Devouge19, Nathalie Gault4, Alexandre Gaymard11, Jade Ghosn4, Tristan Gigante20, Morgane Gilg20, Jérémie Guedj21, Alexandre Hoctin13, Isabelle Hoffmann4, Ikram Houas14, Jean-Sébastien Hulot22, Salma Jaafoura14, Ouifiya Kafif4, Florentia Kaguelidou23, Sabrina Kali4, Antoine Khalil4, Coralie Khan16, Cédric Laouénan4, Samira Laribi4, Minh Le4, Quentin Le Hingrat4, Soizic Le Mestre5, Hervé Le Nagard24, François-Xavier Lescure4, Sophie Letrou4, Yves Levy25, Bruno Lina11, Guillaume Lingas24, Jean Christophe Lucet4, Denis Malvy26, Marina Mambert13, France Mentré4, Amina Meziane9, Hugo Mouquet7, Jimmy Mullaert4, Nadège Neant24, Duc Nguyen26, Marion Noret27, Saad Nseir17, Aurélie Papadopoulos14, Christelle Paul5, Nathan Peiffer-Smadja4, Thomas Perpoint28, Ventzislava Petrov-Sanchez5, Gilles Peytavin4, Huong Pham4, Olivier Picone6, Valentine Piquard4, Oriane Puéchal29, Christian Rabaud30, Manuel Rosa-Calatrava11, Bénédicte Rossignol20, Patrick Rossignol30, Carine Roy4, Marion Schneider4, Richa Su4, Coralie Tardivon4, Marie-Capucine Tellier4, François Téoulé9, Olivier Terrier11, Jean-François Timsit4, Christelle Tual31, Sarah Tubiana4, Sylvie Van Der Werf7, Noémie Vanel32, Aurélie Veislinger31, Benoit Visseaux4, Aurélie Wiedemann25, Yazdan Yazdanpanah4
1INSERM UMR 1163, Paris, France. 2CHU Amiens, Amiens, France. 3Hôpital Necker, Paris, France. 4Hôpital Bichat, Paris, France. 5ANRS, Paris, France. 6Hôpital Louis Mourier, Colombes, France. 7Pasteur Institute, Paris, France. 8F-CRIN Partners Platform, Paris, France. 9INSERM UMR 1136, Paris, France. 10CHU Nantes, France. 11INSERM UMR 1111, Lyon, France. 12CHRU Jean Minjoz, Besançon, France. 13INSERM UMR 1018, Paris, France. 14INSERM Sponsor, Paris, France. 15Centre d’Investigation Clinique, INSERM CIC 1403, Centre Hospitalo universitaire de Lille, Lille, France. 16INSERM UMR 1219, Bordeaux, France. 17CHU Lille, Lille, France. 18CHI de Créteil, Créteil, France. 19CHU Rouen, Rouen, France. 20F-CRIN INI-CRCT, Nancy, France. 21Université de Paris, INSERM, IAME, F-75018 Paris, France. 22Hôpital Européen Georges Pompidou, Paris, France. 23Hôpital Robert Debré, Paris, France. 24INSERM UMR 1137, Paris, France. 25Vaccine Research Institute (VRI), INSERM UMR 955, Créteil, France. 26CHU Bordeaux, Bordeaux, France. 27RENARCI, Annecy, France. 28CHU Lyon, Lyon, France. 29REACTing, Paris, France. 30CHU Nancy, Nancy, France. 31INSERM CIC-1414, Rennes, France. 32Hôpital la Timone, Marseille, France.
Members of CoV-Contact Cohort: Loubna Alavoine1, Sylvie Behillil2, Charles Burdet3, Charlotte Charpentier4, Aline Dechanet5, Diane Descamps6, Xavier Duval7, Jean-Luc Ecobichon1, Vincent Enouf8, Wahiba Frezouls1, Nadhira Houhou5, Ouifiya Kafif5, Jonathan Lehacaut1, Sophie Letrou1, Bruno Lina9, Jean-Christophe Lucet10, Pauline Manchon5, Mariama Nouroudine1, Valentine Piquard5, Caroline Quintin1, Michael Thy11, Sarah Tubiana1, Sylvie van der Werf8, Valérie Vignali1, Benoit Visseaux10, Yazdan Yazdanpanah10, Abir Chahine12, Nawal Waucquier12, Maria-Claire Migaud12, Dominique Deplanque12, Félix Djossou13, Mayka Mergeay-Fabre14, Aude Lucarelli15, Magalie Demar13, Léa Bruneau16, Patrick Gérardin17, Adrien Maillot16, Christine Payet18, Bruno Laviolle19, Fabrice Laine19, Christophe Paris19, Mireille Desille-Dugast19, Julie Fouchard19, Denis Malvy20, Duc Nguyen20, Thierry Pistone20, Pauline Perreau20, Valérie Gissot21, Carole L. E. Goas21, Samatha Montagne22, Lucie Richard23, Catherine Chirouze24, Kévin Bouiller24, Maxime Desmarets25, Alexandre Meunier26, Marilou Bourgeon26, Benjamin Lefévre27, Hélène Jeulin28, Karine Legrand29, Sandra Lomazzi30, Bernard Tardy31, Amandine Gagneux-Brunon32, Frédérique Bertholon33, Elisabeth Botelho-Nevers32, Kouakam Christelle34, Leturque Nicolas34, Layidé Roufai34, Karine Amat35, Sandrine Couffin-Cadiergues34, Hélène Espérou36, Samia Hendou34
1Centre d’Investigation Clinique, INSERM CIC 1425, Hôpital Bichat Claude Bernard, AP-HP, Paris, France. 2Institut Pasteur, Paris, France. 3Université de Paris, IAME, INSERM U1137, Paris, France; Hôpital Bichat Claude Bernard, AP-HP, Paris, France. 4Service de Virologie, Université de Paris, INSERM, IAME, UMR 1137, Hôpital Bichat Claude Bernard, AP-HP, Paris, France. 5Hôpital Bichat Claude Bernard, AP-HP, Paris, France. 6IAME INSERM U1140, Hôpital Bichat Claude Bernard, AP-HP, Paris, France. 7Centre d’Investigation Clinique, INSERM CIC 1425, AP-HP, IAME, Paris University, Paris, France. 8Institut Pasteur, U3569 CNRS, Université de Paris, Paris, France. 9Virpath Laboratory, International Center of Research in Infectiology, Lyon University, INSERM U1111, CNRS U5308, ENS, UCBL, Lyon, France. 10IAME INSERM U1138, Hôpital Bichat Claude Bernard, AP-HP, Paris, France. 11Center for Clinical Investigation, Assistance Publique-Hôpitaux de Paris, Bichat-Claude Bernard University Hospital, Paris, France. 12Centre d’Investigation Clinique, INSERM CIC 1403, Centre Hospitalo universitaire de Lille, Lille, France. 13Service des maladies infectieuses, Centre Hospitalo universitaire de Cayenne, Guyane, France. 14Centre d’Investigation Clinique, INSERM CIC 1424, Centre Hospitalier de Cayenne, Cayenne, Guyane Française. 15Service Hôpital de jour Adulte, Centre Hospitalier de Cayenne, Guyane, France. 16Centre d’Investigation Clinique, INSERM CIC 1410, Centre Hospitalo universitaire de la Réunion, La Réunion, France. 17Centre d’Investigation Clinique, INSERM CIC 1410, CHU Reunion, Saint-Pierre, Reunion Island. 18Centre d’Investigation Clinique, INSERM CIC 1410, Centre de Ressources Biologiques, Centre Hospitalo universitaire de la Réunion, La Réunion, France. 19Centre d’Investigation Clinique, INSERM CIC 1414, Centre Hospitalo universitaire de Rennes, Rennes, France. 20Service des maladies infectieuses, Centre Hospitalo universitaire de Bordeaux, Bordeaux, France. 21Centre d’Investigation Clinique, INSERM CIC 1415, CHRU Tours, Tours, France. 22CRBT, Centre Hospitalo universitaire de Tours, Tours, France. 23Pole de Biologie Médicale, Centre Hospitalo universitaire de Tours, Tours, France. 24Service des maladies infectieuses, Centre Hospitalo universitaire de Besançon, Besançon, France. 25Service des maladies infectieuses, Centre d’investigation clinique, INSERM CIC1431, Centre Hospitalier Universitaire de Besançon, Besançon, France. 26Centre de Ressources Biologiques–Filière Microbiologique de Besançon, Centre Hospitalier Universitaire, Besançon, France. 27Université de Lorraine, CHRU-Nancy and APEMAC, Infectious and Tropical Diseases, Nancy, France. 28Laboratoire de Virologie, CHRU de Nancy Brabois, Vandoeuvre-lès-Nancy, France. 29INSERM CIC-EC 1433, Centre Hospitalo universitaire de Nancy, Nancy, France. 30Centre de ressources Biologiques, Centre Hospitalo universitaire de Nancy, Nancy, France. 31Centre d’Investigation Clinique, INSERM CIC 1408, Centre Hospitalo universitaire de Saint Etienne, Saint Etienne, France. 32Service des maladies infectieuses, Centre Hospitalo universitaire de Saint Etienne, Saint Etienne, France. 33Service des maladies infectieuses, CRB42-BTK, Centre Hospitalo Universitaire de Saint Etienne, Saint Etienne, France. 34Pole Recherche Clinique, INSERM, Paris, France. 35IMEA Fondation Léon M’Ba, Paris, France. 36INSERM Clinical Research Department, Paris, France.
Members of the COVIDeF study group: Serge Bureau1, Yannick Vacher1, Anne Gysembergh-Houal1, Lauren Demerville1, Abla Chachoua1, Sebastien Abad2, Radhiya Abassi3, Abdelrafie Abdellaoui3, Abdelkrim Abdelmalek4, Hendy Abdoul5, Helene Abergel6, Fariza Abeud7, Sophie Abgrall8, Noemie Abisror4, Marylise Adechian9, Nordine Aderdour9, Hakeem Farid Admane4, Frederic Adnet2, Sara Afritt5, Helene Agostini10, Claire Aguilar11, Sophie Agut12, Tommaso Francesco Aiello13, Marc Ait Kaci14, Hafid Ait Oufella4, Gokula Ajeenthiravasan15, Virginie Alauzy3, Fanny Alby-Laurent11, Lucie Allard2, Marie-Alexandra Alyanakian11, Blanca Amador Borrero7, Sabrina Amam6, Lucile Amrouche11, Marc Andronikof16, Dany Anglicheau11, Nadia Anguel9, Djillali Annane15, Mohammed Aounzou3, Caroline Aparicio7, Gladys Aratus4, Jean-Benoit Arlet14, Jeremy Arzoine3, Elisabeth Aslangul13, Mona Assefi3, Adeline Aubry3, Laetitia Audiffred4, Etienne Audureau17, Christelle Nathalie Auger5, Jean-Charles Auregan8, Celine Awotar11, Sonia Ayllon Milla5, Delphine Azan5, Laurene Azemar7, Billal Azzouguen7, Marwa Bachir Elrufaai12, Aïda Badsi7, Prissile Bakouboula11, Coline Balcerowiak3, Fanta Balde12, Elodie Baldivia17, Eliane-Flore Bangamingo18, Amandine Baptiste3, Fanny Baran-Marszak2, Caroline Barau17, Nathalie Barget19, Flore Baronnet3, Romain Barthelemy7, Jean-Luc Baudel4, Camille Baudry2, Elodie Baudry9, Laurent Beaugerie4, Adel Belamri3, Nicolas Belaube12, Rhida Belilita3, Pierre Bellassen3, Rawan Belmokhtar2, Isabel Beltran6, Ruben Benainous2, Mourad Benallaoua2, Robert Benamouzig2, Amélie Benbara19, Jaouad Benhida3, Anis Benkhelouf3, Jihene Benlagha18, Chahinez Benmostafa14, Skander Benothmane18, Miassa Bentifraouine2, Laurence Berard4, Quentin Bernier3, Enora Berti17, Astrid Bertier9, Laure Berton7, Simon Bessis15, Alexandra Beurton20, Celine Bianco4, Clara Bianquis3, Frank Bidar3, Philippe Blanche5, Clarisse Blayau12, Alexandre Bleibtreu3, Emmanuelle Blin12, Coralie Bloch-Queyrat2, Marie-Christophe Boissier2, Diane Bollens4, Marion Bolzoni4, Rudy pierre Bompard12, Nicolas Bonnet2, Justine Bonnouvrier4, Shirmonecrystal Botha3, Wissam Boucenna4, Fatiha Bouchama3, Olivier Bouchaud2, Hanane Bouchghoul9, Taoueslylia Boudjebla12, Noel Boudjema17, Catherine Bouffard6, Adrien Bougle3, Meriem Bouguerra3, Leila Bouras7, Agnes Bourcier3, Anne Bourgarit Durand19, Anne Bourrier4, Fabrice Bouscarat6, Diane Bouvry2, Nesrine Bouziri3, Ons Bouzrara3, Sarah Bribier9, Delphine Brugier3, Melanie Brunel11, Eida Bui4, Anne Buisson21, Iryna Bukreyeva9, Côme Bureau20, Jacques Cadranel12, Johann Cailhol2, Ruxandra Calin12, Clara Campos Vega11, Pauline Canavaggio3, Marta Cancella3, Delphine Cantin22, Albert Cao3, Lionel Carbillon19, Nicolas Carlier5, Clementine Cassard3, Guylaine Castor7, Marion Cauchy7, Olivier Cha4, Benjamin Chaigne5, Salima Challal2, Karine Champion7, Patrick Chariot19, Julie Chas12, Simon Chauveau2, Anthony Chauvin7, Clement Chauvin18, Nathalie Chavarot11, Kamélia Chebbout3, Mustapha Cherai3, Ilaria Cherubini3, Amelie Chevalier5, Thibault Chiarabini4, Thierry Chinet10, Richard Chocron14, Pascaline Choinier12, Juliette Chommeloux3, Christophe Choquet6, Laure Choupeaux11, Benjamin Chousterman7, Dragosmarius Ciocan8, Ada Clarke5, Gaëlle Clavere23, Florian Clavier3, Karine Clement3, Sebastien Clerc14, Yves Cohen2, Fleur Cohen3, Adrien Cohen23, Audrey Coilly24, Hester Colboc25, Pauline Colin3, Magalie Collet7, Chloé Comarmond7, Emeline Combacon5, Alain Combes3, Celine Comparon2, Jean-Michel Constantin3, Hugues Cordel2, Anne-Gael Cordier9, Adrien Costantini10, Nathalie Costedoat Chalumeau5, Camille Couffignal6, Doriane Coupeau4, Alain Creange17, Yannie Cuvillier Lamarre22, Charlène Da Silveira6, Sandrine Dautheville Guibal El Kayani12, Nathalie De Castro18, Yann De Rycke3, Lucie Del Pozo19, Quentin Delannoy3, Mathieu Delay12, Robin Deleris3, Juliette Delforge13, Laëtitia Delphine3, Noemie Demare2, Sophie Demeret3, Alexandre Demoule3, Aurore Deniau2, François Depret18, Sophie Derolez2, Ouda Derradji9, Nawal Derridj10, Vincent Descamps6, Lydia Deschamps6, Celine Desconclois8, Cyrielle Desnos3, Karine Desongins18, Robin Dhote2, Benjamin Diallo12, Morgane Didier2, Myriam Diemer7, Stephane Diez9, Juliette Djadi-Prat14, Fatima-Zohra Djamouri Monnory12, Siham Djebara3, Naoual Djebra2, Minette Djietcheu18, Hadjer Djillali4, Nouara Djouadi4, Severine Donneger19, Catarina Dos Santos5, Nathalie Dournon2, Martin Dres20, Laura Droctove3, Marie Drogrey3, Margot Dropy3, Elodie Drouet4, Valérie Dubosq12, Evelyne Dubreucq12, Estelle Dubus7, Boris Duchemann2, Thibault Duchenoy5, Emmanuel Dudoignon18, Romain Dufau19, Florence Dumas5, Clara Duran10, Emmanuelle Duron24, Antoine Durrbach17, Claudine Duvivier11, Nathan Ebstein2, Jihane El Khalifa6, Alexandre Elabbadi12, Caroline Elie11, Gabriel Ernotte3, Anne Esling11, Martin Etienne9, Xavier Eyer7, Muriel sarah Fartoukh12, Takoua Fayali3, Marion Fermaut19, Arianna Fiorentino4, Souha Fliss2, Marie-Céline Fournier7, Benjamin Fournier11, Hélène Francois12, Olivia Freynet2, Yvann Frigout14, Isaure Fromont7, Axelle Fuentes6, Thomas Furet3, Joris Galand7, Marc Garnier4, Agnes Gaubert3, Stéphane Gaudry2, Samuel Gaugain7, Damien Gauthier3, Maxime Gautier7, Sophie Georgin-Lavialle12, Daniela Geromin14, Mohamed Ghalayini2, Bijan Ghaleh17, Myriam Ghezal21, Aude Gibelin12, Linda Gimeno3, Benoit Girard5, Bénédicte Giroux Leprieur2, Doryan Gomes18, Elisabete Gomes-Pires11, Guy Gorochov3, Anne Gouge18, Amel Gouja17, Helene Goulet12, Sylvain Goupil11, Jeanne Goupil De Bouille2, Julien Gras7, Segolene Greffe10, Lamiae Grimaldi9, Paul Guedeney3, Bertrand Guidet4, Matthias Guillo18, Mariechristelle Gulczynski26, Tassadit Hadjam7, Didier Haguenauer13, Soumeya Hammal3, Nadjib Hammoudi3, Olivier Hanon23, Anarole Harrois9, Pierre Hausfater3, Coraline Hautem14, Guillaume Hekimian3, Nicholas Heming15, Olivier Hermine11, Sylvie Ho3, Marie Houllier9, Benjamin Huot7, Tessa Huscenot7, Wafa Ibn Saied12, Ghilas Ikherbane3, Meriem Imarazene11, Patrick Ingiliz4, Lina Iratni17, Stephane Jaureguiberry9, Jean-Francois Jean-Marc10, Deleena Jeyarajasingham18, Pauline Jouany14, Veronique Jouis7, Clement Jourdaine7, Ouifiya Kafif6, Rim Kallala24, Sandrine Katsahian14, Lilit Kelesyan27, Vixra Keo3, Flora Ketz21, Warda Khamis2, Enfel Khelili3, Mehdi Khellaf17, Christy Gaëlla Kotokpo Youkou10, Ilias Kounis24, Gaelle Kpalma3, Jessica Krause4, Vincent Labbe12, Karine Lacombe4, Jean-Marc Lacorte3, Anne Gaelle Lafont4, Emmanuel Lafont11, Lynda Lagha27, Lionel Lamhaut11, Aymeric Lancelot3, Cecilia Landman4, Fanny Lanternier11, Cecile Larcheveque3, Caroline Lascoux Combe18, Ludovic Lassel12, Benjamin Laverdant12, Christophe Lavergne18, Jean-Rémi Lavillegrand4, Pompilia Lazureanu7, Loïc Le Guennec3, Lamia Leberre4, Claire Leblanc19, Marion Leboyer28, Francois Lecomte5, Marine Lecorre3, Romain Leenhardt4, Marylou Lefebvre4, Bénédicte Lefebvre4, Paul Legendre5, Anne Leger3, Laurence Legros24, Justyna Legrosse3, Sébastien Lehuunghia5, Julien Lemarec3, Jeremie Leporrier-Ext11, Manon Lesein5, Hubert Lesur24, Vincent Levy2, Albert Levy14, Edwige Lopes7, Amanda Lopes7, Vanessa Lopez11, Julien Lopinto12, Olivier Lortholary11, Badr Louadah7, Bénédicte Loze18, Marie-Laure Lucas22, Axelle Lucasamichi8, Liem Binh Luong5, Arouna Magazimama-Ext7, David Maingret7, Lakhdar Mameri18, Philippe Manivet7, Cylia Mansouri4, Estelle Marcault6, Jonathan Marey5, Nathalie Marin5, Clémence Marois3, Olivier Martin2, Lou Martineau3, Cannelle Martinez-Lopez15, Pierre Martyniuck4, Pauline Mary De Farcy29, Nessrine Marzouk12, Rafik Masmoudi14, Alexandre Mebazaa7, Frédéric Mechai2, Fabio Mecozzi11, Chamseddine Mediouni10, Bruno Megarbane7, Mohamed Meghadecha22, Élodie Mejean12, Arsene Mekinian4, Nour Mekki Abdelhadi6, Rania Mekni3, Thinhinan Sabrina Meliti3, Breno Melo Lima18, Paris Meng12, Soraya Merbah3, Fadhila Messani2, Yasmine Messaoudi3, Baboo-Irwinsingh Mewasing12, Lydia Meziane3, Carole Michelot-Burger11, Françoise Mignot18, Fadi Hillary Minka7, Makoto Miyara3, Pierre Moine15, Jean-Michel Molina18, Anaïs Montegnies-Boulet5, Alexandra Monti21, Claire Montlahuc18, Anne-Lise Montout3, Alexandre Moores5, Caroline Morbieu5, Helene Mortelette14, Stéphane Mouly7, Rosita Muzaffar18, Cherifa Iness Nacerddine3, Marine Nadal12, Hajer Nadif3, Kladoum Nassarmadji7, Pierre Natella17, Sandrine Ndingamondze3, Stefan Neraal5, Caroline Nguyen6, Bao N'Guyen3, Isabelle Nion Larmurier4, Luc Nlomenyengue14, Nicolas Noel9, Hilario Nunes2, Edris Omar3, Zineb Ouazene4, Elise Ouedraogo2, Wassila Ouelaa3, Anissa Oukhedouma3, Yasmina Ould Amara3, Herve Oya3, Johanna Oziel2, Thomas Padilla3, Elena Paillaud26, Solenne Paiva3, Beatrice Parfait5, Perrine Parize11, Christophe Parizot3, Antoine Parrot12, Arthur Pavot9, Laetitia Peaudecerf5, Frédéric Pene5, Marion Pepin10, Julie Pernet3, Claire Pernin7, Mylène Petit2, Olivier Peyrony18, Marie-Pierre Pietri22, Olivia Pietri4, Marc Pineton De Chambrun3, Michelle Pinson13, Claire Pintado18, Valentine Piquard6, Christine Pires3, Benjamin Planquette14, Sandrine Poirier8, Anne-Laure Pomel8, Stéphanie Pons3, Diane Ponscarme18, Annegaelle Pourcelot9, Valérie Pourcher3, Anne Pouvaret11, Florian Prever4, Miresta Previlon18, Margot Prevost3, Marie-Julie Provoost7, Cyril Quemeneur3, Cédric Rafat12, Agathe Rami7, Brigitte Ranque14, Maurice Raphael9, Jean Herle Raphalen11, Anna Rastoin7, Mathieu Raux3, Amani Rebai2, Michael Reby25, Alexis Regent5, Asma Regrag14, Matthieu Resche-Rigon18, Quentin Ressaire18, Christian Richard9, Mariecaroline Richard3, Maxence Robert3, Benjamin Rohaut3, Camille Rolland-Debord12, Jacques Ropers3, Anne-Marie Roque-Afonso24, Charlotte Rosso30, Mélanie Rousseaux4, Nabila Rousseaux3, Swasti Roux26, Lorène Roux4, Claire Rouzaud11, Antoine Rozes3, Emma Rubenstein7, Jean-Marc Sabate2, Sheila Sabet12, Sophie-Caroline Sacleux24, Nathalie Saidenberg Kermanach2, Faouzi Saliba24, Dominique Salmon22, Laurent Savale31, Guillaume Savary3, Rebecca Sberro11, Anne Scemla11, Frederic Schlemmer17, Mathieu Schwartz7, Saïd Sedfi3, Samia Sefir-Kribel5, Philippe Seksik4, Pierre Sellier7, Agathe Selves3, Nicole Sembach14, Luca Semerano2, Marie-Victoire Senat9, Damien Sene7, Alexandra Serris11, Lucile Sese2, Naima Sghiouar15, Johanna Sigaux2, Martin Siguier12, Johanne Silvain3, Noémie Simon3, Tabassome Simon4, Lina Innes Skandri2, Miassa Slimani2, Aurélie Snauwaert6, Harry Sokol4, Heithem Soliman4, Nisrine Soltani9, Benjamin Soyer7, Gabriel Steg6, Lydia Suarez7, Tali-Anne Szwebel5, Kossi Taffame3, Yacine Tandjaoui-Lambiotte2, Claire Tantet2, Mariagrazia Tateo18, Igor Theodose18, Pierre clement Thiebaud4, Caroline Thomas4, Kelly Tiercelet18, Julie Tisserand9, Carole Tomczak18, Krystel Torelino3, Fatima Touam-Ext11, Lilia Toumi11, Gustave Toury14, Mireille Toy-Miou3, Olivia Tran Dinh Thanh Lien7, Alexy Trandinh6, Jean-Marc Treluyer5, Baptiste Trinque7, Jennifer Truchot5, Florence Tubach3, Sarah Tubiana6, Simone Tunesi19, Matthieu Turpin12, Agathe Turpin3, Tomas Urbina4, Rafael Usubillaga Narvaez22, Yurdagul Uzunhan2, Prabakar Vaittinadaayar27, Arnaud Valent18, Maelle Valentian12, Nadia Valin4, Hélène Vallet4, Marina Vaz3, Miguel-Alejandro Vazquezibarra7, Benoit Vedie14, Laetitia Velly3, Celine Verstuyft9, Cedric Viallette3, Eric Vicaut7, Dorothee Vignes8, Damien Vimpere11, Myriam Virlouvet9, Guillaume Voiriot12, Lena Voisot21, Emmanuel Weiss27, Nicolas Weiss3, Anaïs Winchenne2, Youri Yordanov4, Lara Zafrani18, Mohamad Zaidan9, Wissem Zaidi4, Cathia Zak12, Aida Zarhrate-Ghoul3, Ouassila Zatout6, Suzanne Zeino9, Michel Zeitouni3, Naïma Zemirli3, Lorene Zerah3, Ounsa Zia3, Marianne Ziol19, Oceane Zolario4, Julien Zuber11
1DRCI-APHP, Paris, France, 2Hôpital Avicenne, Bobigny, France, 3Hôpital Pitié-Salpêtrière, Paris, France, 4Hôpital Saint-Antoine, Paris, France, 5Hôpital Cochin, Paris, France, 6Hôpital Bichat, Paris, France, 7Hôpital Lariboisière, Paris, France, 8Hôpital Antoine Béclère, Clamart, France, 9Hôpital Kremlin Bicêtre, Le Kremlin-Bicêtre, France, 10Hôpital Ambroise-Paré, Boulogne Billancourt, France, 11Hopital Necker Enfants malades, Paris, France, 12Hôpital Tenon, Paris, France, 13Hôpital Louis Mourier, Colombes, France, 14Hôpital Européen Georges Pompidou, Paris, France, 15Hôpital Raymond Poincaré, Garches, France, 16Hôpital Antoine Béclère, Calmart, France, 17Hôpital Henri Mondor, Créteil, France, 18Hôpital Saint Louis, Paris, France, 19Hôpital Jean Verdier, Bondy, France, 20Université Paris-Sorbonne, Hôpital Pitié-Salpêtrière, INSERM, Paris, France, 21Hôpital Charles Foix, Ivry-sur-Seine, France, 22Hôpital Hôtel Dieu, Paris, France, 23Hôpital Broca, Paris, France, 24Hôpital Paul-Brousse, Villejuif, France, 25Hôpital Rothschild, Paris, France, 26Hôpital Corentin Celton, Issy-les-Moulineaux, France, 27Hôpital Beaujon, Clichy, France, 28Hôpital Albert Chenevier, Créteil, France, 29Hôpital Sainte-Périne, Paris, France, 30Université Paris-Sorbonne, Hôpital Pitié-Salpêtrière, INSERM, CNRS, Paris, France, 31Université Paris-Saclay, Hôpital Kremlin Bicêtre, INSERM, Le Kremlin-Bicêtre, France
Members of Amsterdam UMC Covid-19 Biobank: Michiel van Agtmael2, Anne Geke Algera1, Brent Appelman2, Frank van Baarle1, Diane Bax3, Martijn Beudel4, Harm Jan Bogaard5, Marije Bomers2, Peter Bonta5, Lieuwe Bos1, Michela Botta1, Justin de Brabander2, Godelieve de Bree2, Sanne de Bruin1, David T. P. Buis1, Marianna Bugiani5, Esther Bulle1, Osoul Chouchane2 Alex Cloherty3, Mirjam Dijkstra12, Dave A. Dongelmans1, Romein W. G. Dujardin1, Paul Elbers1, Lucas Fleuren1, Suzanne Geerlings2 Theo Geijtenbeek3, Armand Girbes1, Bram Goorhuis2, Martin P. Grobusch2, Florianne Hafkamp3, Laura Hagens1, Jorg Hamann7, Vanessa Harris2, Robert Hemke8, Sabine M. Hermans2 Leo Heunks1, Markus Hollmann6, Janneke Horn1, Joppe W. Hovius2, Menno D. de Jong9, Rutger Koning4, Endry H. T. Lim1, Niels van Mourik1, Jeaninne Nellen2, Esther J. Nossent5, Frederique Paulus1, Edgar Peters2, Dan A. I. Pina-Fuentes4, Tom van der Poll2, Bennedikt Preckel6, Jan M. Prins2, Jorinde Raasveld1, Tom Reijnders2, Maurits C. F. J. de Rotte12, Michiel Schinkel2, Marcus J. Schultz1, Femke A. P. Schrauwen12, Alex Schuurmans10, Jaap Schuurmans1, Kim Sigaloff1, Marleen A. Slim1,2, Patrick Smeele5, Marry Smit1, Cornelis S. Stijnis2, Willemke Stilma1, Charlotte Teunissen11, Patrick Thoral1, Anissa M. Tsonas1, Pieter R. Tuinman2, Marc van der Valk2, Denise Veelo6, Carolien Volleman1, Heder de Vries1, Lonneke A. Vught1,2, Michèle van Vugt2, Dorien Wouters12, A. H. (Koos) Zwinderman13, Matthijs C. Brouwer4, W. Joost Wiersinga2, Alexander P. J. Vlaar1, Diederik van de Beek4
1Department of Intensive Care, Amsterdam UMC, Amsterdam, Netherlands. 2Department of Infectious Diseases, Amsterdam UMC, Amsterdam, Netherlands. 3Experimental Immunology, Amsterdam UMC, Amsterdam, Netherlands. 4Department of Neurology, Amsterdam UMC, Amsterdam Neuroscience, Amsterdam, Netherlands. 5Department of Pulmonology, Amsterdam UMC, Amsterdam, Netherlands. 6Department of Anesthesiology, Amsterdam UMC, Amsterdam, Netherlands. 7Amsterdam UMC Biobank Core Facility, Amsterdam UMC, Amsterdam, Netherlands. 8Department of Radiology, Amsterdam UMC, Amsterdam, Netherlands. 9Department of Medical Microbiology, Amsterdam UMC, Amsterdam, Netherlands. 10Department of Internal Medicine, Amsterdam UMC, Amsterdam, Netherlands. 11Neurochemical Laboratory, Amsterdam UMC, Amsterdam, Netherlands. 12Department of Clinical Chemistry, Amsterdam UMC, Amsterdam, Netherlands. 13Department of Clinical Epidemiology, Biostatistics and Bioinformatics, Amsterdam UMC, Amsterdam, Netherlands.
Members of NIAID-USUHS COVID Study Group: Miranda F. Tompkins1, Camille Alba1, Andrew L. Snow2, Daniel N. Hupalo1, John Rosenberger1, Gauthaman Sukumar1, Matthew D. Wilkerson1, Xijun Zhang1, Justin Lack3, Andrew J. Oler4, Kerry Dobbs5, Ottavia M. Delmonte5, Jeffrey J. Danielson5, Andrea Biondi6, Laura Rachele Bettini6, Mariella D’Angio’6, Ilaria Beretta7, Luisa Imberti8, Alessandra Sottini8, Virginia Quaresima8, Eugenia Quiros-Roldan9, Camillo Rossi10
1American Genome Center, Uniformed Services University of the Health Sciences, Bethesda, MD, USA; Henry M. Jackson Foundation for the Advancement of Military Medicine, Bethesda, MD, USA. 2Department of Pharmacology and Molecular Therapeutics, Uniformed Services University of the Health Sciences, Bethesda, MD, USA. 3NIAID Collaborative Bioinformatics Resource, Frederick National Laboratory for Cancer Research, Leidos Biomedical Research Inc., Frederick, MD, USA. 4Bioinformatics and Computational Biosciences Branch, Office of Cyber Infrastructure and Computational Biology, NIAID, NIH, Bethesda, MD, USA. 5Laboratory of Clinical Immunology and Microbiology, Division of Intramural Research, NIAID, NIH, Bethesda, MD, USA. 6Pediatric Departement and Centro Tettamanti-European Reference Network PaedCan, EuroBloodNet, MetabERN-University of Milano-Bicocca-Fondazione MBBM-Ospedale, San Gerardo, Monza, Italy. 7Department of Infectious Diseases, University of Milano-Bicocca, San Gerardo Hospital, Monza, Italy. 8CREA Laboratory, Diagnostic Department, ASST Spedali Civili di Brescia, Brescia, Italy. 9Department of Infectious and Tropical Diseases, University of Brescia and ASST Spedali Civili di Brescia, Brescia, Italy. 10Chief Medical Officer, ASST Spedali Civili di Brescia, Brescia, Italy.
Funding
The Laboratory of Human Genetics of Infectious Diseases is supported by the Howard Hughes Medical Institute, the Rockefeller University, the St. Giles Foundation, the National Institutes of Health (NIH) (R01AI088364 and R01AI63029), the National Center for Advancing Translational Sciences (NCATS), NIH Clinical and Translational Science Award (CTSA) program (UL1 TR001866), a Fast Grant from Emergent Ventures, Mercatus Center at George Mason University, the Yale Center for Mendelian Genomics and the GSP Coordinating Center funded by the National Human Genome Research Institute (NHGRI) (UM1HG006504 and U24HG008956), the Yale High Performance Computing Center (S10OD018521), the Fisher Center for Alzheimer’s Research Foundation, the JPB Foundation, the Meyer Foundation, the French National Research Agency (ANR) under the “Investments for the Future” program (ANR-10-IAHU-01), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), the French Foundation for Medical Research (FRM) (EQU201903007798), the ANR GenMISC (ANR-21-COVR-039), the ANRS-COV05, ANR GENVIR (ANR-20-CE93-003) ANR AABIFNCOV (ANR-20-CO11-0001) projects, the ANR-RHU program COVIFERON (ANR-21-RHUS-08), the European Union’s Horizon 2020 research and innovation program under grant agreement No. 824110 (EASI-genomics), the HORIZON-HLTH-2021-DISEASE-04 program under grant agreement 01057100 (UNDINE), the Square Foundation, Grandir—Fonds de solidarité pour l’enfance, Fondation du Souffle, the SCOR Corporate Foundation for Science, the Battersea & Bowery Advisory Group, The French Ministry of Higher Education, Research, and Innovation (MESRI-COVID-19), Institut National de la Santé et de la Recherche Médicale (INSERM), REACTing-INSERM and the University of Paris Cité. The study was supported by the ORCHESTRA project, which has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No 10101616. P Bastard was supported by the MD-PhD program of the Imagine Institute (with the support of the Fondation Bettencourt-Schueller). The French COVID Cohort study group was sponsored by INSERM and supported by the REACTing consortium and by a grant from the French Ministry of Health (Grant PHRC 20–0424). The Cov-Contact Cohort was supported by the REACTing consortium, the French Ministry of Health, and the European Commission (Grant RECOVER WP 6). The COVIDeF study was supported by the French Ministry of Health, Fondation AP-HP et Programme Hospitalier de Recherche Clinique (PHRC COVID-19–20-0048) and was sponsored by APHP. Y.Z., O.M.D., L.D.N., H.C.S. are supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH. G.N. and A.N. are supported by Regione Lazio (Research Group Projects 2020) No. A0375-2020–36663, GecoBiomark. I Meyts is a Senior Clinical Investigator at the Research Foundation – Flanders, and is supported by the CSL Behring Chair of Primary Immunodeficiencies, by the KU Leuven C1 Grant C16/18/007, by a VIB GC PID Grant, by the FWO Grants G0C8517N, G0B5120N and G0E8420N and by the Jeffrey Modell Foundation. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 948959). This work is supported by the Swiss National Science Foundation (grant # 310030L_197721 to JF). This work is supported by ERN-RITA. The Canarian Sequencing Hub is funded by Instituto de Salud Carlos III (COV20_01333, and COV20_01334, and PI20/00876) and Spanish Ministry of Science and Innovation (RTC-2017–6471-1; AEI/FEDER, UE), co-financed by the European Regional Development Funds, “A way of making Europe” from the European Union, and Cabildo Insular de Tenerife (CGIEU0000219140 and “Apuestas científicas del ITER para colaborar en la lucha contra la COVID-19”). This work was funded, at least in part, by grant AJF202059 from Al Jalila Foundation, Dubai, United Arab Emirates. Sample processing at IrsiCaixa was possible thanks to the crowdfunding initiative YoMeCorono. We thank I Erkizia, E Grau, M Massanella, and J Guitart from the IrsiCaixa and Hospital Germans Trias i Pujol (Badalona, Spain) for sample collection, handling and processing.
Author information
Authors and Affiliations
Consortia
Contributions
D Matuozzo, ET, AM, JM, YS, YZ, A Bolze, MC, BM, P Zhang, LA, and AC performed computational analysis. D Matuozzo, AG, P Bastard, TA, LB, I Meyts, SYZ, A Puel, SBD, BB, EJ, and QZ performed or supervised experiments, generated and analyzed data, and contributed to the manuscript by providing figures and tables. P Bastard, FB, HA, AAT, AA, IAD, LMA, RAA, AAA, GA, P Bergman, SB, YTB, IGB, OCM, SC, PC, GC, KC, RC, CAN, OMD, LEZ, CF, PKG, MG, FH, RH, SH, LH, NH, AK, SK, CK, RLL, JLF, D Mansouri, JMP, OMA, I Migeotte, PEM, GM, AMN, GN, AN, TO, FP, QPH, RP, LPS, DEP, CP, A Pujol, LFR, JGR, CRG, JR, PRQ, MS, A Sobh, PSP, YTL, IT, CT, JT, MZ, P Zawadzki, SZAM, MFA, FMA, HBF, MJB, SNC, MAC, CLD, JF, JRH, YLL, RPL, TM, THM, HVB, AL, MV, A Boland, JFD, FM, ST, GG, FT, PH, LDN, and HCS evaluated and recruited patients and /or controls. CRG, CF, A Schlüter, MS, MZ, P Zawadzki, SZAM, HBF, MJB, SNC, MAC, CLD, JF, JRH, YLL, RPL, TM, THM, HVB, AL, MV, A Boland, JFD, RN, and AKK performed sequencing. D Matuozzo, BB, JLC, QZ, LA, and AC wrote the manuscript. JLC, QZ, LA and AC supervised the project. All the authors edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All the enrolled participants provided written informed consent for participation and were recruited through protocols conforming to local ethics requirements. For patients enrolled in the French COVID cohort (ClinicalTrials.gov NCT04262921), ethics approval was obtained from the Comité de Protection des Personnes Ile De France VI (ID RCB, 2020-A00256-33) or the Ethics Committee of Erasme Hospital (P2020/203). For participants enrolled in the COV-Contact study (ClinicalTrials.gov NCT04259892), ethics approval was obtained from the CPP IDF VI (ID RCB, 2020-A00280-39). For patients enrolled in the Italian cohort, ethics approval was obtained from the University of Milano-Bicocca School of Medicine, San Gerardo Hospital, Monza–Ethics Committee of the National Institute of Infectious Diseases Lazzaro Spallanzani (84/2020) (Italy), and the Comitato Etico Provinciale (NP 4000–Studio CORONAlab). STORM-Health care workers were enrolled in the STudio OsseRvazionale sullo screening dei lavoratori ospedalieri per COVID-19 (STORM-HCW) study, with approval from the local institutional review board (IRB) obtained on June 18, 2020. Patients and relatives from San Raffaele Hospital (Milan) were enrolled in COVID-BioB/Gene-COVID protocols and, for additional studies, TIGET-06, with the approval of the local ethics committee. Patients and relatives from Rome were enrolled in Protocol no. 50/20 (Tor Vergata University Hospital). Informed consent was obtained from each patient. For the patients enrolled in the COVIDeF Study Group (ClinicalTrials.gov NCT04352348), ethics approval was obtained from the Comité de Protection des Personnes Ile de France XI (ID RCB, 2020-A00754-35). For patients enrolled in Spain, the study was approved by the Committee for Ethical Research of the Infanta Leonor University Hospital, code 008–20; the Committee for Ethical Research of the 12 de Octubre University Hospital, code 16/368; the Bellvitge University Hospital, code PR127/20; the University Hospital of Gran Canaria Dr. Negrín, code 2020–200-1 COVID-19; and the Vall d’Hebron University Hospital, code PR(AMI)388/2016. Anonymized samples were sequenced at the National Institute of Allergy and Infectious Diseases (NIAID) through the Uniformed Services University of the Health Sciences (USUHS)/the American Genome Center (TAGC) under nonhuman subject research conditions; no additional IRB consent was required at the National Institutes of Health (NIH). For patients enrolled in the Swedish COVID cohort, ethics approval was obtained from the Swedish Ethical Review Agency (2020–01911 05).
Consent for publication
Not applicable.
Competing interests
RN and AKK are employees of Invitae and hold equities in the company. RPL is a member of the board of directors of Roche and its subsidiary Genentech. I Meyts holds a chair in Primary Immunodeficiencies and receives research grant from CSL Behring, paid to KUL. JLC reported a patent to PCT/US2021/042741 pending. FT is head of the Centre de Pharmacoépidémiologie (Cephepi) of the Assistance Publique – Hôpitaux de Paris and of the Clinical Research Unit of Pitié-Salpêtrière hospital, both these structures have received unrestricted research funding and grants for the research projects handled and fees for consultant activities from a large number of pharmaceutical companies, that have contributed indiscriminately to the salaries of its employees. FT is not employed by these structures and did not receive any personal remuneration from these companies. The remaining authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been updated to correct an author name.
Supplementary Information
Additional file 1:
Supplementary Methods. Fig S1. Luciferase assay on HEK293T cells transfected with the pGL4.32 luciferase reporter construct and an expression vector for Renilla luciferase together with no vector (mock), EV, WT, or 4 TLR7 variants found in our cohort. After 24 h, transfected cells were left untreated or were treated by incubation with 1 μg/mL R848 for 24 h. These data were established from two independent experiments. The y-axis represents NF-κB transcriptional activity as a percentage of the WT. The x-axis indicates the alleles used for transfection. Fig S2. Age distribution as boxplot and violin plot of the critical COVID-19 cases according to the carrier status of pLOF/bLOF at 15 type I IFN-related loci. Mean age of the patients for each category is shown in red. T-test was used to compare the means, showing a significant difference between non-carriers and carriers of heterozygous or homozygous/hemizygous variants (P = 2.2 × 10−6) and between heterozygous carriers and homozygous/hemizygous carriers (P = 0.008). Fig S3. Empirical power of our sample to detect an association at the 2.5 × 10−6 exome-wide significance threshold for various relative risks and proportion of carriers of at least one disease causing variant in the general population (PD), as estimated by simulation study (N = 1000 replicates).
Additional file 2
: Table S1. Number of genes tested and Genomic inflation factor for each model and variant set. Table S2. Complete results of the genome-wide burden joint analysis, trans-pipeline meta-analysis and trans-ethnic meta-analysis on rare variants. Table S3. Best results of the genome-wide burden analysis on rare variants under a co-dominant and dominant model. Table S4. TLR7 homozygous and hemizygous variants (AF < 0.01). Table S5. Results of the genome-wide burden analysis on common and rare variants under a co-dominant model. Table S6. Results of the genome-wide burden analyses for the candidate genes identified by GWAS under co-dominant model. Table S7. Characteristics of patients and controls in the full sample and according to the inclusion in the Zhang Q. et al., Science 2020 paper. Table S8. Carriers of rare pLOF/bLOF variants in genes involved in type I IFN immunity to influenza virus. Table S9. Branchpoint variants identified by BPHunter and characteristics of the carriers. Table S10. Age and sex stratified analysis for the 15 type I IFN-related loci. Table S11. Enrichment analysis of rare variants, including missense and inframe variants, in genes involved in type I IFN immunity in the full cohort of 3269 cases and 1373 controls. Table S12. pLI and CoNeS distribution of the analyzed genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Matuozzo, D., Talouarn, E., Marchal, A. et al. Rare predicted loss-of-function variants of type I IFN immunity genes are associated with life-threatening COVID-19. Genome Med 15, 22 (2023). https://doi.org/10.1186/s13073-023-01173-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-023-01173-8