
Abstract
Antimicrobial resistance (AMR) is a major global public health threat, which has been largely driven by the excessive use of antimicrobials. Control measures are urgently needed to slow the trajectory of AMR but are hampered by an incomplete understanding of the interplay between pathogens, AMR encoding genes, and mobile genetic elements at a microbial level. These factors, combined with the human, animal, and environmental interactions that underlie AMR dissemination at a population level, make for a highly complex landscape. Whole-genome sequencing (WGS) and, more recently, metagenomic analyses have greatly enhanced our understanding of these processes, and these approaches are informing mitigation strategies for how we better understand and control AMR. This review explores how WGS techniques have advanced global, national, and local AMR surveillance, and how this improved understanding is being applied to inform solutions, such as novel diagnostic methods that allow antimicrobial use to be optimised and vaccination strategies for better controlling AMR. We highlight some future opportunities for AMR control informed by genomic sequencing, along with the remaining challenges that must be overcome to fully realise the potential of WGS approaches for international AMR control.
Similar content being viewed by others


Background
Antimicrobial resistance (AMR) is one of the greatest current threats in international public health [1]. The rapid increase and worldwide spread of AMR threaten the advances in modern medicine, compromising the treatment of common infections such as pneumonia, urinary tract infections, and tuberculosis as well as the care of patients needing organ transplantation, complex surgery, cancer chemotherapy, and intensive care [2]. There are also significant economic and societal costs associated with AMR infections [3, 4]. These costs are attributed with longer hospital stays, higher medical bills, and increased mortality. Although AMR is a global problem, the burden of AMR falls disproportionately on low- and middle-income countries (LMICs), where it threatens sustainable development [4,5,6].
The first wave of antimicrobials was derived from naturally occurring compounds; therefore, the emergence of AMR is largely a natural process, and AMR genes have been detected in samples originating millions of years before the widespread use of antimicrobials [7,8,9]. However, an exponential growth in antimicrobial use in recent decades has exerted enormous selective pressures on bacterial populations, which has dramatically accelerated the evolution of AMR [10]. Indeed, as soon as mankind develops a new class of small molecules to kill bacteria, organisms evolve to resist their action, leading to an increasing prevalence of multi-drug-resistant (MDR), extended-drug-resistant (XDR), and pan-drug-resistant (PDR) organisms.
The detection of AMR has been traditionally reliant on culture-based antimicrobial susceptibility testing (AST), which remains the mainstay of clinical microbiology and patient management. Whilst phenotyping provides direct visual evidence of how a bacterium will interact with an antimicrobial, it generally provides little or no data regarding the resistance mechanisms, with disparate genetic clones often demonstrating identical resistance profiles [11]. Genetic typing methods, such as multi-locus sequence typing (MLST), provide a higher level of pathogen resolution compared to AST but are highly restrictive as they only describe a small fraction of a genome. Whole-genome sequencing (WGS), in contrast, provides genome-wide information at the single nucleotide level that can be used to identify the presence and mechanisms of AMR, as well as pathogen identity, virulence, and ancestry [12,13,14]. The advent of next-generation sequencing (NGS), via high-throughput, parallel sequencing of DNA fragments, has allowed pathogen genomes to be determined rapidly and at comparatively low cost [13, 15, 16]. The universality of the genetic code allows a unified approach to be applied to all organisms, and a range of technologies and platforms can provide the same data output [11]. Comparative phylogenetic analysis can be exploited to determine the degree of relatedness between different isolates based on the extent of the similarity between genomes and, when overlaid with epidemiological and clinical data, can inform our understanding of the specific temporospatial dynamics of AMR and transmission [11]. Additionally, the recent optimisation of metagenomic sequencing approaches circumvents the necessity for culture entirely. Therefore, by incorporating all available genomic material in a sample, metagenomic analysis facilitates a shift in focus from an individual pathogen to the community, microbiome landscape, generating a highly detailed model of how pathogens interact, and how they mobilise and access AMR genes [17].
The power of WGS is being increasingly employed to address the public health challenge of AMR, supporting surveillance, outbreak investigation, and contributing to improved diagnostics and therapeutics as highlighted in Table 1 [12, 27]. This review highlights some of the successes and advances supported by WGS in these areas and outlines future directions and remaining challenges associated with using WGS technologies to support public health efforts for AMR.
AMR surveillance
Surveillance is the cornerstone of public health efforts in controlling AMR. AMR surveillance has traditionally relied on phenotypic AST, but different testing methods, variation in interpretation, the extent to which thresholds are clinically validated, and changes in interpretive guidelines limit standardisation. WGS data overcomes many of these limitations, providing detailed insights that can greatly augment the value of AMR surveillance. Such data can inform an understanding of AMR evolution and spread, inform control strategies, facilitate the detection of new and emerging threats, and support new diagnostic and therapeutic approaches [4, 10, 12, 28,29,30].
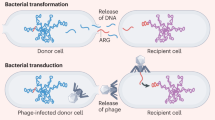
Underlying the expansion of AMR is a dynamic and complex interaction between microbes, AMR encoding genes, and mobile genetic elements that act as vehicles for AMR via horizontal gene transfer (HGT) [7, 10, 11, 31]. Once horizontally transferred AMR genes have become chromosomally integrated, clonal expansion can lead to the rapid dissemination of these genes. This phenomenon has been observed with methicillin-resistant Staphylococcus aureus (MRSA), penicillin-resistant Streptococcus pneumoniae, vancomycin-resistant enterococci, and fluroquinolone-resistant Clostridium difficile [11, 32,33,34,35,36,37]. The granular resolution afforded by WGS allows inferences on the nature of AMR evolution and dissemination [38], providing insights that can help contain AMR and protect public health.
International surveillance
Increased accessibility to WGS has significantly enhanced our understanding of the global evolution and spread of AMR. High-throughput WGS methods have facilitated the sequencing of large, geographically representative collections of isolates, overcoming many of the biases associated with historic, small-scale studies that were often skewed by the domination of local clonal dissemination. This paradigm is illustrated by a modified understanding of AMR in E. coli ST131, which has rapidly spread to become a frequent cause of healthcare and community-acquired infection since it was first described in 2008 [33]. ST131 E. coli frequently exhibit cephalosporin (most commonly due to a CTX-M-15 encoding gene) and fluroquinolone resistance [33, 39]. The development of AMR in ST131 E. coli was initially speculated to have arisen from frequent and independent acquisitions of mobile genetic elements [40]. However, a study from the USA concluded that the success of AMR in ST131 E. coli was associated with a sustained clonal expansion. However, it was not until a more comprehensive global study that the true diversity of AMR in ST131 was revealed, which identified the chromosomal integrations of various resistance genes, the persistence and evolution of mobile elements within sub-lineages, and the sporadic acquisition of several different resistance elements [41]. As well as enhancing the mechanistic understanding of AMR, this study highlighted the need for multifaceted control strategies that could limit the spread of ST131, as well as the spread of mobile genetic elements to other pathogens [42]. These studies were foundational for additional work specifically investigating risk factors for ST131 infection and spread. ST131 is now known to be a common gut commensal, with opportunistic infections occurring mainly in functionally compromised hosts such as the elderly, particularly those having prior antimicrobial use and living in long-term care facilities [43,44,45,46,47]. Infection control measures for ST131 E. coli in long-term care facilities focusing on contact precautions have been largely ineffective [45, 48], leading to the strategy of limiting multi-bedded rooms and communal dining facilities [49]. The targeted screening of hospitalised patients with specific risk factors for ST131 facilitated patient isolation to prevent nosocomial transmission, as well as better management of empirical broad-spectrum antimicrobial therapy in specific high-risk patient groups [46, 47].
Global AMR surveillance has been bolstered by programmes such as the Global Antimicrobial Resistance and Use Surveillance System (GLASS) from the World Health Organization (WHO) [4], and the European Antimicrobial Resistance Surveillance Network (EARS-Net) [50]. These networks encourage data sharing and provide standardised templates for reporting AST data to facilitate comparative analysis. Recently, such networks have documented a rapid increase in MDR Gram-negative bacteria (GNB) that have become recognised as leading AMR threats, including third-generation cephalosporin-resistant and carbapenem-resistant Enterobacterales (CRE), carbapenem-resistant Acinetobacter baumannii and Pseudomonas aeruginosa, [1, 22, 51,52,53,54,55]. These networks are expanding their evidence base for other key threats, such as Klebsiella pneumoniae, which can cause a wide range of infections, including pneumonia, bacteraemia, and urinary tract infections [56], and have been identified as a crucial entry point for MDR into Enterobacterales [57,58,59]. Carbapenem-resistant K. pneumoniae (CR-Kp) is considered to be the fastest-growing AMR threat in Europe [60] and has a high attributable mortality rate (estimated as 30–70%) [61, 62]. Systematic surveillance of CR-Kp across Europe demonstrated that nosocomial spread was driving this epidemic, with carbapenemase acquisition occurring across diverse phylogenetic backgrounds [18]. As well as describing the epidemiology of CR-Kp, this study found that isolates were concentrated into four clonal lineages: sequence types (ST) 11, 15, 101, and 258/512. These data contrast with studies in other settings that found high genomic plasticity in CRE, with frequent poly-clonal and poly-species horizontal gene transfer accounting for much of the acquired resistance [54, 63, 64]. This inconsistency highlights the complex interplay between bacteria and MDR genes, with the role of clonal expansion [18] and HGT [54, 63, 64] potentially contributing variably across different timeframes and geographies.
Embedding WGS into prospective, cross-national surveillance will further strengthen AMR surveillance, as highlighted by the recent outbreak genomic investigation of blaNDM-1 and blaOXA-48 CR-Kp in Germany [65]. In the absence of routine collection and reporting of European-wide WGS data, investigators pooled data from 13 national surveillance systems to investigate the spread of the CR-Kp outbreak, identifying cross-border transmission of several unrelated clusters of blaNDM-1- and blaOXA-48-positive CR-Kp. It is anticipated that the planned incorporation of WGS into the European Antimicrobial Resistance Genes Surveillance Network (EURGen-Net) will facilitate the routine detection of such transmission events and inform targeted control measures [65].
National surveillance
The inclusion of WGS can similarly strengthen national AMR surveillance. The introduction of WGS into AMR surveillance in the Philippines, for example, showed that HGT was key for driving carbapenem resistance in Klebsiella, with mobile genetic element (MGE) acquisition often leading to MDR [11]. WGS data also identified a localised plasmid-driven outbreak of CR-Kp, where various infection control measures, including patient isolation, were initiated [11]. By contributing WGS data to the WHO international surveillance network, national surveillance data also enhanced the global understanding of some high-risk clones of interest, notably ST147 CR-Kp, of which a previously uncharacterised clade was described [11].
National surveillance can directly guide the need for AMR control measures that can be driven, resourced, and monitored centrally, as was demonstrated in Israel following the introduction of CR-Kp ST258 in 2005. The initial measures were unsuccessful at controlling the spread of CR-Kp ST258, and infections rapidly rose to a peak incidence of 41.9 per 100,000 patient-days [61]. Nationally mandated control measures were subsequently introduced in 2007, successfully reducing the incidence of infections by 79%, as well as the rate of asymptomatic carriage [52, 66,67,68,69].
The routine inclusion of WGS into national surveillance can also be used to evaluate the risk from emerging AMR threats, such as transferable colistin resistance [70]. Phenotypic colistin resistance testing is not routinely performed and therefore is not readily detectable without WGS [70]. Routine WGS of GNB in the UK generated a substantial data resource over time, which was mined to determine the extent of the spread of plasmid-encoded colistin resistance after it emerged as an AMR threat [70]. Although colistin resistance can be detected in organisms from a range of environments, this retrospective study found that it remained uncommon in the UK, leading to the monitoring of colistin prescribing in a bid to minimise any potential selective pressure within the UK.
Local surveillance
Healthcare facilities house patients with serious and complex infections in proximity to patients with compromised immunity, in settings that are characterised by high antimicrobial use [71, 72]. In this context, healthcare-associated infections (HCAIs), frequently with pathogens that are highly drug-resistant, are a significant challenge requiring rigorous infection prevention and control practices to reduce their incidence [72]. The ability to perform WGS in a clinically relevant turnaround time has made genomics an actionable front-line tool to investigate HCAIs [73].
A key strength of WGS is that it permits the degree of relatedness between selected isolates to be determined and, in the context of HCAIs, allows the reconstruction of nosocomial outbreaks [14, 39]. One of the earliest applications of this process was in a retrospective investigation of a protracted outbreak of MRSA in a neonatal unit [73]. By reconstructing the full outbreak, the source was eventually identified, ultimately, supporting infection control measures that eventually led to its resolution [19]. Other studies of S. aureus HCAI have similarly shown the improved granularity of WGS compared to other approaches, with more conventional S. aureus-specific staphylococcal protein A (spa) typing failing to detect transmission events, as well as the false attribution of unrelated isolates as HCAIs [74].
By exploiting a ‘molecular clock’ (the mutation rate in the genome of a specific organism), WGS can also infer directionality in outbreaks [13, 14, 19, 27]. This approach was key to understanding an outbreak of MDR Acinetobacter baumannii in a UK hospital treating civilian patients and injured military personnel returning from the Middle East [20]. Pre-admission colonisation of the wounds of military personnel with distinct lineages of A. baumannii was described, with the onward transmission of one specific clone leading to the infection of a civilian. Importantly, an additional civilian infection was not associated with the military-related infection, highlighting that other distantly related A. baumannii were also circulating. Similarly, the utility of WGS in determining the chains of transmission has also been demonstrated for MRSA and CR-Kp [75].
By generating detailed insights into HCAI, WGS surveillance can help identify the main sources of AMR, providing an opportunity for targeted infection control measures that can reduce the risk of further transmission events [73]. A large study of HCAI CR-Kp showed that the majority of inpatient infections resulted from the transfer of a relatively small number of patients from specific, high-risk facilities and that targeting interventions at such facilities could significantly reduce HCAIs across the healthcare system [75]. WGS-based investigations can similarly limit the use of unnecessary or ineffective measures that can be costly to implement. For example, studies of MRSA transmission have shown that patient-to-patient transmission is relatively uncommon and that persistently colonised staff are more frequent sources of infection than patients [74]. Targeting infection control at the staff rather than patients may therefore be a more efficient and more cost-effective control mechanism [76].
Prospectively applying WGS for local HCAI control has also been suggested for early outbreak detection, and thereby early intervention and source control, with a substantial cost saving [4]. This approach may be especially true for problematic pathogens in high-risk settings, such as MDR K. pneumoniae in ICUs [77]. WGS of routinely collected MRSA samples, obtained from both hospital and community settings over a year in the US identified extensive and previously unrecognised transmission events, highlighting the utility of routine WGS screening in high-risk patients and/or settings [76].
Community surveillance and outbreak investigation
WGS is also contributing to community public health efforts, such as contact tracing and the detection of secondary cases of TB. Contact tracing is difficult as there is often a long interval between the initial infection and the diagnosis. Several countries, such as the UK, have established a nationwide WGS database of TB to facilitate the identification of transmission events. This system enhances contact tracing as well as informs a wider understanding of TB control. Using WGS in preference to variable number tandem repeat (MIRU-VNTR) typing has been shown to reduce the number of false case clusters and has improved the detection of cases where potential clusters are identified [21, 78,79,80,81].
Understanding the drivers of AMR
The paradigm that the rapid expansion in AMR is a response to selective pressure from antimicrobial use drives AMR is well accepted [82]. WGS is being used to interrogate this selective evolution at the molecular level, across wide geographical areas, and between genetically diverse organisms. WGS was exploited to investigate AMR and the clinical epidemiology of atypical enteropathogenic E. coli isolated from symptomatic and asymptomatic children in South Asia and sub-Saharan Africa [22]. Despite broad geographical, symptomatic, and phylogenetic diversity, 65% of the bacterial isolates were resistant to three or more classes of antimicrobials. In this study, the best predictor of resistance profile was not the presence or absence of clinical symptoms or genetic lineage, but the geographical patterns of antimicrobial usage [22].
On an individual patient level, the likelihood of developing infection with extended-spectrum beta-lactamase-producing bacteria is associated with an increased length of hospital stay prior to infection, exposure to antimicrobials, and recent overseas travel [83]. Predictors of MDR GNB infection more broadly include male sex, older age, and co-morbidities [84]. Changes in the human microbiota occur in response to illness, particularly when associated with frequent and/or prolonged antimicrobial exposure. The Enterobacterales are habitual colonisers of the gastrointestinal tract, where they can act as a major reservoir for mobile AMR genes [22]. Metagenomic studies have shown that commensal bacteria in healthy individuals help maintain pathogenic bacteria at a low density, meaning that carriage is rarely problematic [71]. However, when patients undergo an invasive procedure, there is a loss in microbial diversity followed by colonisation with pathogenic bacteria [85]. This effect is can be exacerbated by antimicrobial use, which frequently results in the selection of drug-resistant pathogens [71] and facilitates HGT of AMR genes between bacterial lineages and species [7, 86]. WGS studies are now being used to determine the colonisation factors that facilitate the rapid growth and persistence of certain pathogens in such circumstances, with the hypothesis that therapeutics targeting persistent organisms may be developed to control pathogen colonisation.
AMR and ‘One Health’
One Health focuses on understanding the interconnectivity between ecosystems [87], recognising that human health is connected with, and dependent on, the health of animals, plants, and the wider environment [4, 17, 88]. AMR transmission occurs both within and across different ecosystems, facilitated by close animal and human contact, food, and water systems, all of which are influenced by culture and economics [87]. Understanding this complex interplay is crucial for the control of AMR. WGS is being increasingly used to support One Health aspects of AMR, such as by interrogating the spread of AMR via food and farming [88, 89], building on an established food-borne disease surveillance [27], and outbreak investigation [90, 91]. A WGS-based investigation of commensal E. coli isolates in livestock in China after a rapid increase of colistin resistance, for example, showed that this increase was due to the emergence of a plasmid associated colistin resistance gene in E. coli, designated MCR-1, and led to international efforts to control the dissemination of MCR-1 via food supply networks [23].
Optimising antimicrobial use through WGS-based rapid diagnostic tests
Rapid, accurate, low-cost diagnostic tests can aid in optimising and limiting antimicrobial use, thereby minimising the potential selective pressures. Culture-based microbiological diagnostics and AST are widely utilised for bacterial infections but are not rapid and not applicable to viral infections or fastidious organisms. WGS-based diagnostic approaches are being used to overcome these limitations, notably for HIV and tuberculosis, and offer the prospect of improved outcomes.
The prognosis of HIV infection has been transformed by combination anti-retroviral therapy (cART), but these treatments are complicated by the emergence of viral resistance. Genomic replication of RNA viruses utilises a reverse transcriptase lacking proofreading capacity, leading to the accumulation of mutations, with in host infection existing as a population of closely related genomes (quasispecies) [92]. Further in host genetic variability can result from the recombination of viruses infecting the same cell and the accumulation of variants over time [93]. WGS can detect viral variants at a prevalence of ~ 1%, and WGS-based resistance testing is used prior to initiating cART to detect mutations conferring drug resistance in the infecting quasispecies [94,95,96,97]. The full clinical implications of minority variants are unclear [98], but they predict failure of first-line regimens [99] and have been shown to be increasing in some locations [24], highlighting the need for the global monitoring of drug resistance in HIV.
Treatment success in M. tuberculosis declines from 83% for susceptible isolates to 54% and 30% for MDR- and XDR-TB isolates, respectively [100]. M. tuberculosis is slow growing, with culture-based diagnosis and AST often taking weeks and requiring a high-level biosafety laboratory. Poor reproducibility, uncertainty regarding appropriate drug concentrations for susceptibility testing [101], and a propensity for laboratory contamination can lead to false positives [102,103,104]. The M. tuberculosis genome comprised a single chromosome with drug resistance mediated through mutations in core genes or promoters [105], making the organism perfectly suited to WGS diagnostic approaches [105]. WGS diagnostics significantly decrease the time to a confirmatory TB diagnosis [106, 107], plus it reliably predicts the drug resistance profile [108], whilst facilitating the detection, monitoring, and diagnosis of de novo drug resistance [109]. These approaches have revolutionised TB control and are being increasingly used routinely in TB management.
Informing vaccination strategies to control AMR
Vaccination has an established role in AMR reduction. Conjugate vaccines for Haemophilus influenzae and Streptococcus pneumoniae have been shown to not only decrease the incidence of disease, but also reduce AMR [110, 111]. Increasing access to these vaccines would likely have a further impact on decreasing both disease and AMR [112, 113]. Vaccines against viral infections including influenza, rotavirus, and varicella zoster have also been shown to reduce antimicrobial use by decreasing the incidence of secondary bacterial infections and syndromic presentations for illnesses that would lead to antimicrobial use [112,113,114].
More recently, vaccination has been used as a direct mechanism for controlling AMR in typhoid fever, the result of many years of effort at understanding typhoid fever, in which WGS has played a significant role. Typhoid fever is caused by the bacterium Salmonella enterica serovar Typhi (S. Typhi) and causes an estimated 10.9 million cases and 116,800 deaths annually [115]. Whilst most cases of typhoid fever can be treated with antimicrobials, the emergence and spread of AMR have posed an increasing threat to typhoid control, particularly in South Asia, where resistance to each class of oral antimicrobials used to treat typhoid fever has emerged [25]. S. Typhi is highly clonal [116], and MDR S. Typhi is heavily associated with a single haplotype (H58) [117]. The global spread of H58 has largely replaced other non-H58 haplotypes and is causing sustained ongoing typhoid transmission in east and southern Africa [117]. MDR within H58 is linked with a single mobile element that was introduced via a specific plasmid (IncHI1-PST6) but has since transferred to the chromosome. As new resistance phenotypes have emerged, WGS has facilitated the further investigation of the underlying molecular mechanisms, including the recent emergence of azithromycin resistant typhoid in Bangladesh, Pakistan, Nepal, and India [26, 118, 119]. WGS-based investigation of an outbreak of ceftriaxone-resistant typhoid in Hyderabad, Pakistan, revealed the emergence of an extensively drug-resistant (XDR) variant that was not only resistant to the first-line antimicrobials chloramphenicol, ampicillin, and trimethoprim-sulfamethoxazole, but also fluoroquinolones and third-generation cephalosporins [120]. This variant, which was within the H58 clade, had acquired a plasmid encoding additional resistance elements, including a blaCTX-M-15 and a qnrS fluoroquinolone resistance gene [121].
Following the initial reports of XDR typhoid, intensified surveillance was undertaken to monitor its spread of typhoid into Karachi and beyond [122]. Reactive vaccination campaigns were initiated in Hyderabad and Karachi, and vaccine safety and effectiveness data were generated [123, 124]. All of these efforts played a major role in the decision of the Federal Expanded Program on Immunization to introduce typhoid conjugate vaccine (TCV) into their national immunisation programme, making Pakistan the first country to do so [26, 125]. The phased introduction began with a vaccination campaign targeting ~ 10 million children in the urban areas of Sindh province in November 2019, which were the areas hardest hit by XDR typhoid, and a subsequent campaign was conducted in Islamabad and Punjab province, where XDR had spread, that covered over 13 million children [126]. Consequently, WGS and phenotypic AMR data had a direct impact on the decision to introduce TCV and on the vaccine introduction strategy itself.
In addition to elucidating the molecular mechanisms of AMR and providing well-defined, standardised AMR data where mechanisms of resistance are known, WGS data can also provide important information regarding typhoid transmission pathways. The 2018 WHO Position Paper on typhoid vaccines indicated that TCV introduction should be prioritised “in countries with the highest burden of typhoid disease or a high burden of antimicrobial-resistant S. Typhi” [127]. Given the ubiquitous nature of drug-resistant typhoid, the anticipated country demand for TCVs, and the presence of only two WHO prequalified manufacturers of TCV, additional selection criteria may be required [127]. Globally representative WGS data show that drug-resistant typhoid typically emerges and spreads from South Asia [117]; therefore, prioritising TCV introduction in this region may reduce the burden of AMR in the region and prevent the spread of new resistance phenotypes internationally. Such phylodynamic data can inform optimal vaccine introduction strategies for other key highly drug-resistant pathogens, as and when new vaccines become available.
Limitations, challenges, and future directions
Technical limitations
The concept of relatedness between bacterial isolates is based on a process of continuous evolution, but prospectively identifying genetic signals of AMR evolution from the background noise of genetic variation and sequencing error is difficult. Using sequence data to infer relatedness and transmission is highly variable, and mutation rates can vary because of different lifestyles/conditions such as biofilm formation, antimicrobial exposure, disease states, and environmental pressures such as starvation [128,129,130]. The threshold number of SNPs above which relatedness is unlikely is therefore highly context-dependent [21, 80, 131]; if not recognised, then selection biases within isolates in collections can lead to false epidemiological inferences. Despite these challenges, defined cut-off points for the relatedness of specific pathogens, such as MRSA and M. tuberculosis, have been suggested in order to establish a threshold beyond which recent transmission is unlikely [132].
Inferring or excluding transmission events for pathogens that accumulate mutations slowly can be difficult [133], which presents a further challenge of differentiating genuine variants from sequencing error [134]. Interpreting the significance of WGS variation for pathogens that can establish asymptomatic carriage, have a period of latency, or cause a chronic, indolent illness can also prove challenging, as a degree of in-host genetic variation occurs with time, and different lineages may be intermittently transmitted at different points from a single host [13]. Bayesian mathematical approaches that integrate and model pathogen evolution with contact and temporal data on symptom onset, and that permit sequencing error, are being used to improve the inference of transmission chains, but understanding the sequence variance in real-world scenarios remains a major challenge [13, 133, 134].
Highly transmissible MGEs (such as plasmids) facilitate non-mutational AMR gene acquisition by HGT and are highly implicated in AMR dissemination [135]. In comparison, AMR that is driven by mutations on the bacterial chromosome is stable, and transmission is generally confined largely to progeny [136]. Determining the genetic location of AMR genes is therefore a key aspect of understanding AMR evolution and transmission, but is not readily determined by short-read sequencing [137,138,139]. Due to an inability to sequence long stretches of DNA and a failure to generate sufficient overlap in DNA fragments to allow accurate assembly of repetitive DNA sequences that are longer than the read length, these approaches are less well suited to describe the genetic environments of AMR genes [138, 139]. Additionally, AMR genes are commonly associated with long, repetitive insertion sequences, and short-read sequencing techniques cannot resolve if these are carried on a plasmid or chromosomally located [38, 54, 140, 141]. Most outbreak investigations have largely assumed the relative conservation of plasmid structures over the limited time period of an outbreak and the extent to which HGT events contribute to outbreaks of AMR is not fully understood [54]. However, more affordable long-read sequencing technology is becoming increasingly available and can generate reads that can span repetitive areas, allowing for the complete reconstruction of genome structure. This approach, however, is generally less accurate and has lower throughput compared to short-read sequencing [139, 140]. Hybrid approaches whereby isolates are sequenced en masse with short-read sequencing to determine identity, gene content, and relatedness, with selective addition of long-read sequencing to resolve the structure of MGEs, have been used to harness the benefits of both approaches [135, 139, 140, 142, 143].
Challenges in implementing WGS for routine use
The routine, prospective inclusion of WGS into microbiological surveillance has the potential to greatly enhance and strengthen public health efforts to combat AMR but comes with significant logistical and financial implications [144]. Unfortunately, prospective data demonstrating and quantifying the beneficial impact of routine WGS implementation are limited [145, 146]; most data are derived from retrospective studies, specifically from studies reconstructing chains of transmission during outbreak investigations [145]. Cost-effectiveness is even harder to determine, reflecting marked uncertainties and challenges in estimating the costs for the implementation of WGS. Generating accurate, contemporary cost estimates is difficult in the context of the marked decrease in costs that has accompanied the rapid technological advances in recent years, a problem compounded by the challenge of estimating costs for complex workflows that include several steps, including downstream analysis [147]. Further, routine diagnostics frequently requires a rapid turnaround time to support the management of individual cases, necessitating the processing of individual samples. This approach contrasts with many research and surveillance studies that lend themselves to batch processing of samples [147]. Despite these challenges, studies showing the cost-effectiveness of WGS for infection control and outbreak detection and management are starting to emerge and will help support implementation decisions [148,149,150,151].
Ensuring the reproducibility and validity of results through standardisation and quality assurance (QA) processes is an essential part of routine medical laboratory processing [152]. However, implementing QA processes for WGS is challenging. Variations in DNA extraction methods and reagents, sequencing technologies, analysis pipelines, and bioinformatic approaches can all impact WGS analysis [152], making the standardisation and quality assurance methods challenging [147, 153]. Nevertheless, efforts at quality assurance and result standardisation across laboratories are currently being attempted for WGS workflows [154]. Ring trials showing high performance and consistency across laboratories of WGS methods applied to S. aureus are highly encouraging [152, 153] and suggest that developing isolate collections that can act as biological standards, against which reproducibility and robustness of methods can be measured, might be feasible.
The ability to share, integrate, and compare the vast wealth of data derived from WGS across laboratories, settings, and time is key to harnessing the power of WGS for global AMR surveillance [4, 154]. Barriers to data sharing include the reluctance of academics to share data before publication (a process that is itself often slow and time-consuming), political sensitivities when competing interests are at play (for example tourism), legal and requirements to protect personal data [154], and the need for standardised methodological and analytical approaches to generate comparable data [155]. Databases such as GenBank facilitate sharing of existing genomes, but their utility is again compromised by a lack of data standardisation, as well as deficiencies in accompanying metadata [154]. The Global Microbial Identifier (GMI) initiative is in the early stages of trying to address these issues, aiming to develop standardised identification and characterisation approaches in order to form a global interactive network of genomic databases [154]. The absence of validated globally standardised systems for genomic typing, defining clusters and determining AMR genes, further intensifies the challenges of interpreting genomic data [4, 11, 147]. Clinical and epidemiological metadata are essential for public health solutions to AMR, but are associated with standardisation, computational, and technical challenges [154]. The collaborative approach to sharing of the SARS-CoV-2 genomic data demonstrates that such an approach is possible [156, 157], but to date, scalable solutions to genomic data sharing have remained elusive.
Introducing WGS in LMICs
Despite AMR being a major problem in LMICs, there is currently limited understanding of AMR dynamics in these settings [22, 28] and a notable lack of sustained access to WGS surveillance [11]. The capital investment required to establish and maintain WGS platforms is substantial. Supply, procurement, and maintenance of WGS reagents and equipment can be highly challenging [11, 158], particularly, in locations with inconsistent supply chains, where short shelf-life consumables and long intervals between procurement and delivery may create wastage or significant project delays. Support for data analysis is also indispensable, along with investment in training and retention of people to develop and sustain the necessary proficiency. Even simple analyses of bacterial WGS data require access to high-performance compute (HPC) clusters, stable internet access, and expertise [11].
Conclusions
The rapid emergence and spread of AMR in recent decades are a major threat to global public health, and current antimicrobial development efforts cannot keep pace with pathogen evolutionary dynamics. Addressing this threat will require diverse, cross-sectoral interventions at all levels of public health and political systems. Improvements in genomic technologies in recent years have made these systems more widely accessible and affordable, and they are now being implemented as front-line tools in the battle against AMR. By enhancing AMR surveillance, WGS has greatly improved understanding of how, when, where, and why AMR emerges and spreads and has helped quantify temporal and geographical variations in AMR epidemiology. Genomic insights have also been applied to facilitate outbreak detection and control, improve diagnostic tests, optimise antimicrobial use, and inform vaccination strategies. Although logistical and technological barriers to the universal implementation of genomics in public health remain (particularly in LMICs), the use of these approaches is likely to further expand in the coming years and will hopefully help restrict the adverse public health impacts of AMR.
Availability of data and materials
Not applicable
References
World Health Organization. Antimicrobial resistance: global report on surveillance. World Health Organization. 2014. https://apps.who.int/iris/handle/10665/112642.
Baker S. Infectious disease. A return to the pre-antimicrobial era? Science (New York, NY). 2015;347(6226):1064–6.
Jonas O, Team W: Drug-resistant infections: a threat to our economic future; 2017.
Global antimicrobial resistance and use surveillance system (GLASS) report 2021. Geneva: World Health Organization; 2021. Licence: CC BY-NC-SA 3.0 IGO.
Jasovsky D, Littmann J, Zorzet A, Cars O. Antimicrobial resistance-a threat to the world’s sustainable development. Ups J Med Sci. 2016;121(3):159–64.
Bloom G, Merrett GB, Wilkinson A, Lin V, Paulin S. Antimicrobial resistance and universal health coverage. BMJ Glob Health. 2017;2(4):e000518.
van Schaik W. The human gut resistome. Philos Trans R Soc Lond Ser B Biol Sci. 2015;370(1670):20140087.
Forsberg KJ, Patel S, Gibson MK, Lauber CL, Knight R, Fierer N, et al. Bacterial phylogeny structures soil resistomes across habitats. Nature. 2014;509(7502):612–6.
Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010;74(3):417–33.
Baker S, Thomson N, Weill F-X, Holt KE. Genomic insights into the emergence and spread of antimicrobial-resistant bacterial pathogens. Science (New York, NY). 2018;360(6390):733–8.
Argimón S, Masim MAL, Gayeta JM, Lagrada ML, Macaranas PKV, Cohen V, et al. Integrating whole-genome sequencing within the National Antimicrobial Resistance Surveillance Program in the Philippines. Nat Commun. 2020;11(1):2719.
NIHR Global Health Research Unit on Genomic Surveillance of AMR. Whole-genome sequencing as part of national and international surveillance programmes for antimicrobial resistance: a roadmap. BMJ Glob Health. 2020;5(11):e002244.
Didelot X, Gardy J, Colijn C. Bayesian inference of infectious disease transmission from whole-genome sequence data. Mol Biol Evol. 2014;31(7):1869–79.
Price J, Gordon NC, Crook D, Llewelyn M, Paul J. The usefulness of whole genome sequencing in the management of Staphylococcus aureus infections. Clin Microbiol Infect. 2013;19(9):784–9.
Ashton PM, Nair S, Peters TM, Bale JA, Powell DG, Painset A, et al. Identification of Salmonella for public health surveillance using whole genome sequencing. PeerJ. 2016;4:e1752.
Wainwright C, Altamirano L, Cheney M, Cheney J, Barber S, Price D, et al. A multicenter, randomized, double-blind, controlled trial of nebulized epinephrine in infants with acute bronchiolitis. N Engl J Med. 2003;349(1):27–35.
Blake KS, Choi J, Dantas G. Approaches for characterizing and tracking hospital-associated multidrug-resistant bacteria. Cell Mol Life Sci. 2021;78(6):2585–606.
David S, Reuter S, Harris SR, Glasner C, Feltwell T, Argimon S, et al. Epidemic of carbapenem-resistant Klebsiella pneumoniae in Europe is driven by nosocomial spread. Nat Microbiol. 2019;4(11):1919–29.
Harris SR, Cartwright EJ, Török ME, Holden MT, Brown NM, Ogilvy-Stuart AL, et al. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis. 2013;13(2):130–6.
Lewis T, Loman NJ, Bingle L, Jumaa P, Weinstock GM, Mortiboy D, et al. High-throughput whole-genome sequencing to dissect the epidemiology of Acinetobacter baumannii isolates from a hospital outbreak. J Hosp Infect. 2010;75(1):37–41.
Jajou R, Neeling A, Hunen R, Vries G, Schimmel H, Mulder A, et al. Epidemiological links between tuberculosis cases identified twice as efficiently by whole genome sequencing than conventional molecular typing: a population-based study. PLoS One. 2018;13(4):e0195413.
Ingle DJ, Levine MM, Kotloff KL, Holt KE, Robins-Browne RM. Dynamics of antimicrobial resistance in intestinal Escherichia coli from children in community settings in South Asia and sub-Saharan Africa. Nat Microbiol. 2018;3(9):1063–73.
Liu YY, Wang Y, Walsh TR, Yi LX, Zhang R, Spencer J, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2016;16(2):161–8.
Baxter JD, Dunn D, Tostevin A, Marvig RL, Bennedbaek M, Cozzi-Lepri A, et al. Transmitted HIV-1 drug resistance in a large international cohort using next-generation sequencing: results from the Strategic Timing of Antiretroviral Treatment (START) study. HIV Med. 2021;22(5):360–71.
Hooda Y, Saha S, Sajib MSI, Rahman H, Luby SP, Bondy-Denomy J, et al. Molecular mechanism of azithromycin resistance among typhoidal Salmonella strains in Bangladesh identified through passive pediatric surveillance. PLoS Negl Trop Dis. 2019;13(11):e0007868.
Iqbal J, Dehraj IF, Carey ME, Dyson ZA, Garrett D, Seidman JC, et al. A race against time: reduced azithromycin susceptibility in Salmonella enterica serovar Typhi in Pakistan. mSphere. 2020;5(4):e00215–20 /msphere/00215/00214/mSphere00215-00220.atom.
Besser J, Carleton HA, Gerner-Smidt P, Lindsey RL, Trees E. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin Microbiol Infect. 2018;24(4):335–41.
Perez F, Villegas MV. The role of surveillance systems in confronting the global crisis of antibiotic-resistant bacteria. Curr Opin Infect Dis. 2015;28(4):375–83.
O’Brien TF, Stelling J. Integrated multilevel surveillance of the world’s infecting microbes and their resistance to antimicrobial agents. Clin Microbiol Rev. 2011;24(2):281–95.
Mendelson M, Matsoso MP. The World Health Organization global action plan for antimicrobial resistance. South Afr Med J. 2015;105(5):325.
Partridge SR, Kwong SM, Firth N, Jensen SO. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin Microbiol Rev. 2018;31(4):e00088–17.
Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med. 2005;353(23):2442–9.
Banerjee R, Johnson JR. A new clone sweeps clean: the enigmatic emergence of Escherichia coli sequence type 131. Antimicrob Agents Chemother. 2014;58(9):4997–5004.
McDonald LC, Killgore GE, Thompson A, Owens RC Jr, Kazakova SV, Sambol SP, et al. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353(23):2433–41.
Bozdogan B, Bogdanovich T, Kosowska K, Jacobs MR, Appelbaum PC. Macrolide resistance in Streptococcus pneumoniae: clonality and mechanisms of resistance in 24 countries. Curr Drug Targets-Infect Disord. 2004;4(3):169–76.
Stefani S, Varaldo P. Epidemiology of methicillin-resistant staphylococci in Europe. Clin Microbiol Infect. 2003;9(12):1179–86.
Willems RJ, Top J. Marga van Santen D, Coque TM, Baquero F, Grundmann H, Bonten MJ: Global spread of vancomycin-resistant Enterococcus faecium from distinct nosocomial genetic complex. Emerg Infect Dis. 2005;11(6):821.
Orlek A, Stoesser N, Anjum MF, Doumith M, Ellington MJ, Peto T, et al. Plasmid classification in an era of whole-genome sequencing: application in studies of antibiotic resistance epidemiology. Front Microbiol. 2017;8:182.
Woodford N, Turton JF, Livermore DM. Multiresistant Gram-negative bacteria: the role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol Rev. 2011;35(5):736–55.
Naseer U, Sundsfjord A. The CTX-M conundrum: dissemination of plasmids and Escherichia coli clones. Microb Drug Resist. 2011;17(1):83–97.
Stoesser N, Sheppard AE, Pankhurst L, De Maio N, Moore CE, Sebra R, et al. Evolutionary history of the global emergence of the Escherichia coli epidemic clone ST131. mBio. 2016;7(2):e02162.
Lemmen S, Lewalter K. Antibiotic stewardship and horizontal infection control are more effective than screening, isolation and eradication. Infection. 2018;46(5):581–90.
Banerjee R, Johnston B, Lohse C, Chattopadhyay S, Tchesnokova V, Sokurenko EV, et al. The clonal distribution and diversity of extraintestinal Escherichia coli isolates vary according to patient characteristics. Antimicrob Agents Chemother. 2013;57(12):5912–7.
Johnson JR, Thuras P, Johnston BD, Weissman SJ, Limaye AP, Riddell K, et al. The pandemic H 30 subclone of Escherichia coli sequence type 131 is associated with persistent infections and adverse outcomes independent from its multidrug resistance and associations with compromised hosts. Clin Infect Dis. 2016;62(12):1529–36.
Martischang R, François P, Cherkaoui A, Gaïa N, Renzi G, Agostinho A, et al. Epidemiology of ESBL-producing Escherichia coli from repeated prevalence studies over 11 years in a long-term-care facility. Antimicrob Resist Infect Control. 2021;10(1):148.
Rodríguez-Villodres Á, Martín-Gandul C, Peñalva G, Guisado-Gil AB, Crespo-Rivas JC, Pachón-Ibáñez ME, et al. Cisneros JM: Prevalence and Risk Factors for Multidrug-Resistant Organisms Colonization in Long-Term Care Facilities Around the World: A Review. Antibiotics (Basel). 2021;10(6):680.
Kanamori H, Parobek CM, Juliano JJ, Johnson JR, Johnston BD, Johnson TJ, et al. Genomic analysis of multidrug-resistant Escherichia coli from North Carolina community hospitals: ongoing circulation of CTX-M-producing ST131-H30Rx and ST131-H30R1 strains. Antimicrob Agents Chemother. 2017;61(8):e00912–17.
Tschudin-Sutter S, Lucet J-C, Mutters NT, Tacconelli E, Zahar JR, Harbarth S. Contact precautions for preventing nosocomial transmission of extended-spectrum β lactamase–producing Escherichia coli: a point/counterpoint review. Clin Infect Dis. 2017;65(2):342–7.
Terveer E, Fallon M, Kraakman M, Ormond A, Fitzpatrick M, Caljouw M, et al. Spread of ESBL-producing Escherichia coli in nursing home residents in Ireland and the Netherlands may reflect infrastructural differences. J Hosp Infect. 2019;103(2):160–4.
European Antimicrobial Resistance Surveillance Network [https://ecdc.europa.eu/en/about-us/partnerships-and-networks/disease-and-laboratory-networks/ears-net].
Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis. 2018;18(3):318–27.
Munoz-Price LS, Poirel L, Bonomo RA, Schwaber MJ, Daikos GL, Cormican M, et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis. 2013;13(9):785–96.
Cornaglia G. Fighting infections due to multidrug-resistant Gram-positive pathogens: Elsevier; 2009.
Sheppard AE, Stoesser N, Wilson DJ, Sebra R, Kasarskis A, Anson LW, et al. Nested Russian doll-like genetic mobility drives rapid dissemination of the carbapenem resistance gene blaKPC. Antimicrob Agents Chemother. 2016;60(6):3767.
Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48(1):1–12.
Broberg CA, Palacios M, Miller VL. Klebsiella: a long way to go towards understanding this enigmatic jet-setter. F1000Prime Rep. 2014;6:64.
Yigit H, Queenan AM, Anderson GJ, Domenech-Sanchez A, Biddle JW, Steward CD, et al. Novel carbapenem-hydrolyzing β-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob Agents Chemother. 2001;45(4):1151.
Poirel L, Héritier C, Tolün V, Nordmann P. Emergence of oxacillinase-mediated resistance to imipenem in Klebsiella pneumoniae. Antimicrob Agents Chemother. 2004;48(1):15–22.
Yong D, Toleman MA, Giske CG, Cho HS, Sundman K, Lee K, et al. Characterization of a new metallo-β-lactamase gene, bla NDM-1, and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother. 2009;53(12):5046–54.
Cassini A, Högberg LD, Plachouras D, Quattrocchi A, Hoxha A, Simonsen GS, et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect Dis. 2019;19(1):56–66.
Borer A, Saidel-Odes L, Riesenberg K, Eskira S, Peled N, Nativ R, et al. Attributable mortality rate for carbapenem-resistant Klebsiella pneumoniae bacteremia. Infect Control Hosp Epidemiol. 2009;30(10):972–6.
DeLeo FR, Chen L, Porcella SF, Martens CA, Kobayashi SD, Porter AR, et al. Molecular dissection of the evolution of carbapenem-resistant multilocus sequence type 258 Klebsiella pneumoniae. Proc Natl Acad Sci. 2014;111(13):4988–93.
Marimuthu K, Venkatachalam I, Khong WX, Koh TH, Cherng BPZ, Van La M, et al. Clinical and molecular epidemiology of carbapenem-resistant Enterobacteriaceae among adult inpatients in Singapore. Clin Infect Dis. 2017;64(suppl_2):S68–s75.
Stoesser N, Phan HTT, Seale AC, Aiken Z, Thomas S, Smith M, et al. Genomic Epidemiology of Complex, Multispecies, Plasmid-Borne bla KPC Carbapenemase in Enterobacterales in the United Kingdom from 2009 to 2014. Antimicrob Agents Chemother. 2020;64(5):e02244–19.
Ludden C, Lötsch F, Alm E, Kumar N, Johansson K, Albiger B, et al. Cross-border spread of blaNDM-1-and blaOXA-48-positive Klebsiella pneumoniae: a European collaborative analysis of whole genome sequencing and epidemiological data, 2014 to 2019. Eurosurveillance. 2020;25(20):2000627.
Ben-David D, Masarwa S, Adler A, Mishali H, Carmeli Y, Schwaber MJ. A national intervention to prevent the spread of carbapenem-resistant Enterobacteriaceae in Israeli post-acute care hospitals. Infect Control Hosp Epidemiol. 2014;35(7):802–9.
Schwaber MJ, Lev B, Israeli A, Solter E, Smollan G, Rubinovitch B, et al. Containment of a country-wide outbreak of carbapenem-resistant Klebsiella pneumoniae in Israeli hospitals via a nationally implemented intervention. Clin Infect Dis. 2011;52(7):848–55.
Schwaber MJ, Carmeli Y. An ongoing national intervention to contain the spread of carbapenem-resistant enterobacteriaceae. Clin Infect Dis. 2014;58(5):697–703.
Marchaim D, Navon-Venezia S, Schwaber MJ, Carmeli Y. Isolation of imipenem-resistant Enterobacter species: emergence of KPC-2 carbapenemase, molecular characterization, epidemiology, and outcomes. Antimicrob Agents Chemother. 2008;52(4):1413–8.
Doumith M, Godbole G, Ashton P, Larkin L, Dallman T, Day M, et al. Detection of the plasmid-mediated mcr-1 gene conferring colistin resistance in human and food isolates of Salmonella enterica and Escherichia coli in England and Wales. J Antimicrob Chemother. 2016;71(8):2300–5.
Jung H-J, Littmann ER, Seok R, Leiner IM, Taur Y, Peled J, et al. Genome-wide screening for enteric colonization factors in carbapenem-resistant ST258 Klebsiella pneumoniae. mBio. 2019;10(2):e02663–18.
Morris S, Cerceo E. Trends, Epidemiology, and Management of Multi-Drug Resistant Gram-Negative Bacterial Infections in the Hospitalized Setting. Antibiotics (Basel). 2020;9(4):196.
Köser CU, Holden MT, Ellington MJ, Cartwright EJ, Brown NM, Ogilvy-Stuart AL, et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med. 2012;366(24):2267–75.
Price JR, Golubchik T, Cole K, Wilson DJ, Crook DW, Thwaites GE, et al. Whole-genome sequencing shows that patient-to-patient transmission rarely accounts for acquisition of Staphylococcus aureus in an intensive care unit. Clin Infect Dis. 2014;58(5):609–18.
Snitkin ES, Won S, Pirani A, Lapp Z, Weinstein RA, Lolans K, et al. Integrated genomic and interfacility patient-transfer data reveal the transmission pathways of multidrug-resistant Klebsiella pneumoniae in a regional outbreak. Sci Transl Med. 2017;9(417):eaan0093.
Coll F, Harrison EM, Toleman MS, Reuter S, Raven KE, Blane B, et al. Longitudinal genomic surveillance of MRSA in the UK reveals transmission patterns in hospitals and the community. Sci Transl Med. 2017;9(413).
Ferrari C, Corbella M, Gaiarsa S, Comandatore F, Scaltriti E, Bandi C, et al. Multiple Klebsiella pneumoniae KPC clones contribute to an extended hospital outbreak. Front Microbiol. 2019;10:2767.
Robinson ER, Walker TM, Pallen MJ. Genomics and outbreak investigation: from sequence to consequence. Genome Med. 2013;5(4):1–9.
Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13(2):137–46.
Walker TM, Lalor MK, Broda A, Ortega LS, Morgan M, Parker L, et al. Assessment of Mycobacterium tuberculosis transmission in Oxfordshire, UK, 2007–12, with whole pathogen genome sequences: an observational study. Lancet Respir Med. 2014;2(4):285–92.
Gardy JL, Johnston JC, Sui SJH, Cook VJ, Shah L, Brodkin E, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011;364(8):730–9.
Turnidge J, Christiansen K. Antibiotic use and resistance--proving the obvious. Lancet (London, England). 2005;365(9459):548–9.
Osthoff M, McGuinness SL, Wagen AZ, Eisen DP. Urinary tract infections due to extended-spectrum beta-lactamase-producing Gram-negative bacteria: identification of risk factors and outcome predictors in an Australian tertiary referral hospital. Int J Infect Dis. 2015;34:79–83.
Leal HF, Azevedo J, Silva GEO, Amorim AML, de Roma LRC, Arraes ACP, et al. Bloodstream infections caused by multidrug-resistant gram-negative bacteria: epidemiological, clinical and microbiological features. BMC Infect Dis. 2019;19(1):1–11.
Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, Gobourne A, et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis. 2012;55(7):905–14.
Bell BG, Schellevis F, Stobberingh E, Goossens H, Pringle M. A systematic review and meta-analysis of the effects of antibiotic consumption on antibiotic resistance. BMC Infect Dis. 2014;14(1):1–25.
Hernando-Amado S, Coque TM, Baquero F, Martínez JL. Defining and combating antibiotic resistance from One Health and Global Health perspectives. Nat Microbiol. 2019;4(9):1432–42.
Collineau L, Boerlin P, Carson CA, Chapman B, Fazil A, Hetman B, et al. Integrating whole-genome sequencing data into quantitative risk assessment of foodborne antimicrobial resistance: a review of opportunities and challenges. Front Microbiol. 2019;10:1107.
Madec J-Y, Haenni M. Antimicrobial resistance plasmid reservoir in food and food-producing animals. Plasmid. 2018;99:72–81.
Moura A, Tourdjman M, Leclercq A, Hamelin E, Laurent E, Fredriksen N, et al. Real-time whole-genome sequencing for surveillance of Listeria monocytogenes, France. Emerg Infect Dis. 2017;23(9):1462.
Jackson BR, Tarr C, Strain E, Jackson KA, Conrad A, Carleton H, et al. Implementation of nationwide real-time whole-genome sequencing to enhance listeriosis outbreak detection and investigation. Rev Infect Dis. 2016;63(3):380–6.
Andino R, Domingo E. Viral quasispecies. Virology. 2015;479-480:46–51.
Angus B, Brook G, Awosusi F, Barker G, Boffito M, Das S, Dorrell L, Dixon-Williams E, Hall C, Howe B. (2019 interim update). https://www.bhiva.org/file/DqZbRxfzlYtLg/Monitoring-Guidelines pdf.2019.
Baxter JD, Mayers DL, Wentworth DN, Neaton JD, Hoover ML, Winters MA, et al. A randomized study of antiretroviral management based on plasma genotypic antiretroviral resistance testing in patients failing therapy. CPCRA 046 Study Team for the Terry Beirn Community Programs for Clinical Research on AIDS. Aids. 2000;14(9):F83–93.
Geretti AM, Paredes R, Kozal MJ. Transmission of HIV drug resistance: lessons from sensitive screening assays. Curr Opin Infect Dis. 2015;28(1):23–30.
Van Laethem K, Theys K, Vandamme AM. HIV-1 genotypic drug resistance testing: digging deep, reaching wide? Curr Opin Virol. 2015;14:16–23.
Cingolani A, Antinori A, Rizzo MG, Murri R, Ammassari A, Baldini F, et al. Usefulness of monitoring HIV drug resistance and adherence in individuals failing highly active antiretroviral therapy: a randomized study (ARGENTA). Aids. 2002;16(3):369–79.
Ávila-Ríos S, Parkin N, Swanstrom R, Paredes R, Shafer R, Ji H, et al. Next-generation sequencing for HIV drug resistance testing: laboratory, clinical, and implementation considerations. Viruses. 2020;12(6):617.
Li JZ, Paredes R, Ribaudo HJ, Svarovskaia ES, Metzner KJ, Kozal MJ, et al. Low-frequency HIV-1 drug resistance mutations and risk of NNRTI-based antiretroviral treatment failure: a systematic review and pooled analysis. Jama. 2011;305(13):1327–35.
Chakaya J, Khan M, Ntoumi F, Aklillu E, Fatima R, Mwaba P, et al. Global Tuberculosis Report 2020 - Reflections on the Global TB burden, treatment and prevention efforts. Int J Infect Dis. 2021;113 Suppl 1(Suppl 1):S7–S12.
Heyckendorf J, Andres S, Köser CU, Olaru ID, Schön T, Sturegård E, et al. What is resistance? Impact of phenotypic versus molecular drug resistance testing on therapy for multi-and extensively drug-resistant tuberculosis. Antimicrob Agents Chemother. 2018;62(2):e01550–17.
Lange C, Aarnoutse R, Chesov D, Van Crevel R, Gillespie SH, Grobbel H-P, et al. Perspective for precision medicine for tuberculosis. Front Immunol. 2020;11:2442.
Cabibbe AM, Trovato A, De Filippo MR, Ghodousi A, Rindi L, Garzelli C, et al. Countrywide implementation of whole genome sequencing: an opportunity to improve tuberculosis management, surveillance and contact tracing in low incidence countries. Eur Respir J. 2018;51(6):1800387.
Cabibbe AM, Sotgiu G, Izco S, Migliori GB. Genotypic and phenotypic M. tuberculosis resistance: guiding clinicians to prescribe the correct regimens. Eur Respir J. 2017;50(6):1702292.
Meehan CJ, Goig GA, Kohl TA, Verboven L, Dippenaar A, Ezewudo M, et al. Whole genome sequencing of Mycobacterium tuberculosis: current standards and open issues. Nat Rev Microbiol. 2019;17(9):533–45.
Pankhurst LJ, Del Ojo EC, Votintseva AA, Walker TM, Cole K, Davies J, et al. Rapid, comprehensive, and affordable mycobacterial diagnosis with whole-genome sequencing: a prospective study. Lancet Respir Med. 2016;4(1):49–58.
Doyle RM, Burgess C, Williams R, Gorton R, Booth H, Brown J, et al. Direct whole-genome sequencing of sputum accurately identifies drug-resistant mycobacterium tuberculosis faster than MGIT culture sequencing. J Clin Microbiol. 2018;56(8):e00666–18.
Allix-Beguec C, Arandjelovic I, Bi L, Beckert P, Bonnet M, Bradley P, et al. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N Engl J Med. 2018;379(15):1403–15.
Trauner A, Liu Q, Via LE, Liu X, Ruan X, Liang L, et al. The within-host population dynamics of Mycobacterium tuberculosis vary with treatment efficacy. Genome Biol. 2017;18(1):1–17.
Tomczyk S, Lynfield R, Schaffner W, Reingold A, Miller L, Petit S, et al. Prevention of antibiotic-nonsusceptible invasive pneumococcal disease with the 13-valent pneumococcal conjugate vaccine. Clin Infect Dis. 2016;62(9):1119–25.
Hampton LM, Farley MM, Schaffner W, Thomas A, Reingold A, Harrison LH, et al. Prevention of antibiotic-nonsusceptible Streptococcus pneumoniae with conjugate vaccines. J Infect Dis. 2012;205(3):401–11.
Jansen KU, Knirsch C, Anderson AS. The role of vaccines in preventing bacterial antimicrobial resistance. Nat Med. 2018;24(1):10–9.
Doherty TM, Hausdorff WP, Kristinsson KG. Effect of vaccination on the use of antimicrobial agents: a systematic literature review. Ann Med. 2020;52(6):283–99.
Lewnard JA, Lo NC, Arinaminpathy N, Frost I, Laxminarayan R. Childhood vaccines and antibiotic use in low- and middle-income countries. Nature. 2020;581(7806):94–9.
Stanaway JD, Reiner RC, Blacker BF, Goldberg EM, Khalil IA, Troeger CE, et al. The global burden of typhoid and paratyphoid fevers: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Infect Dis. 2019;19(4):369–81.
Holt KE, Parkhill J, Mazzoni CJ, Roumagnac P, Weill FX, Goodhead I, et al. High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nat Genet. 2008;40(8):987–93.
Wong VK, Baker S, Pickard DJ, Parkhill J, Page AJ, Feasey NA, et al. Phylogeographical analysis of the dominant multidrug-resistant H58 clade of Salmonella Typhi identifies inter- and intracontinental transmission events. Nat Genet. 2015;47(6):632–9.
Hooda Y, Sajib MSI, Rahman H, Luby SP, Bondy-Denomy J, Santosham M, et al. Molecular mechanism of azithromycin resistance among typhoidal Salmonella strains in Bangladesh identified through passive pediatric surveillance. PLoS Negl Trop Dis. 2019;13(11):e0007868.
Duy PT, Dongol S, Giri A, Nguyen To NT, Dan Thanh HN, Nhu Quynh NP, et al. The emergence of azithromycin-resistant Salmonella Typhi in Nepal. JAC-Antimicrobial Resistance. 2020;2(4):dlaa109.
Yousafzai MT, Qamar FN, Shakoor S, Saleem K, Lohana H, Karim S, et al. Ceftriaxone-resistant Salmonella Typhi outbreak in Hyderabad City of Sindh, Pakistan: high time for the introduction of typhoid conjugate vaccine. Clin Infect Dis. 2019;68(Supplement_1):S16–21.
Klemm EJ, Shakoor S, Page AJ, Qamar FN, Judge K, Saeed DK, et al. Emergence of an extensively drug-resistant Salmonella enterica serovar Typhi clone harboring a promiscuous plasmid encoding resistance to fluoroquinolones and third-generation cephalosporins. mBio. 2018;9(1):e00105–18.
Field Epidemiology & Laboratory Training Program National Institute of Health Islamabad: Weekly field epidemiology report update on extensive drug typhoid fever. 2019;2(18):1–7.
Qamar FN, Yousafzai MT, Khaliq A, Karim S, Memon H, Junejo A, et al. Adverse events following immunization with typhoid conjugate vaccine in an outbreak setting in Hyderabad, Pakistan. Vaccine. 2020;38(19):3518–23.
Batool R. Story of Lyari. Am J Trop Med Hyg. 2020;102(4):705–6.
World Health Organization. Regional Office for the Eastern Mediterranean. The work of WHO in the Eastern Mediterranean Region: annual report of the Regional Director 2019. World Health Organization. Regional Office for the Eastern Mediterranean. 2020. https://apps.who.int/iris/handle/10665/335963. License: CC BY-NCSA 3.0 IGO.
Over 13 million children vaccinated with typhoid conjugate vaccine in Punjab and Islamabad [http://www.emro.who.int/pak/pakistan-news/over-13-million-children-vaccinated-with-typhoid-conjugate-vaccine-in-punjab-and-islamabad.html].
Typhoid vaccines. WHO position paper, March 2018–Recommendations. Vaccine. 2019;37(2):214–6.
Fitzgerald DM. Bacterial evolution: the road to resistance. Elife. 2019;8:e52092.
Croucher NJ, Didelot X. The application of genomics to tracing bacterial pathogen transmission. Curr Opin Microbiol. 2015;23:62–7.
Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, Van Der Linden M, et al. Rapid pneumococcal evolution in response to clinical interventions. Science (New York, NY). 2011;331(6016):430–4.
Schürch A, Arredondo-Alonso S, Willems R, Goering R. Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene–based approaches. Clin Microbiol Infect. 2018;24(4):350–4.
Coll F, Raven KE, Knight GM, Blane B, Harrison EM, Leek D, et al. Definition of a genetic relatedness cutoff to exclude recent transmission of meticillin-resistant Staphylococcus aureus: a genomic epidemiology analysis. Lancet Microbe. 2020;1(8):e328–35.
Campbell F, Cori A, Ferguson N, Jombart T. Bayesian inference of transmission chains using timing of symptoms, pathogen genomes and contact data. PLoS Comput Biol. 2019;15(3):e1006930.
De Maio N, Worby CJ, Wilson DJ, Stoesser N. Bayesian reconstruction of transmission within outbreaks using genomic variants. PLoS Comput Biol. 2018;14(4):e1006117.
Juraschek K, Borowiak M, Tausch SH, Malorny B, Käsbohrer A, Otani S, et al. Outcome of different sequencing and assembly approaches on the detection of plasmids and localization of antimicrobial resistance genes in commensal Escherichia coli. Microorganisms. 2021;9(3):598.
Thomas CM, Nielsen KM. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat Rev Microbiol. 2005;3(9):711–21.
Inouye M, Dashnow H, Raven L-A, Schultz MB, Pope BJ, Tomita T, et al. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014;6(11):90.
Mardis ER. DNA sequencing technologies: 2006–2016. Nat Protoc. 2017;12(2):213–8.
De Maio N, Shaw LP, Hubbard A, George S, Sanderson ND, Swann J, et al. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microb Genom. 2019;5(9):e000294.
Ashton PM, Nair S, Dallman T, Rubino S, Rabsch W, Mwaigwisya S, et al. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat Biotechnol. 2015;33(3):296–300.
Arredondo-Alonso S, van Schaik W, Willems R, Schürch A. On the (im)possibility of reconstructing plasmids from whole-genome short-read sequencing data. Microb Genom. 2017;3(10):e000128.
Wick RR, Judd LM, Gorrie CL, Holt KE. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb Genom. 2017;3(10):e000132.
George S, Pankhurst L, Hubbard A, Votintseva A, Stoesser N, Sheppard AE, et al. Resolving plasmid structures in Enterobacteriaceae using the MinION nanopore sequencer: assessment of MinION and MinION/Illumina hybrid data assembly approaches. Microb Genom. 2017;3(8):e000118.
Peacock SJ, Parkhill J, Brown NM. Changing the paradigm for hospital outbreak detection by leading with genomic surveillance of nosocomial pathogens. Microbiology. 2018;164(10):1213–9.
Köser CU, Ellington MJ, Peacock SJ. Whole-genome sequencing to control antimicrobial resistance. Trends Genet. 2014;30(9):401–7.
Köser CU, Ellington MJ, Cartwright EJ, Gillespie SH, Brown NM, Farrington M, et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. 2012;8(8):e1002824.
Rossen JWA, Friedrich AW, Moran-Gilad J. Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin Microbiol Infect. 2018;24(4):355–60.
Dymond A, Davies H, Mealing S, Pollit V, Coll F, Brown NM, et al. Genomic Surveillance of Methicillin-resistant Staphylococcus aureus: a mathematical early modeling study of cost-effectiveness. Clin Infect Dis. 2020;70(8):1613–9.
Gordon LG, Elliott TM, Forde B, Mitchell B, Russo PL, Paterson DL, et al. Budget impact analysis of routinely using whole-genomic sequencing of six multidrug-resistant bacterial pathogens in Queensland, Australia. BMJ Open. 2021;11(2):e041968.
Kumar P, Sundermann AJ, Martin EM, Snyder GM, Marsh JW, Harrison LH, et al. Method for economic evaluation of bacterial whole genome sequencing surveillance compared to standard of care in detecting hospital outbreaks. Clin Infect Dis. 2021;73(1):e9–e18.
Elliott TM, Lee XJ, Foeglein A, Harris PN, Gordon LG. A hybrid simulation model approach to examine bacterial genome sequencing during a hospital outbreak. BMC Infect Dis. 2020;20(1):72.
Mellmann A, Andersen PS, Bletz S, Friedrich AW, Kohl TA, Lilje B, et al. High interlaboratory reproducibility and accuracy of next-generation-sequencing-based bacterial genotyping in a ring trial. J Clin Microbiol. 2017;55(3):908–13.
Dylus D, Pillonel T, Opota O, Wüthrich D, Seth-Smith H, Egli A, et al. NGS-based S. aureus typing and outbreak analysis in clinical microbiology laboratories: lessons learned from a Swiss-wide proficiency test. Front Microbiol. 2020;11:2822.
Wielinga PR, Hendriksen RS, Aarestrup FM, Lund O, Smits SL, Koopmans MP, et al. Global microbial identifier. In: Applied genomics of foodborne pathogens: Springer; 2017. p. 13–31.
Connor TR, Barker CR, Baker KS, Weill F-X, Talukder KA, Smith AM, et al. Species-wide whole genome sequencing reveals historical global spread and recent local persistence in Shigella flexneri. eLife. 2015;4:e07335.
Snell LB, Fisher CL, Taj U, Stirrup O, Merrick B, Alcolea-Medina A, et al. Combined epidemiological and genomic analysis of nosocomial SARS-CoV-2 infection early in the pandemic and the role of unidentified cases in transmission. Clin Microbiol Infect. 2022;28(1):93–100.
du Plessis L, McCrone JT, Zarebski AE, Hill V, Ruis C, Gutierrez B, et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science (New York, NY). 2021;371(6530):708–12.
Okeke IN, Feasey N, Parkhill J, Turner P, Limmathurotsakul D, Georgiou P, et al. Leapfrogging laboratories: the promise and pitfalls of high-tech solutions for antimicrobial resistance surveillance in low-income settings. BMJ Glob Health. 2020;5(12):e003622.
Acknowledgements
We thank the editors and guest editors at Genome Medicine for the invitation to write this review.
Funding
This work was supported by a Wellcome senior research fellowship to Stephen Baker (215515/Z/19/Z). The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
Conceptualisation: CSW, MEC, and SB. Writing original draft: CSW, SB, CB, EH, MEC, and VB. Review and editing: CSW, MEC, and SB. Read and approved the final version of the manuscript: CSW, MEC, CB, EH, VB, and SB
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Waddington, C., Carey, M.E., Boinett, C.J. et al. Exploiting genomics to mitigate the public health impact of antimicrobial resistance. Genome Med 14, 15 (2022). https://doi.org/10.1186/s13073-022-01020-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-022-01020-2