
Abstract
Neoantigens are newly formed peptides created from somatic mutations that are capable of inducing tumor-specific T cell recognition. Recently, researchers and clinicians have leveraged next generation sequencing technologies to identify neoantigens and to create personalized immunotherapies for cancer treatment. To create a personalized cancer vaccine, neoantigens must be computationally predicted from matched tumor–normal sequencing data, and then ranked according to their predicted capability in stimulating a T cell response. This candidate neoantigen prediction process involves multiple steps, including somatic mutation identification, HLA typing, peptide processing, and peptide-MHC binding prediction. The general workflow has been utilized for many preclinical and clinical trials, but there is no current consensus approach and few established best practices. In this article, we review recent discoveries, summarize the available computational tools, and provide analysis considerations for each step, including neoantigen prediction, prioritization, delivery, and validation methods. In addition to reviewing the current state of neoantigen analysis, we provide practical guidance, specific recommendations, and extensive discussion of critical concepts and points of confusion in the practice of neoantigen characterization for clinical use. Finally, we outline necessary areas of development, including the need to improve HLA class II typing accuracy, to expand software support for diverse neoantigen sources, and to incorporate clinical response data to improve neoantigen prediction algorithms. The ultimate goal of neoantigen characterization workflows is to create personalized vaccines that improve patient outcomes in diverse cancer types.
Similar content being viewed by others


Background
The adaptive immune system has inherent antitumor properties that are capable of inducing tumor-specific cell death [1, 2]. CD8+ and CD4+ T cells, two immune cell types that are critical to this process, recognize antigens bound by class I and II major histocompatibility complexes (MHC) on the cell surface, respectively. After antigen recognition, T cells have the ability to signal growth arrest and cell death to tumor cells displaying the antigen, and also release paracrine signals to propagate an antitumor response. Neoantigens are specifically defined here as peptides derived from somatic mutations that provide an avenue for tumor-specific immune cell recognition and that are important targets for cancer immunotherapies [3,4,5]. Studies have shown that, in addition to tumor mutational burden (TMB), high neoantigen burden can be a predictor of response to immune checkpoint blockade (ICB) therapy [6, 7]. This treatment strategy targets the signaling pathways that suppress antitumor immune responses, allowing the activation of neoantigen-specific T cells and promoting immune-mediated tumor cell death. Therefore, accurate neoantigen prediction is vital for the success of personalized vaccines and for the prioritization of candidates underlying the mechanism of response to ICB. These approaches have great therapeutic potential because neoantigen-specific T cells should not be susceptible to central tolerance.
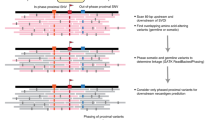
With the advent of next generation sequencing (NGS), researchers can now rapidly sequence a patient’s DNA and RNA before analyzing these sequencing data to predict neoantigens computationally. This process requires several steps, each involving the use of bioinformatics tools and complex analytical pipelines (Fig. 1; Table 1). Matched tumor–normal DNA sequencing data are processed and analyzed to call somatic mutations of various types. Human leukocyte antigen (HLA) haplotyping is performed to determine a patient’s HLA alleles and the corresponding MHC complexes. Finally, RNA sequencing (RNA-seq) data are used to quantify gene and transcript expression, and can verify variant expression prior to neoantigen prediction. Multiple pipelines exist to identify candidate neoantigens that have high binding affinities to MHC class I or II. Additional steps are subsequently required to prioritize them for clinical use in personalized vaccines and to address manufacturing and delivery issues [8, 9].

Overview of the bioinformatic characterization of neoantigens. Major analysis steps in a comprehensive workflow for neoantigen characterization are depicted in a simplified form. For each component, critical concepts and analysis considerations are indicated. Specific exemplar bioinformatics tools for each step are indicated in italics. Starting at the top left, patient sequences are analyzed to determine human leukocyte antigen (HLA) types and to predict the corresponding major histocompatibility complexes (MHC) for each tumor. Somatic variants of various types, including single nucleotide variants (SNVs; blue), deletions (red), insertions (green), and fusions (pink), are detected and the corresponding peptide sequences are analyzed with respect to their predicted expression, processing, and ability to bind the patient’s MHC complexes. Candidates are then selected for vaccine design and additional analyses are performed to assess the T cell response. Abbreviations: CDR3 complementarity-determining region 3, FFPE formalin-fixed paraffin-embedded, IEDB Immune Epitope Database, TCR T cell receptor
The general concept of neoantigens and their role in personalized immunotherapies have been extensively reviewed elsewhere [10,11,12]. Although experimental methods exist to assess neoantigens (e.g., mass spectrometry (MS)), the focus of this review is a comprehensive survey of computational approaches (tools, databases, and pipelines) for neoantigen characterization. The ultimate goal is to discover neoepitopes, the part of the neoantigen that is recognized and bound by T cells, but current workflows are largely focused on predicting MHC-binding antigens with limited prediction of recognition by T cells or therapeutic potential. We have been particularly inspired by the use of computational approaches in human clinical trials involving personalized neoantigen vaccines alone or in combination with ICB. A rapid expansion of the number and diversity of these trials has occurred over the past few years, but there is limited community consensus on approaches for neoantigen characterization. Adoption of standards for the accurate identification of neoantigens and for the reporting of their features will be critical for the interpretation of results from early-stage trials and for the optimization of future trials. This review is focused on human clinical data; nevertheless, neoantigen characterization work involving model organisms (such as mice) will be critical to advance the field, and many of the tools and approaches described herein may be applied to these model systems with appropriate modifications. In addition to describing emerging best practices, we highlight the current limitations and critical areas for the improvement of the computational approaches needed to understand the immunogenicity of neoantigens.
Neoantigen identification
Two types of antigens that can induce an antitumor response are tumor-specific antigens (or neoantigens) and tumor-associated antigens (TAA). Neoantigens contain altered amino-acid sequences that result from non-silent somatic mutations, whereas TAAs, which may originate from endogenous proteins or retroviruses, are selectively expressed or overexpressed by tumor cells but may also be expressed by non-tumor cell populations [13]. This review focuses on the detection and selection of neoantigens, but many analytical steps that are used can apply to other antigen types. Considerations such as sample type (fresh frozen, formalin-fixed paraffin-embedded (FFPE) tissue or circulating tumor DNA (ctDNA)), tumor type (solid or blood), biopsy site, and sequencing approach (DNA, RNA, or targeted sequencing) can impact somatic variant detection and interpretation, and should be taken into account during data processing and downstream analysis [13,14,15,16]. In addition, tumors that exhibit high intratumoral heterogeneity can require alternative methods, such as collecting multiple biopsies per tumor [17].
Somatic variant callers identify single nucleotide variants (SNVs) from tumor and matched non-tumor DNA sequence data, such as whole genome, or more commonly, whole exome sequencing (WES) data [18]. Three common limitations to SNV calling—low frequency variant detection, distinguishing germline variants from tumor in normal contamination, and removing sequencing artifacts—have been addressed by the variant callers discussed below. MuTect2 [19] and Strelka [20] have high sensitivity in detecting SNVs at low allele fractions, enabling accurate subclonal variant detection. VarScan2 [21] and SomaticSniper [22] require higher allele fractions for recognizing variants but can improve performance in cases of tumor in normal contamination [23, 24]. MuTect2 can further exclude sequencing or alignment artifacts by implementing a panel-of-normals file, containing false positives detected across normal samples. Running multiple variant calling algorithms simultaneously is recommended and can result in higher detection accuracy. For example, Callari et al. [25] achieved 17.1% higher sensitivity without increasing the false-positive rate by intersecting a single variant caller’s results from multiple alignment pipelines and then combining the intersected results from two callers, MuTect2 and Strelka, to achieve a final consensus. The list of variant callers mentioned here is not exhaustive (see Table 1 for additional options) and high-quality pipelines using different combinations are certainly possible. Regardless of the combination of callers used, manual review of matched tumor–normal samples in Integrative Genomics Viewer (IGV) [26], with a documented standard operating procedure, is recommended to further reduce false positives [27]. In addition to IGV, targeted sequencing approaches such as custom capture reagents can be utilized for further variant validation.
Recently, neoantigen vaccine trials for melanoma demonstrated that SNV-derived neoantigens can expand T cell populations [28] and induce disease regression [29, 30]. However, recent studies have also increased appreciation for diverse neoantigen sources beyond simple SNVs, including short insertions and deletions (indels) [31], fusions [32, 33], intron retentions [34], non-coding expressed regions [35], exon–exon junction epitopes [36], B cell receptor (BCR) and T cell receptor (TCR) sequences for B and T cell malignancies, respectively [37], and more [38].
Frameshift mutations resulting from insertions and deletions create alternative open reading frames (ORFs) with novel tumor-specific sequences that are completely distinct from those that encode wild-type antigens. A pan-cancer analysis of 19 cancer types from The Cancer Genome Atlas demonstrated that frameshift-derived neoantigens were present in every cancer type [31]. This mutation type also occurs frequently in microsatellite instability high (MSI-H) colon and other cancers and correlates with higher CD8+ T cell infiltrate in the tumors [31, 39]. For calling indels, in addition to Strelka, EBCall [40] demonstrates the least sensitivity to coverage variability [41, 42]. Pindel [43] specializes in calling larger indels, from 0.50–10 kilobases in length, and structural variants. Though these are popular indel callers, they are only a subset of the available tools (see Table 1 for additional options).
Translocations may result in tumor-specific fusion genes, which can alter the reading frame and provide novel junction sequences. Researchers recently investigated the presence of translocations in osteosarcoma, characterized by high genomic instability [44], and discovered multiple fusion-derived junction-spanning neoantigens [45]. The identification of novel sequences resulting from inter- and intrachromosomal rearrangements in mesothelioma also resulted in the prediction of multiple neoantigens for each patient [46]. Many tools have been developed to predict fusion genes from RNA-seq and/or whole genome sequencing (WGS) data; recent tools include pizzly [47], STAR-fusion [48], JAFFA [49], GFusion [50], and INTEGRATE [51] (refer to Table 1). The main limitation of these fusion callers is the low level of overlap between tools; they largely achieve high sensitivity at the cost of low specificity. The presence of many false positives makes accurate detection difficult, but this can be mitigated by using multiple tools [52] and by requiring predictions to be supported by multiple callers and/or data types (e.g., WGS and RNA-seq).
In addition to mutation-derived neoantigens from known protein-coding genes, noncoding regions have immunogenic potential. Noncoding transcripts can be created from noncoding exons, introns, and untranslated regions (UTRs), as well as from non-canonical reading frames in the coding region [53]. Laumont et al. [35] investigated traditionally noncoding sequences using liquid chromatography tandem-MS (LC-MS/MS) and RNA sequencing (RNA-seq) in leukemia and lung cancer patients and found an abundance of antigens, both mutated and unmutated, from noncoding regions.
Recent publications have shown that aberrant tumor-specific splicing patterns can create neoantigens. Smart et al. [54] found an approximately 70% increase in total predicted neoantigens after including retained intron sequences along with SNVs in the prediction pipeline. Novel junctions created by exon skipping events, or neojunctions, have been shown to create neoantigens [36]. Tumor-specific splicing patterns can also cause distinct alternative 3′ or 5′ splice sites, known as splice-site-creating mutations, and these mutations are predicted to create an average of 2.0–2.5 neoantigens per mutation [55].
In addition to the neoantigen sources discussed above, many alternative sources can create neoantigens. For example, V(D) J recombination and somatic hypermutation generate immunoglobulin (Ig) variable region diversity in B and T lymphocytes, and the resulting unique receptor sequences can function as neoantigens in heme malignancies [37, 56]. Further, researchers have demonstrated that peptides with post translational modifications, including phosphorylation and O-GlcNAcylation, in primary leukemia samples can serve as MHC-I restricted neoantigens [57, 58]. Alternative translation events resulting from non-AUG start codons and viral sequences that are associated with tumors (e.g., human papilloma virus (HPV)) are also a source of neoantigens [59,60,61,62,63]. Overall, neoantigen identification requires a sensitive, accurate, and comprehensive somatic variant calling pipeline that is capable of robustly detecting all of the variant classes that are relevant for a tumor type (Table 2).
HLA typing, expression, and mutation analysis
T cell priming depends in part on neoantigen presentation on the surface of dendritic cells, a type of professional antigen presenting cells (APCs). Dendritic cells engulf extracellular proteins, process the peptides, and present the neoantigens on MHC I or II molecules. MHC in humans is encoded by the HLA gene complex, which is located on chromosome 6p21.3. This locus is highly polymorphic, with over 12,000 established alleles and more in discovery [64]. Because HLA genes are extensively individualized, precise HLA haplotyping is essential for accurate neoantigen prediction. The gold standard for this process is clinical HLA typing using sequence-specific PCR amplification [65]. More recently, NGS platforms such as Illumina MiSeq and PacBio RSII have been combined with PCR amplification to sequence the HLA locus [66]. However, clinical typing can be laborious and expensive, so a common alternative approach is computational HLA typing using the patient’s WGS, WES, and/or RNA-seq datasets, which are typically created from a peripheral blood sample, except in heme malignancies, where a skin sample is often used (Table 2).
HLA class I typing algorithms (Table 1) have reached up to 99% prediction accuracy when compared to curated clinical typing results [67, 68]. Although many class I typing algorithms exist, OptiType [69], Polysolver [70], and PHLAT [71] currently have the highest reported accuracies [67, 68, 70]. Despite the high precision of class I tools, class II HLA typing algorithms remain less reliable and require additional development to improve their prediction accuracy. Few benchmarking studies that consider class II algorithm accuracy have been performed, but a combined class I and II comparison demonstrated that PHLAT [71], HLA-VBSeq [72], and seq2HLA [73] performed well with WES and RNA-seq data [67]. Additional HLA typing algorithms, xHLA [74] and HLA-HD [75], have recently been published and show comparable accuracies to those of the tools described above.
Tumor-specific T cell recognition relies on efficient antigen presentation by tumor cells, so one mechanism of resistance to immunotherapies is the loss or attenuated expression of the HLA gene loci. Recently, researchers have identified transcriptional HLA repression in a patient with Merkel cell carcinoma (MCC) following treatment with autologous T cell therapy and ICB [76]. The authors found that the transcriptional silencing can be reversed in ex vivo cultures by treatment with 5-aza and other hypomethylating agents, indicating that reversing the epigenetic silencing of the HLA genes could sensitize tumors that exhibit HLA downregulation in response to immunotherapies [77].
Genetic changes at the HLA locus can be determined by Polysolver [70], an algorithm that detects HLA-specific somatic mutations from computational HLA typing and variant calling of the tumor HLA locus. Somatic mutation analysis of head and neck squamous cell carcinoma (HNSCC), lung cancer, and gastric adenocarcinoma cohorts demonstrated that HLA mutations are prevalent in all three cancer types [78,79,80]. In addition, HLA mutations (particularly frameshifts, nonsense, and splicing mutations) are enriched towards the beginning of the genes or within functional domains, where they would be expected to result in a loss-of-function phenotype [70]. Another tool, LOHHLA, can identify copy number variations in the HLA locus that result in loss of heterozygosity [81].
Additional components of the antigen presenting machinery, including B2M and TAP (Transporter associated with antigen processing), have been shown to accrue mutations and to exhibit altered expression patterns in tumors. In lung cancer and MSI-CRC, mutations or biallelic loss of B2M causes lack of class I HLA presentation [82, 83]. Downregulation of B2M, TAP1, and TAP2 expression has also been shown to inhibit tumor antigen presentation [84, 85] and correlate with metastatic breast cancer phenotypes [86]. Identifying and characterizing altered HLA and associated presentation genes will allow clinicians to prioritize neoantigens that bind to expressed and unmutated alleles.
Predicting peptide processing
Recognition of a peptide-MHC (pMHC) complex by the T cell is a complex process with many steps and requirements. Most of the attention in the field has been focused on predicting the binding affinity between the patient’s MHC molecule and a given peptide sequence, as this is believed to provide much of the specificity of the overall recognition [87]. However, even if a peptide has strong MHC binding prediction, the prediction may be meaningless if upstream processing prevents the actual loading of that peptide. In general, pipelines generate k-mer peptides using a sliding window that is applied to the mutant protein sequence, and these peptide sequences are subsequently fed into algorithms that predict the affinity of the peptide to the corresponding MHC. However, not all of the k-mers can be generated in vivo due to the limitations of the immune proteasome. In addition, only a subset of generated peptides will be transported into the appropriate cellular compartments and will interact with MHC molecules. These aspects of peptide processing, specifically immune proteasome processing and peptide cleavage, must be considered and several tools have been developed to address this component specifically [88].
For both the MHC class I and II pathways, an important upstream step prior to pMHC interaction is proteolysis, which refers to the degradation of proteins into peptides, particularly by the immunoproteasome. Multiple tools are now available to capture the specificity of proteasomes and to predict the cleavage sites that are targeted by different proteases. These tools include NetChop20S [89], NetChopCterm [89], and ProteaSMM [89, 90] for MHC class I antigens, and the more recent PepCleaveCD4 and MHC NP II for MHC class II antigens [91, 92]. Algorithms that have been developed in this area are generally trained on two different types of data, in vitro proteasome digestion data or in vivo MHC-I and -II ligand elution data. The neural network-based prediction method NetChop-3.0 Cterm has been shown to have the best performance in predicting in vivo proteolysis that provides peptide sources for MHC class I antigen presentation [88]. Cleavage site predictions for MHC class II epitopes show promise, but have yet to be validated for predicting immunogenicity [88, 92].
For MHC class I antigen processing, peptide fragments are generated from proteins that are present in the cytoplasm and transported by the TAP protein into the endoplasmic reticulum (ER), where the peptide is loaded onto an MHC molecule. Thus, in addition to tools focusing on the process of proteolysis, other tools have also been developed to predict the efficiency of peptide transportation on the basis of affinity to TAP proteins. Different methods have been employed in an attempt to determine which peptides have high affinity for TAP binding, including simple/cascade support vector machine (SVM) models [93, 94] and weight matrix models [95]. To address the entirety of this process, the Immune Epitope Database (IEDB) has also developed a predictor for the combination of these processes (proteasomal cleavage/TAP transport/MHC class I) [90, 96].
In the MHC class II pathway, the peptides are mostly exogenous and enter the endosome of APCs through endocytosis. As endosomes mature into late endosomal compartments, acidity levels increase and serine, aspartic, and cysteine proteases are activated. Proteins, upon exposure to a series of proteases, are degraded into potential antigens for presentation. MHC class II molecules are assembled in the ER and transported to these high acidity late endosomes, also known as MHC-II compartments (MIIC). Here, peptides can bind to class II molecules and are protected from destructive processing [97, 98]. In contrast to the protein denaturation in the MHC class I processing pathway, cleavage in the MHC class II pathway occurs on folded proteins. Predictors for class II peptide preprocessing prior to MHC binding show the important role that secondary structures play in such reactions, as multiple measures related to secondary structures were found to be highly correlated with the predicted cleavage score [91]. Consideration of secondary structure will be critical to the future development of tools predicting class II processed peptides. However, although the class I antigen processing pathway has been studied extensively, researchers have only recently started to focus on class-II-specific neoantigens as promising results have been shown in cancer immunotherapies [99,100,101]. There remains a great need to develop supporting tools and algorithms to characterize class-II-specific neoantigens.
For the purposes of neoantigen prioritization, it is important to take into account processing steps such as peptide cleavage and TAP transport when using binding prediction algorithms that were trained on in vitro binding data. Recently, published binding prediction algorithms have been transitioning to training on data generated in vivo, in which case the processing steps are accounted for intrinsically.
MHC binding prediction
Neoantigen characterization pipelines have been established specifically to predict the binding of neoantigens to the patient’s specific class I and II MHC molecules (based on HLA typing). Algorithmic development and the refinement of reference data sets are very active in this area. Here, we describe the current state of the art with respect to algorithmic innovation and refinement of the major classes of data that are used to train these algorithms (largely from in vitro binding assays involving specific MHCs and peptide libraries or from MS-based approaches) [87, 102,103,104].
Peptides bind MHC molecules at a membrane-distal groove that is formed by two antiparallel α-helices overlaying an eight-strand β-sheet [97]. The peptide-binding region of the MHC protein is encoded by exons 2 and 3 of the corresponding HLA gene [105]. High allelic polymorphism allows the binding pocket of MHC molecules to recognize a range of different peptides sequences, and the positions that are involved in anchoring the peptide to the MHC molecule in particular vary for each HLA allele. The algorithms and training datasets for predicting pMHC binding remain an active area of development. Various methods have been employed in an attempt to capture the characteristics of peptide and MHC molecules that have a high probability of binding (Table 1).
Early algorithms have mostly focused on training using in vitro pMHC binding affinity measurement datasets. MHC peptide binding is thought to be the most selective step in the antigen presentation process, but sole consideration of peptide binding predictions still results in high rates of false-positive predictions of neoantigens for applications in personalized immunotherapy [28, 29]. This insufficiency probably results from the influence of other factors including the preprocessing of peptides, the stability of the pMHC complex [106, 107], and peptide immunogenicity [108]. Recently published MHC binding algorithms use either only peptidome data, generated from in vivo immunoprecipitation of pMHC complexes followed by MS characterization, or an integration of MS and binding-affinity data [87, 102, 104]. By directly examining ligands that are eluted from pMHC complexes identified in vivo, predictive models can capture features unique to peptides that have undergone the entire processing pathway. Over 150 HLA alleles have corresponding binding-affinity datasets available in IEDB (with highly variable amounts of data for each allele) [96]. By contrast, MS peptidome datasets are available for only approximately 55 HLA alleles [87], probably because of the lack of high-throughput characterization assays. However, continuous development in MS profiling techniques [109] may soon close the gap between the two types of data. Zhao and Sher [110] recently performed systematic benchmarking for 12 of the most popular pMHC class I binding predictors, with NetMHCpan4 and MHCflurry determined to have the highest accuracy in binding versus non-binding classifications. The analysis also revealed that the incorporation of peptide elution data from MS experiments has indeed improved the accuracy of recent predictors when evaluated using high-quality naturally presented peptides [110].
Different types of algorithmic approaches have been used to model and make predictions for the binding affinity of MHC class I molecules. Initially, predictors relied on linear regression algorithms and more specifically on stabilized matrix methods, such as SMM [111], SMMPMBEC [112], and Pickpocket [113]. However, recently published or updated predictors almost exclusively employ variations of neural networks [87, 102, 104, 114], as shown in Table 3. Linear regression assumes a linear contribution of individual residues to the overall binding affinity; however, while artificial neural networks require more training data, they are able to capture the nonlinear relationship between the peptide sequence and the binding affinity for the corresponding MHC molecules through hidden layers in their network architecture. Given the growing number of available training datasets, applications of artificial neural networks have been able to achieve higher accuracy than that provided by linear regression predictive methods [110].
While prediction algorithms for MHC class I molecules are well developed, algorithms for MHC class II are fewer, less recently developed, and trained with smaller datasets. Unlike MHC class I molecules, class II molecules are heterodimeric glycoproteins that include an ɑ-chain and a β-chain; thus, MHC II molecules are more variable than MHC I molecules as a result of the dimerization of highly polymorphic alpha and beta chains. The binding pocket for class II molecules is open on both ends, which allows a larger range of peptides to bind. The most frequently observed lengths of peptides that bind to class II MHCs are between 13 and 25 amino acids [115], whereas those for class I typically fall between 8 and 15 amino acids [87]. Nevertheless, for any one particular MHC allele, the preferred number of amino acids may be much more constrained to one or two lengths. Algorithms built for class II predictions generally rely on matrix-based methods and ensembles of artificial networks. A selection of popular MHC class II binding prediction algorithms are summarized in Table 1 [116].
There is an extensive list of MHC binding prediction tools for both class I and class II molecules, but there remains a need not only to expand the training data for a larger range of HLA alleles but also to refine the type of training data being used in these algorithms. Although in vivo MS data capture the features of peptides that are naturally presented by MHC molecules, they cannot confirm whether such peptides are able to induce an immune response. Algorithms should ideally incorporate experimentally and clinically validated immunogenic peptides in their training and validation datasets. As ongoing neoantigen clinical trials produce more of such data, tool development and refinement in this area will also become possible.
Neoantigen prioritization and vaccine design pipelines
Owing to the numerous factors that are involved in the process of antigen generation, processing, binding, and recognition, a number of bioinformatic pipelines have emerged with the goal of assembling the available tools in order to streamline the neoantigen identification process for different clinical purposes (such as predicting the response to ICB, designing peptide- or vector-based vaccines, and so on). Table 1 includes a selection of these pipelines and Table 2 provides extensive practical guidance for their use in clinical studies. These pipelines address multiple factors that should be given careful consideration when attempting to predict neoantigens for effective cancer treatments. These considerations include: the use of multiple binding prediction algorithms (variability among binding predictions); the integration of both DNA and RNA data (expression of neoantigen candidate genes or transcripts and expression of variant alleles); the phasing of variants (proximal variants detected on the same allele will influence neoantigen sequences) [32, 117]; the interpretation of variants in the context of clonality or heterogeneity [118]; the HLA expression and somatic mutations of patient tumors; and the prediction of tumor immunogenicity [119, 120]. These pipelines are able to provide a comprehensive summary of critical information for each neoantigen prediction, including: variant identity (genomic coordinates, ClinGen allele registry ID, and Human Genome Variation Society (HGVS) variant name); predicted consequence of the variant on the amino acid sequence; corresponding gene and transcript identifiers; peptide sequence; position of the variant within the candidate neoantigen peptide; binding affinity predictions for mutant peptides and the corresponding wild-type peptide sequences; agretopicity value (mutant versus wild-type peptide binding affinity) [121]; DNA variant allele frequency (VAF); RNA VAF; and gene expression values for the gene harboring the variant. Additional data on whether peptides are generated from oncogenic genes, peptide stability, peptide processing and cleavage, and peptide manufacturability should also be considered for final assessment of neoantigens (Table 2).
Several pipelines attempt to integrate DNA and RNA sequencing data by evaluating the VAFs and the gene or transcript expression values of the mutations. Most pipelines currently take into account SNVs and indels, with only a subset considering gene fusion events [8, 32, 122]. Consistent use of the same build or assembly of the genome throughout analysis pipelines, as well as an emphasis on quality control (QC) when performing variant detection and expression analysis, is important for ensuring high confidence in the variants that are detected (Table 2). Once the mutations are confirmed to exist and be expressed, the pipelines then generate a list of neoantigen candidates and consider the probability of cleavage, the location of cleavage, and the TAP transport efficiency of each candidate [8, 123, 124]. The binding affinities of the peptides to the patient-specific MHC molecules are subsequently predicted by using one or more algorithms (Table 1). However, binding-affinity predictions that are made by multiple prediction algorithms vary, and best practices for determining a consensus are poorly articulated at this time. Furthermore, the gold-standard independent validation datasets that exist to evaluate the accuracy of divergent predictions are limited. It remains to be determined whether combining multiple prediction algorithms increases the true positive rate of neoantigen predictions. Some pipelines also consider: (i) manufacturability by measuring peptide characteristics [9]; (ii) immunogenicity by comparing either self-antigens defined by the reference or by the wild-type proteome or known epitopes from viruses and bacteria provided by IEDB [119]; and (iii) pMHC stability [8, 107].
Pipelines vary in their choices of how to rank neoantigens and which specific type of algorithm to use when performing such calculations. Thus, a major challenge lies in how each component should be weighted to create an overall ranking of neoantigens in terms of their potential effectiveness. Kim et al. [125] have attempted to capture the contributions of nine immunogenicity features through the training of machine-learning-based classifiers. Nevertheless, high-quality and experimentally validated neoantigens for training such models remain extremely sparse. In other words, there is no consensus on the features of a ‘good’ neoantigen that would be capable of inducing T cell responses in patients. Furthermore, clinicians may need to consider customized filtering and ranking criteria for individual patient cases, tumor types, or clinical trial designs, details that are not well supported by the existing pipelines. For these reasons, clinical trial efforts should establish an interdisciplinary team of experts analogous to a molecular tumor board for formal quantitative and qualitative review of each patient’s neoantigens. Pipelines such as pVACtools and Vaxrank are designed to support such groups, but there are many important areas in current pipelines that could be improved upon, including: i) consideration of whether the mutation is located within anchor residues for each HLA allele; ii) somatic mutation and expression of patient-specific HLA alleles; iii) the expression level of important cofactors such as genes that are involved in processing, binding, and presentation; and iv) additional factors that influence the manufacturing and delivery of the predicted neoantigens.
Peptide creation, delivery mechanisms, and related analysis considerations for vaccine design
Once neoantigen prioritization is complete, personalized vaccines are designed from predicted immunogenic candidate sequences. Multiple delivery mechanisms exist for use in clinical trials; these include synthetic peptides, DNA, mRNA, viral vectors, and ex-vivo-loaded dendritic cell vaccines [126, 127]. Cancer vaccine delivery is an extensive topic beyond the scope of this review, but other reviews discuss this topic in detail [126,127,128]. Once a mechanism is chosen and the vaccine is delivered to the patient, professional APCs endocytose the neoantigen sequences. Then, they are processed to generate class-I- and II-restricted MHC peptides for presentation and T cell activation. To design a successful delivery vector, additional analysis steps are necessary to assess peptide manufacturability and to avoid potential incidental DNA vector junctional epitope sequences, or junctions spanning neoantigen sequences that create unintended immunogenic epitopes [8, 129].
Synthetic long peptides (SLPs) are an effective neoantigen delivery mechanism in personalized immunotherapy preclinical studies and clinical trials [30, 101, 130, 131]. These peptides are created from sequences of 15–30 amino acids that contain a core predicted neoantigen. SLPs have greater efficacy than short synthetic peptides, of 8–11 amino acids, because longer peptides require internalization and processing by professional APCs, whereas short peptides can induce immunological tolerance by binding directly to MHC-I on non-professional APCs [132,133,134]. One limitation of SLPs is manufacturability. Certain chemical properties of the amino acid sequence can make peptides difficult to synthesize, and longer peptides can encounter solubility problems (i.e., they become insoluble). Vaxrank [9] aims to address these concerns by incorporating a manufacturability prediction step in the neoantigen prioritization pipeline. This step measures nine properties that contribute to manufacturing difficulty, including the presence of hydrophobic sequences, cysteine residues, and asparagine-proline bonds. The algorithm then uses this information to choose an ideal window surrounding the somatic mutation for optimum synthesis.
DNA vectors have also delivered neoantigens successfully in a recent preclinical study [135], and DNA neoantigen vaccine clinical trials are currently ongoing in pancreatic and triple-negative breast cancer [136]. Neoantigen encoding DNA sequences can be either directly injected via plasmid vectors using electroporation or incorporated into viral vectors for delivery into patient cells. Adenovirus and vaccinia are the most common viral vectors for personalized vaccines; both are double-stranded DNA (dsDNA) viruses that can incorporate foreign DNA [137]. To maximize neoantigen effectiveness for both vectors, researchers must design sequences with effective junctions and/or spacers. This ensures correct cleavage of the combined sequence by the proteasome as well as the avoidance of inadvertent immunogenic junction antigens. Multiple methods exist to address these challenges.
Furin is a peptidase in the trans-Golgi network that cleaves immature proteins at sequence-specific motifs [138]. Recently, furin-sensitive cleavage sequences were incorporated into a neoantigen DNA vaccine to cleave the sequence into functional neoantigens [135]. EpiToolKit [123] addresses incorrect peptide cleavage in its pipeline by incorporating NetChop [89]. This tool predicts the proteasomal cleavage sites for each neoantigen and can be used to exclude candidates that would undergo inappropriate cleavage. pVACvector, an algorithm included in pVACtools [8], optimizes neoantigen sequence order by running pVACseq on the junction sequences and prioritizing those with low immunogenicity. If high junction immunogenicity cannot be avoided, spacer sequences are included to decrease the potential for inadvertent neoantigens. Taking such analytical considerations into account during personalized vaccine design ensures maximum treatment efficacy in patients.
T cell recognition, TCR profiling, and immune cell profiling to evaluate response
The ultimate objective of introducing a neoantigen-derived vaccine is to elicit and/or expand a tumor-specific T cell response. This can be evaluated by experimental methods that measure T cell activation and activity, or by computational methods that characterize the patient’s TCR repertoire prior to and after immunotherapy. Standard methods such as IFN-γ ELISPOT assays [139] or MHC multimer assays [140] are beyond the scope of this review, but have been used widely for neoantigen validation purposes [28, 141]. T cells individually undergo complex combinatorial rearrangements in the T cell receptor gene loci in order to create unique clonotypes that are responsible for recognizing antigens. This process occurs within the V(D) J region of the gene, particularly the complementarity-determining region 3 (CDR3), which encodes a region of the TCR that is important for recognizing the pMHC complex. Thus, attempts to characterize the TCR repertoire focus on the identification and characterization of CDR3 sequences, which are representative of the unique T cell clones. This process, termed TCR clonotyping, has been used to identify clonal T cell responses to neoantigens following vaccination with a personalized cancer vaccine or after checkpoint blockade therapy [28]. Researchers have also established an association between the size and diversity of a patient’s TCR repertoire and their response to cancer immunotherapies [142]. Changes in the clonality and diversity of the TCR repertoire, observed from either peripheral blood or tumor-infiltrating lymphocytes (TIL), suggest that an antitumor T cell response is occurring, but they are global metrics that do not successfully identify the T cell clonotypes responsible for tumor rejection.
A variety of available technologies and tools allow sequencing and subsequent analysis of the TCR repertoire. Commercial services such as Adaptive, ClonTech, and iRepertoire differ in a number of aspects, including the required starting material, their library preparation methods, the targeted TCR chains and/or CDR regions for sequencing, the supported organisms, and the sequencing platforms used [143]. Several tools exist to identify TCR CDR3 sequences using various types of data, such as output data from focused assays (e.g., Adaptive, ClonTech or CapTCR), bulk tumor RNA-seq [144], and single cell RNA-seq [144, 145], particularly from the TCR alpha and beta genes (TRA, TRB). Challenges associated with TCR profiling include the diversity of the repertoire itself, correctly determining the pairing of TRA and TRB clonotypes, and the subsequent analysis or validation necessary to pair T cell clones with their target neoantigens. Studies have quantified or predicted the T cell richness, or total number of T cell clones, in the peripheral blood of a healthy individual as up to 1019 cells [146]. Thus, there is a sampling bias—based upon the blood draw that was taken, the sample used for sequencing, and the input material for library preparation—that prevents complete evaluation of the global T cell repertoire.
TCR profiling requires the alignment of sequencing reads to the reference TCR genes and the assembly of the rearranged clonotypes. MixCR has been used for TCR alignment and assembly in both bulk and single-cell methods [144, 147]. MIGEC [148] is utilized for methods involving the use of unique molecular identifiers, whereas TraCeR is designed specifically for single-cell methods [145]. MiXCR recovers TCR sequences from raw data through alignment and subsequent clustering, which allows the grouping of identical sequences into clonotypes. If sequences are generated from bulk material (e.g., whole blood or bulk TIL), TRA and TRB sequences cannot be paired to define the T cell clonotypes specifically. They may be inferred on the basis of frequency, but due to the very high diversity of the T cell repertoire, there are often many clonotypes at similar or low frequencies that make deconvolution of TRA–TRB pairs difficult. With the advent of single-cell sequencing data, tools such as TraCeR are now able to identify paired alpha–beta sequences within individual cells that have the same receptor sequences and thus have been derived from the same clonally expanded cells [145].
The identification of clonally expanded neoantigen-specific TCRs complements neoantigen prediction and characterization by indicating whether an active T cell response has been stimulated by an immunotherapeutic intervention. Lu et al. [149] recently developed a single cell RNA-seq approach that identifies neoantigen-specific TCRs by culturing TILs with tandem minigene (TMG)-transfected or peptide-pulsed autologous APCs. Experimental validation data for individual neoantigens can then be utilized to train and improve current neoantigen prioritization strategies.
The clonality of the TCR repertoire can be further evaluated to identify T cell clones that may recognize the same neoantigen. Studies have identified oligoclonal T cell populations that converge, with consistent CDR3 motif sequences, to recognize the same neoantigen [150]. Taking into account the diversity of the repertoire, these findings suggest that oligoclonal events are more likely than monoclonal events, and that there is not likely to be one-to-one mapping between T cell clones and neoantigens. Oligoclonal events and the convergence of the T cell repertoire can be better studied with tools such as GLIPH, which was developed to identify consistent CDR3 motifs across [151] T cells in bulk TCR sequencing.
Antitumor T cell responses have been correlated with changes in the infiltrating immune microenvironment. Methods such as CIBERSORT have been developed to characterize cell compositions on the basis of gene expression profiles from tumor samples [152]. Association between immune cell infiltrates and various factors, including somatic mutation, copy number variation, and gene expression, can be explored interactively through TIMER [153]. This topic has been reviewed in more depth elsewhere [154]. A larger selection of available tools related to T cell and immune cell profiling are listed in Table 1. Overall, few studies have focused on the integration of T cell profiling with neoantigen detection, with the exception of that reported in Li et al. [155], in which TCR clones that were identified from RNAseq samples across Cancer Genome Atlas samples were compared to the mutational profiles of tumors, successfully identifying several public neoantigens that are shared across individuals. Owing to the limited availability of peripheral blood samples and TCR sequencing data with matched tumor DNA or RNA sequencing, one major area for development in the field remains the aggregation of these data and the introduction of an appropriate supervised approach to identify TCR–neoantigen pairs. Such progress would leverage the available data to enhance the identification of neoantigens and to optimize personalized medicine approaches for cancer immunotherapy.
Conclusions and future directions
Great strides have been made in developing pipelines for neoantigen identification, but there is significant room for improvement. Tools for the rational integration of the myriad complex factors described above are needed. In some cases, useful tools exist but have not been incorporated into analysis workflows. In other cases, factors we believe are important are not being considered because of a lack of tools.
Variant types beyond SNVs and indels have been confirmed as neoantigen sources, but there remains little support for them in current pipelines. Fusions have recently been incorporated into pipelines such as pVACfuse (a tool within pVACtools [8]), INTEGRATE-neo [32], and NeoepitopePred [122]. However, additional genomic variant types that lead to alternative isoforms and to the expression of normally non-coding genomic regions remain unsupported, despite preliminary analyses suggesting their importance. An additional orthogonal, but poorly supported, neoantigen source is the proteasome, which was found to be capable of creating novel antigens by splicing peptides from diverse proteins to create a single antigen [156]. Several computational tools exist to predict post-translational modifications and alternative translation events from sequencing data, such as GPS [157] and KinasePhos [158] for phosphorylation events and altORFev [159] for alternative ORFs. To determine the immunogenicity of these alternative peptides, any tumor-specific predicted sequences could be input into neoantigen prediction software.
The low accuracy of class II HLA typing algorithms has impeded extensive class II neoantigen prediction. When clinical class II HLA typing data are available, they should be used in place of computational HLA predictions in pipelines to improve prediction reliability. In addition, although somatic alterations in HLA gene loci and in the antigen presentation machinery have been implicated in immunotherapeutic resistance, these properties have not been leveraged in predicting neoantigen candidates. HLA gene expression is more often summarized at the gene rather than the allele level. Furthermore, HLA expression is commonly determined from bulk tumor RNAseq data, which are derived from normal, stromal, and infiltrating immune cells, all of which may express HLA genes. The relationship between the present HLA alleles and a predicted neoantigen profile has not been studied, and it remains to be seen whether neoantigens that are restricted to absent or mutant HLA alleles should be specifically filtered out.
For the neoantigen prediction step, mutation positions in the neoantigen should be carefully considered if they occur in anchor residues, since the core sequence of these peptides would be unaffected and identical to that of the wild-type protein. There is also a bias towards class I neoantigen prediction because there are fewer binding-affinity training data and fewer algorithms for class II neoantigens because of their increased MHC binding complexity. Studies have also shown low consensus across MHC binding predictors [8]. pVACtools [8] addresses this challenge by running multiple algorithms simultaneously and reporting the lowest or median score, but a more definitive method for obtaining a binding-affinity consensus remains to be developed. Neoantigen prediction pipelines could also benefit from the inclusion of information on the proposed delivery mechanism to improve prioritization and to streamline vaccine creation.
Although TCR sequences have been recognized to be highly polymorphic, TCRs from T cells that recognize the same pMHC epitope may share conserved sequence features. Researchers have started to quantify these predictive features with the hope of modeling epitope–TCR specificity [160]. Multiple tools (such as TCRex, NetTCR, Repitope) now attempt to predict epitope–TCR binding when given specific TCR sequences. By taking into account the binding specificity of the patient’s existing TCR sequences, neoantigen candidates can be further prioritized according to their immunogenicity. A major advance in optimizing treatment strategies may require the integration of pipelines that perform all of the steps necessary for the generation and processing of neoantigens and for the identification of T cell clones that efficiently recognize them.
Implementing a set of best practices to predict high-quality immunogenic neoantigens can lead to improved personalized patient care in the clinic. Predicting and prioritizing neoantigens is, however, a complicated process that involves many computational steps, each with individualized, adjustable parameters (we provide a specific end-to-end workflow based on our current practices at https://pmbio.org/). Given this complexity, the review of candidates by an immunogenomics tumor board with diverse expertise is highly recommended. We have outlined each step in the neoantigen workflow with human clinical trials in mind, but further research is needed in model organisms to facilitate the development of immunotherapies for human use. Improving neoantigen characterization tools to support the in silico modeling of immune response, model organism systems, human derived samples, and human patient trials is an essential step for improving patient response rates across cancer types.
Abbreviations
- APC:
-
Antigen presenting cell
- CDR3:
-
Complementarity-determining region 3
- FFPE:
-
Formalin-fixed paraffin-embedded
- HLA:
-
Human leukocyte antigen
- ICB:
-
Immune checkpoint blockade
- IEDB:
-
Immune Epitope Database
- Indel:
-
Insertion and deletion
- MHC:
-
Major histocompatibility complex
- MS:
-
Mass spectrometry
- MSI-H:
-
Microsatellite instability-high
- NGS:
-
Next generation sequencing
- ORF:
-
Open reading frame
- pMHC:
-
Peptide-loaded MHC
- QC:
-
Quality control
- RNA-seq:
-
RNA sequencing
- SNV:
-
Single nucleotide variant
- SLP:
-
Synthetic long peptides
- TCR:
-
T cell receptor
- TAP:
-
Transporter associated with antigen processing
- TIL:
-
Tumor-infiltrating lymphocytes
- VAF:
-
Variant allele frequency
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequencing
References
Zitvogel L, Apetoh L, Ghiringhelli F, André F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? J Clin Invest. 2008;118:1991–2001.
Medler TR, Cotechini T, Coussens LM. Immune response to cancer therapy: mounting an effective antitumor response and mechanisms of resistance. Trends Cancer Res. 2015;1:66–75.
Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, et al. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci U S A. 2005;102:16013–8.
Robbins PF, Lu Y-C, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19:747–52.
van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31:e439–42.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–11.
Hundal J, Kiwala S, McMichael J, Miller CA, Wollam AT, Xia H, et al. pVACtools: a computational toolkit to select and visualize cancer neoantigens. bioRxiv. 2018. https://doi.org/10.1101/501817.
Rubinsteyn A, Hodes I, Kodysh J, Hammerbacher J. Vaxrank: a computational tool for designing personalized cancer vaccines. bioRxiv. 2017. https://doi.org/10.1101/142919.
Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125:3413–21.
Liu XS, Mardis ER. Applications of immunogenomics to cancer. Cell. 2017;168:600–12.
Lee C-H, Yelensky R, Jooss K, Chan TA. Update on tumor neoantigens and their utility: why it is good to be different. Trends Immunol. 2018;39:536–48.
Guo Q, Wang J, Xiao J, Wang L, Hu X, Yu W, et al. Heterogeneous mutation pattern in tumor tissue and circulating tumor DNA warrants parallel NGS panel testing. Mol Cancer. 2018;17:131.
Oh E, Choi Y-L, Kwon MJ, Kim RN, Kim YJ, Song J-Y, et al. Comparison of accuracy of whole-exome sequencing with formalin-fixed paraffin-embedded and fresh frozen tissue samples. PLoS One. 2015;10:e0144162.
Robbe P, Popitsch N, Knight SJL, Antoniou P, Becq J, He M, et al. Clinical whole-genome sequencing from routine formalin-fixed, paraffin-embedded specimens: pilot study for the 100,000 genomes project. Genet Med. 2018;20:1196–205.
Griffith M, Miller CA, Griffith OL, Krysiak K, Skidmore ZL, Ramu A, et al. Optimizing cancer genome sequencing and analysis. Cell Syst. 2015;1:210–23.
Fennemann FL, de Vries IJM, Figdor CG, Verdoes M. Attacking tumors from all sides: personalized multiplex vaccines to tackle intratumor heterogeneity. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2019.00824.
Xu C. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput Struct Biotechnol J. 2018;16:15–24.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213.
Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor—normal sample pairs. Bioinformatics. 2012;28:1811–7.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76.
Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–7.
Wang Q, Jia P, Li F, Chen H, Ji H, Hucks D, et al. Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med. 2013;5:91.
Xu H, DiCarlo J, Satya RV, Peng Q, Wang Y. Comparison of somatic mutation calling methods in amplicon and whole exome sequence data. BMC Genomics. 2014;15:244.
Callari M, Sammut S-J, De Mattos-Arruda L, Bruna A, Rueda OM, Chin S-F, et al. Intersect-then-combine approach: improving the performance of somatic variant calling in whole exome sequencing data using multiple aligners and callers. Genome Med. 2017;9:35.
Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92.
Barnell EK, Ronning P, Campbell KM, Krysiak K, Ainscough BJ, Sheta LM, et al. Standard operating procedure for somatic variant refinement of sequencing data with paired tumor and normal samples. Genet Med. 2019;21:972–81.
Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803–8.
Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222.
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217.
Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017;18:1009–21.
Zhang J, Mardis ER, Maher CA. INTEGRATE-neo: a pipeline for personalized gene fusion neoantigen discovery. Bioinformatics. 2017;33:555–7.
Yang W, Lee K-W, Srivastava RM, Kuo F, Krishna C, Chowell D, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25:767–75.
Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, et al. Intron retention is a source of neoepitopes in cancer. Nat Biotechnol. 2018;36:1056–8.
Laumont CM, Vincent K, Hesnard L, Audemard É, Bonneil É, Laverdure J-P, et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med. 2018;10. https://doi.org/10.1126/scitranslmed.aau5516.
Kahles A, Lehmann K-V, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. 2018;34:211–24.
Khodadoust MS, Olsson N, Wagar LE, Haabeth OAW, Chen B, Swaminathan K, et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature. 2017;543:723–7.
Smith CC, Selitsky SR, Chai S, Armistead PM, Vincent BG, Serody JS. Alternative tumour-specific antigens. Nat Rev Cancer. 2019;19:465–78.
Maby P, Galon J, Latouche J-B. Frameshift mutations, neoantigens and tumor-specific CD8(+) T cells in microsatellite unstable colorectal cancers. Oncoimmunology. 2016;5:e1115943.
Shiraishi Y, Sato Y, Chiba K, Okuno Y, Nagata Y, Yoshida K, et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013;41:e89.
Bohnert R, Vivas S, Jansen G. Comprehensive benchmarking of SNV callers for highly admixed tumor data. PLoS One. 2017;12:e0186175.
Krøigård AB, Thomassen M, Lænkholm A-V, Kruse TA, Larsen MJ. Evaluation of nine somatic variant callers for detection of somatic mutations in exome and targeted deep sequencing data. PLoS One. 2016;11:e0151664.
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71.
Kuijjer ML, Hogendoorn PCW, Cleton-Jansen A-M. Genome-wide analyses on high-grade osteosarcoma: making sense of a genomically most unstable tumor. Int J Cancer. 2013;133:2512–21.
Rathe SK, Popescu FE, Johnson JE, Watson AL, Marko TA, Moriarity BS, et al. Identification of candidate neoantigens produced by fusion transcripts in human osteosarcomas. Sci Rep. 2019;9:358.
Mansfield AS, Peikert T, Smadbeck JB, Udell JBM, Garcia-Rivera E, Elsbernd L, et al. Neoantigenic potential of complex chromosomal rearrangements in mesothelioma. J Thorac Oncol. 2019;14:276–87.
Melsted P, Hateley S, Joseph IC, Pimentel H, Bray NL, Pachter L. Fusion detection and quantification by pseudoalignment. bioRxiv. 2017. https://doi.org/10.1101/166322.
Haas B, Dobin A, Stransky N, Li B, Yang X, Tickle T, et al. STAR-fusion: fast and accurate fusion transcript detection from RNA-Seq. bioRxiv. 2017. https://doi.org/10.1101/120295.
Davidson NM, Majewski IJ, Oshlack A. JAFFA: high sensitivity transcriptome-focused fusion gene detection. Genome Med. 2015;7:43.
Zhao J, Chen Q, Wu J, Han P, Song X. GFusion: an effective algorithm to identify fusion genes from cancer RNA-Seq data. Sci Rep. 2017;7:6880.
Zhang J, White NM, Schmidt HK, Fulton RS, Tomlinson C, Warren WC, et al. INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res. 2016;26:108–18.
Liu S, Tsai W-H, Ding Y, Chen R, Fang Z, Huo Z, et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2016;44:e47.
Laumont CM, Perreault C. Exploiting non-canonical translation to identify new targets for T cell-based cancer immunotherapy. Cell Mol Life Sci. 2018;75:607–21.
Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, et al. Intron retention as a novel source of cancer neoantigens. bioRxiv. 2018. https://doi.org/10.1101/309450.
Jayasinghe RG, Cao S, Gao Q, Wendl MC, Vo NS, Reynolds SM, et al. Systematic analysis of splice-site-creating mutations in cancer. Cell Rep. 2018;23:270–81.
Khodadoust MS, Olsson N, Chen B, Sworder B, Shree T, Liu CL, et al. B-cell lymphomas present immunoglobulin neoantigens. Blood. 2019;133:878–81.
Cobbold M, De La Pena H, Norris A, Polefrone JM, Qian J, English AM, et al. MHC class I-associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci Transl Med. 2013;5:203ra125.
Malaker SA, Penny SA, Steadman LG, Myers PT, Loke JC, Raghavan M, et al. Identification of glycopeptides as posttranslationally modified neoantigens in leukemia. Cancer Immunol Res. 2017;5:376–84.
Ronsin C, Chung-Scott V, Poullion I, Aknouche N, Gaudin C, Triebel F. A non-AUG-defined alternative open reading frame of the intestinal carboxyl esterase mRNA generates an epitope recognized by renal cell carcinoma-reactive tumor-infiltrating lymphocytes in situ. J Immunol. 1999;163:483–90.
Wang RF, Johnston SL, Zeng G, Topalian SL, Schwartzentruber DJ, Rosenberg SA. A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J Immunol. 1998;161:3598–606.
Rosenberg SA, Tong-On P, Li Y, Riley JP, El-Gamil M, Parkhurst MR, et al. Identification of BING-4 cancer antigen translated from an alternative open reading frame of a gene in the extended MHC class II region using lymphocytes from a patient with a durable complete regression following immunotherapy. J Immunol. 2002;168:2402–7.
Welters MJP, Kenter GG, Piersma SJ, Vloon APG, Löwik MJG. Berends-van der Meer DMA, et al. induction of tumor-specific CD4+ and CD8+ T-cell immunity in cervical cancer patients by a human papillomavirus type 16 E6 and E7 long peptides vaccine. Clin Cancer Res. 2008;14:178–87.
Kenter GG, Welters MJP, Valentijn ARPM, Lowik MJG. Berends-van der Meer DMA, Vloon APG, et al. vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838–47.
Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SGE. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 2015;43:D423–31.
Bunce M, Passey B. HLA typing by sequence-specific primers. Methods Mol Biol. 2013;1034:147–59.
Cereb N, Kim HR, Ryu J, Yang SY. Advances in DNA sequencing technologies for high resolution HLA typing. Hum Immunol. 2015;76:923–7.
Bauer DC. Zadoorian a, Wilson LOW, Melbourne genomics health Alliance, Thorne NP. Evaluation of computational programs to predict HLA genotypes from genomic sequencing data. Brief Bioinform. 2018;19:179–87.
Kiyotani K, Mai TH, Nakamura Y. Comparison of exome-based HLA class I genotyping tools: identification of platform-specific genotyping errors. J Hum Genet. 2017;62:397–405.
Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 2014;30:3310–6.
Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33:1152–8.
Bai Y, Wang D, Fury W. PHLAT: inference of high-resolution HLA types from RNA and whole exome sequencing. Methods Mol Biol. 1802;2018:193–201.
Nariai N, Kojima K, Saito S, Mimori T, Sato Y, Kawai Y, et al. HLA-VBSeq: accurate HLA typing at full resolution from whole-genome sequencing data. BMC Genomics. 2015;16(Suppl 2):S7.
Boegel S, Löwer M, Schäfer M, Bukur T, de Graaf J, Boisguérin V, et al. HLA typing from RNA-Seq sequence reads. Genome Med. 2012;4:102.
Xie C, Yeo ZX, Wong M, Piper J, Long T, Kirkness EF, et al. Fast and accurate HLA typing from short-read next-generation sequence data with xHLA. Proc Natl Acad Sci U S A. 2017;114:8059–64.
Kawaguchi S, Higasa K, Shimizu M, Yamada R, Matsuda F. HLA-HD: an accurate HLA typing algorithm for next-generation sequencing data. Hum Mutat. 2017;38:788–97.
Paulson KG, Voillet V, McAfee MS, Hunter DS, Wagener FD, Perdicchio M, et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun. 2018;9:3868.
Paulson KG, Tegeder A, Willmes C, Iyer JG, Afanasiev OK, Schrama D, et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol Res. 2014;2:1071–9.
Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60.
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25.
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9.
McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171:1259–71.
Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 2018;8:730–49.
Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017;7:1420–35.
Romero JM, Jiménez P, Cabrera T, Cózar JM, Pedrinaci S, Tallada M, et al. Coordinated downregulation of the antigen presentation machinery and HLA class I/β2-microglobulin complex is responsible for HLA-ABC loss in bladder cancer. Int J Cancer. 2005;113:605–10.
Leone P, Shin E-C, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst. 2013;105:1172–87.
Liu Y, Komohara Y, Domenick N, Ohno M, Ikeura M, Hamilton RL, et al. Expression of antigen processing and presenting molecules in brain metastasis of breast cancer. Cancer Immunol Immunother. 2012;61:789–801.
Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol. 2017;199:3360–8.
Calis JJA, Reinink P, Keller C, Kloetzel PM, Keşmir C. Role of peptide processing predictions in T cell epitope identification: contribution of different prediction programs. Immunogenetics. 2015;67:85–93.
Nielsen M, Lundegaard C, Lund O, Keşmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57:33–41.
Tenzer S, Peters B, Bulik S, Schoor O, Lemmel C, Schatz MM, et al. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol Life Sci. 2005;62:1025–37.
Hoze E, Tsaban L, Maman Y, Louzoun Y. Predictor for the effect of amino acid composition on CD4+ T cell epitopes preprocessing. J Immunol Methods. 2013;391:163–73.
Paul S, Karosiene E, Dhanda SK, Jurtz V, Edwards L, Nielsen M, et al. Determination of a predictive cleavage motif for eluted major histocompatibility complex class II ligands. Front Immunol. 2018;9:1795.
Bhasin M, Lata S, Raghava GPS. TAPPred prediction of TAP-binding peptides in antigens. Methods Mol Biol. 2007;409:381–6.
Bhasin M, Raghava GPS. Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Sci. 2004;13:596–607.
Peters B, Bulik S, Tampe R, Van Endert PM, Holzhütter H-G. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J Immunol. 2003;171:1741–9.
Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2019;47:D339–43.
Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. 2013;31:443–73.
Unanue ER, Turk V, Neefjes J. Variations in MHC class II antigen processing and presentation in health and disease. Annu Rev Immunol. 2016;34:265–97.
Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015;21:81–5.
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4 T cells in a patient with epithelial cancer. Science. 2014;344:641–5.
Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–6.
O’Donnell TJ, Rubinsteyn A, Bonsack M, Riemer AB, Laserson U, Hammerbacher J. MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. 2018;7:129–32.
Phloyphisut P, Pornputtanapong N, Sriswasdi S, Chuangsuwanich E. MHCSeqNet: a deep neural network model for universal MHC binding prediction. bioRxiv. 2018. https://doi.org/10.1101/371591.
Bulik-Sullivan B, Busby J, Palmer CD, Davis MJ, Murphy T, Clark A, et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat Biotechnol. 2018. https://doi.org/10.1038/nbt.4313.
Little AM, Parham P. Polymorphism and evolution of HLA class I and II genes and molecules. Rev Immunogenet. 1999;1:105–23.
Harndahl M, Rasmussen M, Roder G, Pedersen ID, Sørensen M, Nielsen M, et al. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur J Immunol. 2012;42:1405–16.
Rasmussen M, Fenoy E, Harndahl M, Kristensen AB, Nielsen IK, Nielsen M, et al. Pan-specific prediction of peptide–MHC class I complex stability, a correlate of T cell immunogenicity. J Immunol. 2016;197:1517–24.
Calis JJA, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A, et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol. 2013;9:e1003266.
Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. 2017;46:315–26.
Zhao W, Sher X. Systematically benchmarking peptide-MHC binding predictors: from synthetic to naturally processed epitopes. PLoS Comput Biol. 2018;14:e1006457.
Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics. 2007;8:238.
Kim Y, Sidney J, Pinilla C, Sette A, Peters B. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics. 2009;10:394.
Zhang H, Lund O, Nielsen M. The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC-peptide binding. Bioinformatics. 2009;25:1293–9.
Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32:511–7.
Chicz RM, Urban RG, Lane WS, Gorga JC, Stern LJ, Vignali DAA, et al. Predominant naturally processed peptides bound to HLA-DR1 are derived from MHC-related molecules and are heterogeneous in size. Nature. 1992;358:764–8.
Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. 2018;154:394–406.
Hundal J, Kiwala S, Feng Y-Y, Liu CJ, Govindan R, Chapman WC, et al. Accounting for proximal variants improves neoantigen prediction. Nat Genet. 2019;51:175–9.
Schenck RO, Lakatos E, Gatenbee C, Graham TA, Anderson ARA. NeoPredPipe: high-throughput neoantigen prediction and recognition potential pipeline. BMC Bioinformatics. 2019;20:264.
Bjerregaard A-M, Nielsen M, Hadrup SR, Szallasi Z, Eklund AC. MuPeXI: prediction of neo-epitopes from tumor sequencing data. Cancer Immunol Immunother. 2017;66:1123–30.
Tappeiner E, Finotello F, Charoentong P, Mayer C, Rieder D, Trajanoski Z. TIminer: NGS data mining pipeline for cancer immunology and immunotherapy. Bioinformatics. 2017;33:3140–1.
Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, Pawashe AP, et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med. 2014;211:2231–48.
Chang T-C, Carter RA, Li Y, Li Y, Wang H, Edmonson MN, et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017;9:78.
Schubert B, Brachvogel H-P, Jürges C, Kohlbacher O. EpiToolKit—a web-based workbench for vaccine design. Bioinformatics. 2015;31:2211–3.
Larsen MV, Lundegaard C, Lamberth K, Buus S, Brunak S, Lund O, et al. An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol. 2005;35:2295–303.
Kim S, Kim HS, Kim E, Lee MG, Shin E-C, Paik S, et al. Neopepsee: accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann Oncol. 2018;29:1030–6.
Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359:1355–60.
Guo Y, Lei K, Tang L. Neoantigen vaccine delivery for personalized anticancer immunotherapy. Front Immunol. 2018;9:1499.
He X, Abrams SI, Lovell JF. Peptide delivery systems for cancer vaccines. Adv Ther (Weinh). 2018;1. https://doi.org/10.1002/adtp.201800060.
Sharma PK, Dmitriev IP, Kashentseva EA, Raes G, Li L, Kim SW, et al. Development of an adenovirus vector vaccine platform for targeting dendritic cells. Cancer Gene Ther. 2018;25:27–38.
Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512:324–7.
Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565:234–9.
Bijker MS, van den Eeden SJF, Franken KL, Melief CJM, van der Burg SH, Offringa R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur J Immunol. 2008;38:1033–42.
Melief CJM, van der Burg SH. Immunotherapy of established (pre) malignant disease by synthetic long peptide vaccines. Nat Rev Cancer. 2008;8:351–60.
Slingluff CL. The present and future of peptide vaccines for cancer: single or multiple, long or short, alone or in combination. Cancer J. 2011;17:343–50.
Duperret EK, Perales-Puchalt A, Stoltz R, Hiranjith GH, Mandloi N, Barlow J, et al. A synthetic DNA, multi-neoantigen vaccine drives predominately MHC class I CD8 T-cell responses, impacting tumor challenge. Cancer Immunol Res. 2019;7:174–82.
Aurisicchio L, Pallocca M, Ciliberto G, Palombo F. The perfect personalized cancer therapy: cancer vaccines against neoantigens. J Exp Clin Cancer Res. 2018;37:86.
Larocca C, Schlom J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011;17:359–71.
Tian S, Huang Q, Fang Y, Wu J. FurinDB: a database of 20-residue furin cleavage site motifs, substrates and their associated drugs. Int J Mol Sci. 2011;12:1060–5.
Slota M, Lim J-B, Dang Y, Disis ML. ELISpot for measuring human immune responses to vaccines. Expert Rev Vaccines. 2011;10:299–306.
Toebes M, Coccoris M, Bins A, Rodenko B, Gomez R, Nieuwkoop NJ, et al. Design and use of conditional MHC class I ligands. Nat Med. 2006;12:246–51.
Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. 2015;125:3981–91.
Hopkins AC, Yarchoan M, Durham JN, Yusko EC, Rytlewski JA, Robins HS, et al. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight. 2018;3. https://doi.org/10.1172/jci.insight.122092.
Rosati E, Dowds CM, Liaskou E, Henriksen EKK, Karlsen TH, Franke A. Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol. 2017;17:61.
Bolotin DA, Poslavsky S, Davydov AN, Frenkel FE, Fanchi L, Zolotareva OI, et al. Antigen receptor repertoire profiling from RNA-seq data. Nat Biotechnol. 2017;35:908–11.
Stubbington MJT, Lönnberg T, Proserpio V, Clare S, Speak AO, Dougan G, et al. T cell fate and clonality inference from single-cell transcriptomes. Nat Methods. 2016;13:329–32.
Warren RL, Freeman JD, Zeng T, Choe G, Munro S, Moore R, et al. Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res. 2011;21:790–7.
Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods. 2015;12:380–1.
Shugay M, Britanova OV, Merzlyak EM, Turchaninova MA, Mamedov IZ, Tuganbaev TR, et al. Towards error-free profiling of immune repertoires. Nat Methods. 2014;11:653–5.
Lu Y-C, Zheng Z, Robbins PF, Tran E, Prickett TD, Gartner JJ, et al. An efficient single-cell RNA-Seq approach to identify neoantigen-specific T cell receptors. Mol Ther. 2018;26:379–89.
Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375:2255–62.
Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, et al. Identifying specificity groups in the T cell receptor repertoire. Nature. 2017;547:94–8.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7.
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, et al. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77:e108–10.
Finotello F, Trajanoski Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol Immunother. 2018;67:1031–40.
Li B, Li T, Pignon J-C, Wang B, Wang J, Shukla SA, et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat Genet. 2016;48:725–32.
Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, et al. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science. 2016;354:354–8.
Xue Y, Liu Z, Cao J, Ma Q, Gao X, Wang Q, et al. GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng Des Sel. 2011;24:255–60.
Wong Y-H, Lee T-Y, Liang H-K, Huang C-M, Wang T-Y, Yang Y-H, et al. KinasePhos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res. 2007;35:W588–94.
Kochetov AV, Allmer J, Klimenko AI, Zuraev BS, Matushkin YG, Lashin SA. AltORFev facilitates the prediction of alternative open reading frames in eukaryotic mRNAs. Bioinformatics. 2017;33:923–5.
Dash P, Fiore-Gartland AJ, Hertz T, Wang GC, Sharma S, Souquette A, et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017;547:89–93.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–5.
Hansen NF, Gartner JJ, Mei L, Samuels Y, Mullikin JC. Shimmer: detection of genetic alterations in tumors using next-generation sequence data. Bioinformatics. 2013;29:1498–503.
Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R, et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016;44:e108.
Gerstung M, Beisel C, Rechsteiner M, Wild P, Schraml P, Moch H, et al. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun. 2012;3:811.
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–2.
Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84.
Jia W, Qiu K, He M, Song P, Zhou Q, Zhou F, et al. SOAPfuse: an algorithm for identifying fusion transcripts from paired-end RNA-Seq data. Genome Biol. 2013;14:R12.
Rodríguez-Martín B, Palumbo E, Marco-Sola S, Griebel T, Ribeca P, Alonso G, et al. ChimPipe: accurate detection of fusion genes and transcription-induced chimeras from RNA-seq data. BMC Genomics. 2017;18:7.
Murphy C, Elemento O. AGFusion: annotate and visualize gene fusions. bioRxiv. 2016. https://doi.org/10.1101/080903.
Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5.
Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–7.
Liu C, Yang X, Duffy B, Mohanakumar T, Mitra RD, Zody MC, et al. ATHLATES: accurate typing of human leukocyte antigen through exome sequencing. Nucleic Acids Res. 2013;41:e142.
Huang Y, Yang J, Ying D, Zhang Y, Shotelersuk V, Hirankarn N, et al. HLAreporter: a tool for HLA typing from next generation sequencing data. Genome Med. 2015;7:25.
Warren RL, Choe G, Freeman DJ, Castellarin M, Munro S, Moore R, et al. Derivation of HLA types from shotgun sequence datasets. Genome Med. 2012;4:95.
Ka S, Lee S, Hong J, Cho Y, Sung J, Kim H-N, et al. HLAscan: genotyping of the HLA region using next-generation sequencing data. BMC Bioinformatics. 2017;18:258.
Kuttler C, Nussbaum AK, Dick TP, Rammensee HG, Schild H, Hadeler KP. An algorithm for the prediction of proteasomal cleavages. J Mol Biol. 2000;298:417–29.
Karosiene E, Lundegaard C, Lund O, Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics. 2012;64:177–86.
Bhattacharya R, Tokheim C, Sivakumar A, Guthrie VB, Anagnostou V, Velculescu VE, et al. Prediction of peptide binding to MHC class I proteins in the age of deep learning. bioRxiv. 2017; https://www.biorxiv.org/content/10.1101/154757.
Andreatta M, Schafer-Nielsen C, Lund O, Buus S, Nielsen M. NNAlign: a web-based prediction method allowing non-expert end-user discovery of sequence motifs in quantitative peptide data. PLoS One. 2011;6:e26781.
Singh H, Raghava GP. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 2001;17:1236–7.
Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, Sahin U, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol. 1999;17:555–61.
Zhang L, Chen Y, Wong H-S, Zhou S, Mamitsuka H, Zhu S. TEPITOPEpan: extending TEPITOPE for peptide binding prediction covering over 700 HLA-DR molecules. PLoS One. 2012;7:e30483.
Reche PA, Glutting J-P, Reinherz EL. Prediction of MHC class I binding peptides using profile motifs. Hum Immunol. 2002;63:701–9.
Bordner AJ, Mittelmann HD. MultiRTA: a simple yet reliable method for predicting peptide binding affinities for multiple class II MHC allotypes. BMC Bioinformatics. 2010;11:482.
Shen W-J, Zhang S, Wong H-S. An effective and efficient peptide binding prediction approach for a broad set of HLA-DR molecules based on ordered weighted averaging of binding pocket profiles. Proteome Sci. 2013;11:S15.
Wood MA, Nguyen A, Struck AJ, Ellrott K, Nellore A, Thompson RF. Neoepiscope improves neoepitope prediction with multi-variant phasing. bioRxiv. 2018. https://doi.org/10.1101/418129.
Zhou Z, Lyu X, Wu J, Yang X, Wu S, Zhou J, et al. TSNAD: an integrated software for cancer somatic mutation and tumour-specific neoantigen detection. R Soc Open Sci. 2017;4:170050.
Paul S, Sidney J, Sette A, Peters B. TepiTool: a pipeline for computational prediction of T cell epitope candidates. Curr Protoc Immunol. 2016;114:18.19.1–18.19.24.
Wang T-Y, Wang L, Alam SK, Hoeppner LH, Yang R. ScanNeo: identifying indel derived neoantigens using RNA-Seq data. Bioinformatics. 2019. https://doi.org/10.1093/bioinformatics/btz193.
Bais P, Namburi S, Gatti DM, Zhang X, Chuang JH. CloudNeo: a cloud pipeline for identifying patient-specific tumor neoantigens. Bioinformatics. 2017;33:3110–2.
Yu Y, Ceredig R, Seoighe C. LymAnalyzer: a tool for comprehensive analysis of next generation sequencing data of T cell receptors and immunoglobulins. Nucleic Acids Res. 2016;44:e31.
Heiden JAV, Vander Heiden JA, Yaari G, Uduman M, Stern JNH, O’Connor KC, et al. pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics. 2014;30:1930–2.
Li B, Li T, Wang B, Dou R, Zhang J, Liu JS, et al. Ultrasensitive detection of TCR hypervariable-region sequences in solid-tissue RNA–seq data. Nat Genet. 2017;49:482–3.
Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, et al. VDJtools: unifying post-analysis of T cell receptor repertoires. PLoS Comput Biol. 2015;11:e1004503.
Bagaev DV, Zvyagin IV, Putintseva EV, Izraelson M, Britanova OV, Chudakov DM, et al. VDJviz: a versatile browser for immunogenomics data. BMC Genomics. 2016;17:453.
Morin A, Kwan T, Ge B, Letourneau L, Ban M, Tandre K, et al. Immunoseq: the identification of functionally relevant variants through targeted capture and sequencing of active regulatory regions in human immune cells. BMC Med Genet. 2016;9:59.
Finotello F, Mayer C, Plattner C, Laschober G, Rieder D, Hackl H, et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019;11:34.
Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248–62.
Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50.
Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17:13.
Zhang J, Griffith M, Miller CA, Griffith OL, Spencer DH, Walker JR, et al. Comprehensive discovery of noncoding RNAs in acute myeloid leukemia cell transcriptomes. Exp Hematol. 2017;55:19–33.
Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol Res. 2014;2:522–9.
Nunes E, Heslop H, Fernandez-Vina M, Taves C, Wagenknecht DR, Eisenbrey AB, et al. Definitions of histocompatibility typing terms. Blood. 2011;118:e180–3.
Zhang J, Caruso FP, Sa JK, Justesen S, Nam D-H, Sims P, et al. The combination of neoantigen quality and T lymphocyte infiltrates identifies glioblastomas with the longest survival. Commun Biol. 2019;2:135.
Kim Y, Sidney J, Buus S, Sette A, Nielsen M, Peters B. Dataset size and composition impact the reliability of performance benchmarks for peptide-MHC binding predictions. BMC Bioinformatics. 2014;15:241.
Acknowledgments
We are grateful to the research and clinical trial participants and their families, without whom none of this would be possible. We would like to thank Christopher Miller, Jasreet Hundal, Susanna Kiwala, Jason Walker, Thomas Mooney, Adam Coffman, Cody Ramirez, Zachary Skidmore, Michael McLellan, Michelle Becker-Hapak, Simon Goedegebuure, Tammi Vickery, Tanner Johanns, Gavin Dunn, Todd Fehniger, Antoni Ribas, Elaine Mardis, and Robert Schreiber for invaluable discussions on relevant immunology, immunogenomics, bioinformatics and analysis concepts. We also thank Joshua McMichael for guidance on the creation of Fig. 1.
Funding
OLG was supported by the NIH National Cancer Institute (U01CA209936, U01CA231844, and U24CA237719). MG was supported by the NIH National Human Genome Research Institute (R00HG007940), the NIH National Cancer Institute (U01CA209936, U24CA237719), and the V Foundation for Cancer Research.
Author information
Authors and Affiliations
Contributions
MMR, HX, KMC, OLG, and MG wrote the manuscript and prepared the figures and tables with input from WEG. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Richters, M.M., Xia, H., Campbell, K.M. et al. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med 11, 56 (2019). https://doi.org/10.1186/s13073-019-0666-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-019-0666-2