
Abstract
Background
Exome sequencing is now being incorporated into clinical care for pediatric and adult populations, but its integration into prenatal diagnosis has been more limited. One reason for this is the paucity of information about the clinical utility of exome sequencing in the prenatal setting.
Methods
We retrospectively reviewed indications, results, time to results (turnaround time, TAT), and impact of exome results for 146 consecutive “fetal exomes” performed in a clinical diagnostic laboratory between March 2012 and November 2017. We define a fetal exome as one performed on a sample obtained from a fetus or a product of conception with at least one structural anomaly detected by prenatal imaging or autopsy. Statistical comparisons were performed using Fisher’s exact test.
Results
Prenatal exome yielded an overall molecular diagnostic rate of 32% (n = 46/146). Of the 46 molecular diagnoses, 50% were autosomal dominant disorders (n = 23/46), 41% were autosomal recessive disorders (n = 19/46), and 9% were X-linked disorders (n = 4/46). The molecular diagnostic rate was highest for fetuses with anomalies affecting multiple organ systems and for fetuses with craniofacial anomalies. Out of 146 cases, a prenatal trio exome option designed for ongoing pregnancies was performed on 62 fetal specimens, resulting in a diagnostic yield of 35% with an average TAT of 14 days for initial reporting (excluding tissue culture time). The molecular diagnoses led to refined recurrence risk estimates, altered medical management, and informed reproductive planning for families.
Conclusion
Exome sequencing is a useful diagnostic tool when fetal structural anomalies suggest a genetic etiology, but other standard prenatal genetic tests did not provide a diagnosis.
Similar content being viewed by others


Background
Congenital fetal anomalies occur in approximately 3% of pregnancies and are responsible for 20% of infant mortality in the USA [1, 2]. Many of these are thought to have an underlying genetic etiology. Current practice guidelines recommend karyotype and chromosomal microarray analysis (CMA) as first-tier tests when a fetal anomaly has been detected by ultrasound or other fetal imaging [3, 4]. These tests are able to detect aneuploidy, chromosomal rearrangements, or copy number variants (CNVs) in a combined 30–40% of pregnancies studied [5,6,7,8].
While these genetic testing approaches are invaluable for prenatal genetic diagnosis, the potential etiology for fetal anomalies remains unsolved in approximately 60% of cases. A proportion of these unsolved cases may be the result of Mendelian disease due to single-gene defects. Historically, clinicians relied on serial sequencing of single genes or gene panels to explore a potential molecular diagnosis for a Mendelian disease trait. However, such approaches usually require a fairly narrow differential diagnosis and are time consuming. This poses a clinical conundrum in prenatal medicine, where the ability to narrow the differential diagnosis may be limited by incomplete phenotypic information due to the inherent limitations of in utero imaging or gestational age. Even when the clinical phenotype manifested during pregnancy is highly specific, targeted gene tests may yield negative results if the disorder is caused by a variant in a disease gene that is not included in the chosen panel.
One solution to this diagnostic challenge is exome sequencing (ES), which has been shown to provide a valuable diagnostic option in postnatal genetic evaluation because it is not disease- or gene-specific and does not require prior knowledge regarding the potential causative gene(s) for an observed phenotype [9]. Exome sequencing has therefore started to be incorporated into clinical care for pediatric and adult populations. While there have been multiple publications showing the diagnostic and clinical utility of ES in the postnatal setting [3, 4, 10,11,12,13,14,15,16,17,18,19], integration of ES into prenatal diagnosis has been more limited. One reason for this is the paucity of information about the clinical utility of ES in the prenatal setting [20, 21]. Here we present a retrospective analysis of the outcomes of prenatal ES that was performed in a diagnostic laboratory as part of the clinical management of pregnancies, including continuing pregnancies, complicated by fetal structural anomalies.
Methods
Sample inclusion criteria
We performed a retrospective review of the indications, exome results, and clinical impact of molecular diagnoses for all fetal samples that were referred to the Baylor Genetics clinical diagnostic laboratory by a physician for exome testing. We defined the following inclusion criteria: (1) the fetal sample was obtained through an invasive diagnostic procedure (including amniocentesis, chorionic villus sampling, and cordocentesis) or product of conception (POC), (2) the fetus had at least one structural anomaly detected by fetal imaging or autopsy, (3) ES was performed at Baylor Genetics, and (4) a final report was issued between March 2012 and November 2017. The Baylor Genetics clinical diagnostic laboratory is accredited by the College of American Pathologists (CAP) and certified by the US Department of Health and Human Services Clinical Laboratory Improvement Amendments (CLIA). De-identified reporting of demographic and molecular data from this laboratory was approved by the Institutional Review Board at Baylor College of Medicine.
Consent procedures and testing protocols
All exome tests involving a fetal sample required informed consent from parents, relevant patient clinical data, and prior approval by a laboratory genetic counselor that ES was an appropriate testing option. Fetal exomes were processed under one of three testing protocols: proband exome (turnaround time (TAT): 12 weeks, available since 2011), trio exome (TAT: 8 weeks, available since 2014), or prenatal trio exome (TAT: 2–3 weeks excluding tissue culture time, available since 2015). The prenatal trio exome test was intended specifically for ongoing pregnancies and was therefore designed with specialized consenting and reporting procedures. The prenatal trio exome consent form did not include options to report reproductive carrier status [22] or variants in medically actionable genes [23, 24] for the fetus. These options were available for the parents at the time of exome sequencing and for the proband after birth upon request.
Selection of the appropriate prenatal exome test was affected by the availability of the method and parental samples at the time of testing and the degree of urgency of the case. For proband ES, next-generation sequencing (NGS) was only performed on the fetal sample and those sequencing results were interpreted in the context of the clinical indications (Additional file 1: Figure S1). Sanger sequencing of clinically relevant variants was then performed on the fetal and available parental samples before final variant interpretation and reporting. The standard and prenatal trio exome tests required that both parental samples accompany the fetal sample. For these tests, the fetal and parental samples underwent ES simultaneously and results were interpreted in unison, taking into account de novo variants in the fetal DNA sample, inheritance, and allelic configuration of each variant, as well as the clinical indications (Additional file 1: Figure S1).
Laboratory exome analysis, variant interpretation and result reporting
Fetal DNA was extracted from chorionic villus samples (CVS), amniotic fluid, tissue samples, or fetal blood. For samples requiring cell culturing before DNA extraction, the TAT was calculated from the date of DNA extraction. DNA was extracted from peripheral blood or saliva samples from both biological parents. Previously extracted DNA from any of these sources was also accepted. All fetal samples were tested for maternal cell contamination by comparison of maternal and fetal DNA using AmpFlSTR® Identifiler®, which simultaneously amplifies 15 short tandem repeat sites and a gender-determining marker on sex chromosomes. All exome samples were also concurrently tested by an Illumina HumanOmni1-Quad or HumanExome-12 v1 single nucleotide polymorphism (SNP) array for quality control of the exome data and to detect large CNVs, absence of heterozygosity (AOH), and uniparental disomy.
Exome sequencing was performed on DNA samples as previously described [10, 11]. The following metrics were achieved for all samples: mean depth of coverage was ~ 150×, and ~ 98% of target bases (exons and ± 20 intronic nucleotides flanking the exon-intron boundaries of all nuclear genes) were interrogated at > 20× read depth (Additional file 1: Table S1). Variants were assessed for pathogenicity based on the adapted American College of Medical Genetics and Genomics (ACMG) guidelines [25] by a team of American Board of Medical Genetics and Genomics-certified molecular lab directors and medical directors as previously described [10, 26].
On the fetal report, pathogenic and likely pathogenic variants that may be causative of or related to the prenatal indications were included; variants of unknown significance (VUS) were occasionally included when there was a strong indication for reporting (e.g., the VUS was compound heterozygous with a pathogenic variant). Using similar inclusion criteria, variants likely to cause significant, childhood-onset disorders not related to the prenatal indications were also included on the fetal report; reporting of such incidental findings was done on a case-by-case basis based on a consensus decision between the laboratory and the ordering physician.
A fetal exome sample was classified as molecularly diagnosed if the aforementioned variant(s) were detected in a disease gene that was consistent with the clinical phenotype of the fetus and the expected disease inheritance pattern. For biallelic variants in presumed autosomal recessive disorders, the phase of the variants was assessed by parental studies (by exome in the case of trio exomes, or by Sanger sequencing in the case of proband exomes). The variants were considered to constitute a molecular diagnosis only if they were determined to be in trans. In some cases, a pathogenic or likely pathogenic variant in trans with a VUS was considered to constitute a molecular diagnosis depending on the phenotypic specificity and overlap with the fetal findings. For dominant disorders, only de novo variants or those that were inherited from a mosaic or affected parent were considered to contribute to a molecular diagnosis. Rarely, it was possible for a VUS or a partial phenotype overlap to contribute to a molecular diagnosis upon consultation with the ordering physician. Clinical impacts and postnatal outcome data were collected for samples that were referred for genetic testing by a local clinical institution.
Initial fetal reports could be issued without Sanger sequencing confirmation if the NGS variant call(s) on the report were of high confidence (coverage ≥ 20×, minor allele fraction ≥ 30%, and Phred score of variant calling ≥ 30).
Fetal phenotype information was converted into top-branch Human Phenotype Ontology (HPO) categories using Phenomizer [27, 28]. Statistical comparisons were performed using the two-tailed Fisher’s test. The Bonferroni correction was applied for multiple comparisons.
Results
Sample characteristics
One hundred and forty-six prenatal samples fulfilling the designated clinical inclusion criteria were received for fetal exome testing and had a final report issued. The majority of fetal samples were received as extracted DNA (n = 43) or amniotic fluid (n = 35 cultured, n = 32 direct, Additional file 1: Figure S2). The remaining sample types included POC (n = 17 direct, n = 7 cultured), cord blood (n = 5), and CVS (n = 4 direct, n = 3 cultured, Additional file 1: Figure S2). To our knowledge, a CMA and/or karyotype was performed prior to exome analysis for 132 of 146 families, but was non-diagnostic for an etiological molecular diagnosis. In two cases, sex chromosome abnormalities were detected (47,XXY and 47,XYY), but because the chromosomal findings did not explain the fetal phenotype, prenatal exome testing was initiated. One sample was referred for exome testing although there was a previously identified CNV of uncertain clinical significance that could potentially explain the fetal anomalies. In this case, ES did not detect additional pathogenic variants, so it was not considered to have a molecular diagnosis in our current analysis. CMA and/or karyotype results for the proband fetus were not available for the remaining 14/146 families. For 6 of these families, a previous similarly affected fetus had non-diagnostic CMA and/or karyotype results, but such analysis of the proband fetus was either in progress, not performed, or results were not provided to our laboratory at the time of testing.
Cases were referred from Genetics (n = 67), Maternal and Fetal Medicine (n = 45), Obstetrics (n = 26), Pediatrics (n = 3), Pediatric Neurology (n = 2), and Pathology (n = 3) departments (Additional file 1: Figure S3). The majority of samples (n = 123) were referred from an academic institution, while 23 were from a private institution (Additional file 1: Figure S3).
In addition to the cohort of 146 samples with completed prenatal exome testing, exome testing was not completed for 13 samples (Additional file 1: Table S2). In 6 cases, testing was canceled at the request of the referring institution. In 7 cases, we were unable to issue a final report due to insufficient samples.
Reported variants, molecular diagnostic rate and TAT
Of 146 total cases, 46 received a molecular diagnosis from exome sequencing, an overall diagnostic rate of 32% (Table 1). Fifty-nine contributing variants, including 8 frameshift, 11 stopgain, 7 splice site, 2 in-frame insertions/deletions, and 31 nonsynonymous changes, were reported in these 46 cases (Table 2). Both parental samples were available for testing in 142 of the 146 cases. Fetal samples in this cohort underwent exome testing by one of three available testing options as described in the “Methods” section: prenatal trio (n = 62), standard trio (n = 33), or proband exome (n = 51). A molecular diagnosis was reported for 35% of prenatal trio exomes (n = 22/62), 21% of standard trio exomes (n = 7/33), and 33% of proband exomes (n = 17/51; Table 1). There was no statistically significant difference in the diagnostic rates of the three groups (prenatal trio versus standard trio, p = 0. 370; proband versus all trios, p = 0.860). The mean TAT from DNA extraction to initial result reporting was 2.0 weeks (range 1.0–5.4 weeks) for prenatal trio exome, 6.2 weeks (range 1.9–11.1 weeks) for standard trio exome, and 12.6 weeks (range 2.6–20.2 weeks) for proband exome (Table 1). Time required for culturing was excluded from these TAT calculations because an ES test order was received concurrently with the sample for only 22% of samples (n = 33/146) for which culturing was required (Cohorts 1a and 1b, Additional file 1: Figure S2). The remaining samples either did not require any culturing (58%, n = 84/146) or the ES test was ordered sometime after sample receipt, culture initiation, and/or DNA extraction was complete (20%, n = 29/146, Additional file 1: Figure S2).
In the absence of professional practice guidelines for reporting incidental findings from prenatal exome sequencing, we defined an internal policy for such findings specifically for prenatal trio exomes (i.e., ongoing pregnancies). As described above (Methods), we included pathogenic and likely pathogenic variants in disease genes that are expected to cause significant childhood-onset disorders on the fetal report, even when not related to the prenatal indications. Reporting of such incidental findings was decided on a case-by-case basis with input from the ordering physician when necessary, taking into account factors such as disease severity and age of onset. Such findings were reported for 3 of the 62 prenatal trio exome tests (see Additional file 1: Table S3).
Results by clinical indication categories
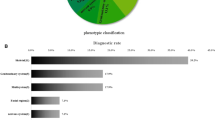
For all samples, the indication was one or more fetal abnormalities detected by prenatal imaging or autopsy. Clinical features that were provided by the referring physician(s) were converted into HPO terms using Phenomizer [27, 28] and grouped into top-branch HPO categories for each fetus (see Additional file 1: Table S4 for reported phenotypes and their corresponding categories). The number of unique top-branch HPO categories was tallied for each fetus. If a fetus had multiple abnormalities within the same top-branch category, that category was only counted once for that fetus. This analysis revealed that the molecular diagnostic rate in fetuses with abnormalities affecting multiple organ systems is higher compared to fetuses with abnormalities in a single organ system (p = 0.018, Fig. 1a and Additional file 1: Table S5). We next investigated whether the diagnostic rate was affected by the nature of the prenatal phenotype (Fig. 1b). Fetuses with craniofacial abnormalities had the highest diagnostic rate (46%, n = 22/48), and this was significantly higher than the rate among fetuses without such abnormalities (24%, n = 24/98, p = 0.013). Nearly half of all fetuses referred for exome sequencing (n = 72/146) had abnormalities affecting the muscular and/or skeletal system. The diagnostic rate for this group was 39% (n = 28/72), while 24% of fetuses without musculoskeletal abnormalities received a molecular diagnosis (n = 18/74, p = 0.075). Although only 14% of fetuses (n = 21/146) had abnormalities involving the respiratory system, this group had a diagnostic rate of 43% (n = 9/21). However, comparison to the diagnostic rate among fetuses without these abnormalities revealed no statistical difference (30%, n = 37/125, p = 0.309). Additional phenotypes that were frequently observed in this cohort affected the nervous system (n = 64/146), the cardiovascular system (n = 37/146), the genitourinary system (n = 38/146), and miscellaneous abnormalities specific to prenatal development (n = 75/146). The diagnostic rates in these groups ranged from 30 to 36%, which is similar to the overall diagnostic rate. Furthermore, the diagnostic rate did not significantly differ between fetuses with these respective abnormalities versus those without (p > 0.05). Abnormalities affecting the abdomen, spleen, thymus, and eye were rarely reported (< 10% of cases, Additional file 1: Table S4).

Molecular diagnostic rates based on phenotype. a Molecular diagnostic rate is higher in fetuses with abnormalities affecting multiple organ systems (p = 0.018; see Additional file 1: Table S5 for non-significant group comparisons). The number of fetuses in each category is indicated on the relevant bar graph. Each top-branch category was only counted once per fetus. b Molecular diagnostic rates are shown for fetuses with (+) or without (−) abnormalities in the stated organ system or top-level HPO category. Fetuses with craniofacial abnormalities were significantly more likely to receive a molecular diagnosis than those without (p = 0.013). Significant p values (p < 0.05) are indicated by (*), Fisher’s exact test
Mendelian inheritance and role of family history
Among the 46 total molecular diagnoses, autosomal dominant (AD) disorders accounted for 50% (n = 23/46), autosomal recessive (AR) for 41% (n = 19/46), and 9% were due to X-linked (XL) disorders (n = 4/46; Table 3). The majority of the autosomal dominant disorders were caused by de novo variants (87%, n = 20/23; Table 3). Mosaicism of the contributing variant was detected in one of these fetuses (Case 85-PRE, Table 2). Inheritance of the variant from a mosaic mother was seen in two cases (Cases 53-P, 67-PRE), and parental samples were not available for one case (Case 45-PRE, Table 2). The majority of the AR disorders were due to compound heterozygous variants (68%, n = 13/19; Table 3). The remaining six AR diagnoses had homozygous contributing variants (32%, n = 6/19; Table 3). Four out of the six cases were determined to be the product of a consanguineous union based on family history and/or AOH data (Case 24-P, 60-T, 84-PRE, 112-PRE; Additional file 1: Table S6). Among the contributing variants in the four XL cases, two were de novo and heterozygous in female fetuses (Case 74-PRE and 122-T) and two were maternally inherited and hemizygous in male fetuses (Cases 111-T and 47-T, Tables 2 and 3).
The diagnostic rate was not different in sporadic cases compared to those with a clinically ascertained significant family history (Table 3). Sporadic cases were defined as those in which the referred proband (fetus) was the first individual in the family to present with the specific phenotype, while cases were considered to have significant family history if a previous fetus or close biological relative had similar clinical features. The majority of cases were sporadic (73%, n = 106/146, Table 3). A diagnosis was made in 33% of sporadic cases (n = 35/106, Table 3). The majority of these were de novo variants associated with either AD (n = 20) or XL (n = 2) disorders (63%, n = 22/35, Table 3). These are associated with a much-reduced recurrence risk (RR), derived from the low likelihood of undetectable somatic or gonadal mosaicism in a parent. The remaining 11 cases had findings that indicated a higher RR of 25% due to homozygous or compound heterozygous variants associated with AR disorders (26%, n = 9/35), up to 50% due to an AD variant inherited from a mosaic parent (6%, n = 2/35), and 50% in males due to a maternally inherited XL variant (3%, n = 1/35; Table 3). Among 40 cases with a significant family history, 11 molecular diagnoses were made (28%, Table 3). All but one of these had biallelic variants associated with AR disorders, indicating a 25% RR (Table 3). The remaining case had a maternally inherited XL hemizygous variant, indicating a 50% RR in males (Table 3).
Genes underlying frequent fetal diagnoses and novel fetal phenotypes of known disease genes
A frameshift (n = 3) or stopgain (n = 1) variant in an internal exon in KMT2D was reported for four fetuses (Case 6-P, 45-P, 48-PRE and 126-PRE, Table 2). These are all predicted to introduce a premature translation termination codon with non-sense mediated decay [29] resulting in a loss-of-function allele, making Kabuki syndrome, caused by haploinsufficiency of KMT2D, the most frequent single-gene disorder in this cohort [30]. In older children, Kabuki syndrome can be clinically diagnosed based on cardinal manifestations including characteristic facial features, abnormal limb/extremity features, microcephaly, short stature, and heart and kidney problems [31] that are neither apparent nor readily recognizable in neonates and infants [32, 33] and are even more challenging prenatally. Comparison of the phenotypes of the four fetuses with a molecular diagnosis of Kabuki syndrome suggests that the co-occurrence of complex cardiac defects (100%, n = 4/4) and renal structural anomalies (75%, n = 3/4) is a common prenatal presentation of this syndrome, which is consistent with described neonatal phenotypes of KMT2D-related Kabuki syndrome [19, 32]. Pathogenic missense variants in COL1A1 or COL1A2 were diagnosed in five fetuses (Case 17-P, 49-T, 90-PRE, 65-PRE, 66-PRE, Table 2) with a skeletal dysplasia phenotype, including shortened long bones and/or abnormalities of the thorax (n = 5), abnormalities of the skull (n = 2), absent fetal nasal bone (n = 1), edema (n = 1), intrauterine growth retardation (n = 1), abnormality of the umbilical cord (n = 1), cardiac abnormalities (n = 1), and genital abnormalities (n = 1) (Table 2). Notably, de novo variants in the DDX3X gene were reported for two female fetuses with cystic hygroma and edema (Case 37-PRE, Additional file 1: Table S3; 122-T, Table 2). Pathogenic variants in DDX3X are known to cause X-linked mental retardation disorder 102 (MRX102, MIM: 300958) in females and rarely in males [34], but have not previously been reported prenatally. The first identified de novo likely pathogenic variant was therefore initially reported as an incidental finding (Case 37-PRE, Additional file 1: Table S3), but the second, in a fetus with identical phenotype, was a known pathogenic variant and reported as a primary finding (Case 122-T, Table 2). One case (101-PRE) carried a de novo frameshift variant in DVL1 previously reported in multiple patients with Robinow syndrome (DRS2, MIM: 616331). This variant is predicted to produce a premature termination codon in the last exon of DVL1 and has been previously described to perturb Wnt signaling through a gain of function or dominant-negative mechanism [35,36,37]. The phenotype of this fetus, absent cavum pellucidum, abnormalities of the genitourinary system, skeletal system, head and neck, and suspected cardiac abnormality is consistent with that of individuals with DVL1 pathogenic variants [35, 36].
Clinical implications of receiving a molecular diagnosis
Information about the clinical implications of receiving an exome diagnosis was available for 14 of the 46 molecular diagnoses and scored as (1) altered medical management, (2) altered reproductive planning, (3) modified recurrence risk estimates, and (4) other impacts. In four cases, medical management was altered either by altering neonatal care or by informing pregnancy termination decisions (Table 2). For example, prenatal detection of a pathogenic COL1A1 variant in case 90-PRE facilitated coordinating an appropriate perinatal care plan and connecting the parents with other families with osteogenesis imperfecta (OI) so they could learn practical skills for caring for a baby with OI. Recurrence risk estimates were modified or refined based on the molecular diagnosis in eight cases (Table 2), with a reported positive psychosocial impact for one family with a history of two previous deceased but undiagnosed infants (Table 2, Other, Case 20-P). Altered reproductive planning for future pregnancies, including targeted prenatal genetic testing or pre-implantation genetic diagnosis, was the most frequent clinical implication (n = 15 cases, Table 2). We are aware of at least 10 cases out of the total 46 molecular diagnoses where the WES result led to targeted testing in a future pregnancy. Additional feedback regarding pregnancy outcomes was provided to the lab for seven local cases (Additional file 1: Table S7).
Discussion
The diagnostic yield of 32% in this cohort of 146 proband and trio exome sequencing tests performed on fetal samples is slightly higher than that of some recent larger series with reported diagnostic rates of 20–24% [38,39,40]. The subset of 62 ongoing pregnancies, with a diagnostic rate of 35%, is one of the first larger series reported to date where exome sequencing was done on still ongoing pregnancies. A prior review of studies, published and presented at international meetings with more than five cases each (range 7–101), indicated a diagnostic yield between 6 and 80% [20, 41,42,43,44,45,46,47,48,49]. This wide range is likely due to a combination of small sample sizes, differences in the a priori likelihood of an underlying Mendelian genetic etiology due to varying inclusion criteria, and variation in interpretation of pathogenicity between reports. Not surprisingly, our diagnostic yield of 32–35% is very close to recently reported 36.7% diagnostic yield from exome sequencing for 278 neonates and infants in intensive care units [19]. Some outcomes of exome sequencing on medical management that were described in this report will likely be applicable to prenatal exome sequencing [19], but more extensive and detailed prenatal studies will be required to further discern its clinical utility and refine clinical inclusion criteria for this advanced testing. We found a higher diagnostic rate for fetuses with structural abnormalities of multiple organ systems and for fetuses with abnormalities of craniofacial morphology, as well as a good diagnostic yield for prenatally detected musculoskeletal, respiratory, nervous system, cardiovascular, and genitourinary anomalies, suggesting clinicians may expect a higher yield in these prenatal presentations, after karyotype studies and chromosome microarray analysis are unrevealing. We further detected multiple large regions of AOH (> 5 Mb) on concurrent SNP array analysis in four fetal samples with homozygous variants [50]. These cases underscore that AOH, particularly as a result of consanguinity, can contribute to autosomal recessive disorders and influence the molecular diagnostic rate [45, 47, 51, 52]. While self-reported family history and SNP arrays provided adequate information to identify a molecular diagnosis for these samples, an alternative approach that could potentially improve sensitivity and reduce cost would be to test for AOH, CNVs, and uniparental disomy simultaneously by calculating the B allele frequency of all single nucleotide variants within the existing exome sequencing data [52].
Reporting VUS and incidental findings in prenatal exome results present a particular challenge because they can create a dilemma for clinicians, genetic counselors, and families who are considering difficult decisions for their pregnancy, delivery and neonatal management in a time-sensitive environment, which must be weighed against the risk of missing a potential molecular diagnosis if a VUS is not reported. Accurate variant interpretation and decisions whether to report a VUS can be compromised by incomplete communication between clinicians and the laboratory about the fetal phenotypic information. Another challenge is that current practice guidelines for reporting incidental findings from diagnostic exomes specifically excludes the prenatal setting [23, 24], although a very recent position statement has begun to address this [21]. To standardize our approach, we defined internal policies for prenatal exomes to report mainly pathogenic or likely pathogenic variants in genes related to prenatal testing indications or known to cause significant disorders during childhood, even if unrelated to the referring indications. We occasionally reported VUS on a case-by-case basis after multidisciplinary consensus decision between the laboratory and the ordering physician when there was a strong indication based on factors such as the presence of a pathogenic variant on the other allele in recessive disorders, good candidate gene based on the fetal phenotype, disease severity, and age of onset.
In the prenatal setting, the timeline for receiving diagnostic testing information is critical as couples may use the test results to support decisions for their pregnancy, including pregnancy continuation or termination, fetal treatment, and delivery management, as well as neonatal treatment. As turnaround time continues to decrease, the diagnostic results of prenatal exome sequencing will increasingly contribute to this decision-making process, in addition to its utility for recurrence risk counseling. We have demonstrated that initial exome results for ongoing pregnancies are routinely reported in ~ 2 weeks excluding cell culture time. Considering that in most cases exome sequencing is not initiated until the result of the CMA, which is usually performed in parallel to the cell culture on DNA directly extracted from the amniotic fluid sample, the need to wait for cell cultures often does not add to the overall time from procedure to the exome result (Additional file 1: Figure S2). Nevertheless, continued reduction in the time to molecular diagnosis remains possible. In addition to time required for specimen culturing, patients often wait for insurance verification, coverage determination, and cost estimates prior to initiating testing [53]. This impacts not only the turnaround time but also the emotional burden on the family.
Currently, little guidance on diagnostic prenatal exome sequencing exists. The ACMG currently recommends ES as an option for fetuses with multiple congenital anomalies suggestive of a genetic disorder for whom genetic tests that are specific to the phenotype have failed to determine a diagnosis [9]. Although the American College of Obstetricians and Gynecologists does not currently recommend the routine use of fetal exomes in the prenatal setting, they state that prenatal exome may be reasonable in select circumstances such as recurrent fetal phenotypes with no diagnosis by standard testing [4]. A recent joint position statement from the International Society for Prenatal Diagnosis, Society for Maternal and Fetal Medicine, and Perinatal Quality Foundation [21] further comments on reasonable indications for fetal exome testing and considers counseling and implementation aspects. Nevertheless, all current professional society statements emphasize the need for more peer-reviewed data regarding implementation of ES for prenatal diagnosis. Our study contributes such valuable information by reporting diagnostic rates, genotype-phenotype correlations, new information regarding prenatal presentations of some molecularly diagnosed disorders, and clinical impact of molecular diagnosis from a cohort of 146 consecutive fetal exomes sequenced and analyzed on a clinical basis.
Conclusions
With rapid mean TAT of 2 weeks, we were able to provide molecular diagnosis for 35% of ongoing pregnancies that underwent prenatal trio exome analysis. An overall diagnostic rate of 32% was achieved including all sub-cohorts of proband, standard trio and prenatal trio exomes. We showed a higher molecular diagnostic rate in fetuses with structural anomalies in multiple organ systems and in fetuses with craniofacial abnormalities. Finally, we demonstrated that prenatal ES can offer substantial advantages for both families and clinicians, in terms of reproductive planning and decision-making, recurrence risk estimation, and medical management. Thus, our study demonstrates compelling evidence for the utility of prenatal exome sequencing as a promising new option in the realm of prenatal genetic diagnostics. We conclude that although more research on its clinical utility for various categories of fetal phenotypes is needed, prenatal exome sequencing can be offered in select cases, but should preferentially be implemented under guidance of experienced multidisciplinary teams that include prenatal genetics experts who work closely with laboratories experienced with both prenatal diagnosis and diagnostic genome-wide sequencing, as previously suggested [21].
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- AD:
-
Autosomal dominant
- AOH:
-
Absence of heterozygosity
- AR:
-
Autosomal recessive
- CAP:
-
College of American Pathologists
- CLIA:
-
Clinical Laboratory Improvement Amendments
- CMA:
-
Chromosomal microarray analysis
- CNV:
-
Copy number variant
- CVS:
-
Chorionic villus samples
- ES:
-
Exome sequencing
- het:
-
Heterozygous
- hom:
-
Homozygous
- hemi:
-
Hemizygous
- HPO:
-
Human Phenotype Ontology
- NGS:
-
Next-generation sequencing
- NR:
-
Information not received from ordering physicians
- OI:
-
Osteogenesis imperfecta
- POC:
-
Product of conception
- RES:
-
Residual recurrence risk due to possibility of parental germline mosaicism
- RR:
-
Recurrence risk
- SNP:
-
Single nucleotide polymorphism
- TAT:
-
Turnaround time
- VUS:
-
Variant of unknown significance
- XL:
-
X-linked
References
Centers for Disease Control and Prevention (CDC). Update on overall prevalence of major birth defects--Atlanta, Georgia, 1978-2005. MMWR Morb Mortal Wkly Rep. 2008;57:1–5.
Matthews TJ, MF MD, Thoma ME. Infant mortality statistics from the 2013 period linked birth/infant death data set. Natl Vital Stat Rep. 2015;64:1–30.
American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Obstetrics, Committee on Genetics, Society for Maternal–Fetal Medicine. Practice bulletin no. 162: prenatal diagnostic testing for genetic disorders. Obstet Gynecol. 2016:e108–22.
Committee on Genetics and the Society for Maternal-Fetal Medicine. Committee Opinion No.682: Microarrays and next-generation sequencing technology: the use of advanced genetic diagnostic tools in obstetrics and gynecology. Obstet Gynecol. 2016:e262–8.
Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, Savage M, Platt LD, Saltzman D, Grobman WA, Klugman S, Scholl T, Simpson JL, McCall K, Aggarwal VS, Bunke B, Nahum O, Patel A, Lamb AN, Thom EA, Beaudet AL, Ledbetter DH, Shaffer LG, Jackson L. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367:2175–84.
Breman A, Pursley AN, Hixson P, Bi W, Ward P, Bacino CA, Shaw C, Lupski JR, Beaudet A, Patel A, Cheung SW, van den Veyver I. Prenatal chromosomal microarray analysis in a diagnostic laboratory; experience with >1000 cases and review of the literature. Prenat Diagn. 2012;32:351–61.
Shaffer LG, Rosenfeld JA, Dabell MP, Coppinger J, Bandholz AM, Ellison JW, Ravnan JB, Torchia BS, Ballif BC, Fisher AJ. Detection rates of clinically significant genomic alterations by microarray analysis for specific anomalies detected by ultrasound. Prenat Diagn. 2012;32:986–95.
Hillman SC, McMullan DJ, Hall G, Togneri FS, James N, Maher EJ, Meller CH, Williams D, Wapner RJ, Maher ER, Kilby MD. Use of prenatal chromosomal microarray: prospective cohort study and systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2013;41:610–20.
ACMG Board of Directors. Points to consider in the clinical application of genomic sequencing. Genet Med. 2012;14:759–61.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, Hardison M, Person R, Bekheirnia MR, Leduc MS, Kirby A, Pham P, Scull J, Wang M, Ding Y, Plon SE, Lupski JR, Beaudet AL, Gibbs RA, Eng CM. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369:1502–11.
Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, Chiang T, Leduc M, Beuten J, Zhang J, He W, Scull J, Willis A, Landsverk M, Craigen WJ, Bekheirnia MR, Stray-Pedersen A, Liu P, Wen S, Alcaraz W, Cui H, Walkiewicz M, Reid J, Bainbridge M, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–9.
Posey JE, Rosenfeld JA, James RA, Bainbridge M, Niu Z, Wang X, Dhar S, Wiszniewski W, Akdemir ZHC, Gambin T, Xia F, Person RE, Walkiewicz M, Shaw CA, Sutton VR, Beaudet AL, Muzny D, Eng CM, Yang Y, Gibbs RA, Lupski JR, Boerwinkle E, Plon SE. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet Med. 2015;18:678–85.
Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, Das K, Toy T, Harry B, Yourshaw M, Fox M, Fogel BL, Martinez-Agosto JA, Wong DA, Chang VY, Shieh PB, Palmer CGS, Dipple KM, Grody WW, Vilain E, Nelson SF. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880–7.
Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, Baxter RM, Zeng W, Mroske C, Parra MC, Gandomi SK, Lu I, Li X, Lu H, Lu H-M, Salvador D, Ruble D, Lao M, Fischbach S, Wen J, Lee S, Elliott A, Dunlop CLM, Tang S. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17:578–86.
Wright CF, Fitzgerald TW, Jones WD, MRes SC, McRae JF, van Kogelenberg M, King DA, Ambridge K, Barrett DM, Bayzetinova T, Bevan AP, Bragin E, Chatzimichali EA, Gribble S, Jones P, Krishnappa N, Mason LE, Miller R, Morley KI, Parthiban V, Prigmore E, Rajan D, Sifrim A, Swaminathan GJ, Tivey AR, Middleton A, Parker M, Carter NP, Barrett JC, Hurles ME, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;308:1–10.
Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, Vertino-Bell A, Smaoui N, Neidich J, Monaghan KG, McKnight D, Bai R, Suchy S, Friedman B, Tahiliani J, Pineda-Alvarez D, Richard G, Brandt T, Haverfield E, Chung WK, Bale S. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18:696–704.
Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, Bedford HM, Bernard G, Bernier FP, Brais B, Bulman DE, Warman Chardon J, Chitayat D, Deladoëy J, Fernandez BA, Frosk P, Geraghty MT, Gerull B, Gibson W, Gow RM, Graham GE, Green JS, Heon E, Horvath G, Innes AM, Jabado N, Kim RH, Koenekoop RK, Khan A, Lehmann OJ, et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet. 2015;89:275–84.
Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, Weiss ME, Köster J, Marais A, Paknia O, Schröder R, Garcia-Aznar JM, Werber M, Brandau O, Calvo Del Castillo M, Baldi C, Wessel K, Kishore S, Nahavandi N, Eyaid W, Rifai Al MT, Al-Rumayyan A, Al-Twaijri W, Alothaim A, Alhashem A, Al-Sannaa N, Al-Balwi M, Alfadhel M, Rolfs A, Abou Jamra R. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet. 2017;25:176–82.
Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, Zhang J, He W, Dharmadhikari AV, Qu C, Ward P, Braxton A, Narayanan S, Ge X, Tokita MJ, Santiago-Sim T, Dai H, Chiang T, Smith H, Azamian MS, Robak L, Bostwick BL, Schaaf CP, Potocki L, Scaglia F, Bacino CA, Hanchard NA, Wangler MF, Scott D, Brown C, et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 2017;171:e173438.
Best S, Wou K, Vora N, Van der Veyver IB, Wapner R, Chitty LS: Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat Diagn 2018, 38:10–19.
The International Society for Prenatal Diagnosis, The Society for Maternal and Fetal Medicine, and the perinatal Quality Foundation. Joint position statement from the International Society of Prenatal Diagnosis (ISPD), the Society of Maternal Fetal Medicine (SMFM) and the perinatal Quality Foundation (PQF) on the use of genome-wide sequencing for fetal diagnosis. Prenat Diagn. 2018;38:6–9.
American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Obstetrics. Committee opinion no. 691 summary: carrier screening for genetic conditions. Obstet Gynecol. 2017;129:597–9.
Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, McKelvey KD, Ormond KE, Richards CS, Vlangos CN, Watson M, Martin CL, Miller DT. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2016;19:249–55.
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, Biesecker LG. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
Yang Y, Eng C, Wong L-J, Xia F, Walkiewicz M, Zhang J, Scull JC, Wang J, Schmitt E, Liu L. Adaptation of the ACMG/AMP standards and guidelines for variant interpretation: experience within a clinical laboratory. Abstract #1818; poster presentation at the American College of Medical Genetics and Genomics Annual Clinical Genetics Meeting; Tampa, FL March 9-11, 2016.
Köhler S, Schulz MH, Krawitz P, Bauer S, Dölken S, Ott CE, Mundlos C, Horn D, Mundlos S, Robinson PN. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet. 2009;85:457–64.
Köhler S, Doelken SC, Mungall CJ, Bauer S, Firth HV, Bailleul-Forestier I, Black GCM, Brown DL, Brudno M, Campbell J, FitzPatrick DR, Eppig JT, Jackson AP, Freson K, Girdea M, Helbig I, Hurst JA, Jähn J, Jackson LG, Kelly AM, Ledbetter DH, Mansour S, Martin CL, Moss C, Mumford A, Ouwehand WH, Park S-M, Riggs ER, Scott RH, Sisodiya S, et al. The human phenotype ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014;42(Database issue):D966–74.
Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet. 2006;14:1074–81.
Micale L, Augello B, Maffeo C, Selicorni A, Zucchetti F, Fusco C, De Nittis P, Pellico MT, Mandriani B, Fischetto R, Boccone L, Silengo M, Biamino E, Perria C, Sotgiu S, Serra G, Lapi E, Neri M, Ferlini A, Cavaliere ML, Chiurazzi P, Monica MD, Scarano G, Faravelli F, Ferrari P, Mazzanti L, Pilotta A, Patricelli MG, Bedeschi MF, Benedicenti F, et al. Molecular analysis, pathogenic mechanisms, and readthrough therapy on a large cohort of Kabuki syndrome patients. Hum Mutat. 2014;35:841–50.
Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A, Adam MP, Hudgins L, Hannibal M. Kabuki Syndrome. Seattle: University of Washington, Seattle; 1993.
Dentici ML, Di Pede A, Lepri FR, Gnazzo M, Lombardi MH, Auriti C, Petrocchi S, Pisaneschi E, Bellacchio E, Capolino R, Braguglia A, Angioni A, Dotta A, Digilio MC, Dallapiccola B: Kabuki syndrome: clinical and molecular diagnosis in the first year of life. Arch Dis Child 2015, 100:158–164.
Vaux KK, Hudgins L, Bird LM, Roeder E, Curry CJR, Jones M, Jones KL. Neonatal phenotype in Kabuki syndrome. Am J Med Genet A. 2005;132A:244–7.
Snijders Blok L, Madsen E, Juusola J, Gilissen C, Baralle D, Reijnders MRF, Venselaar H, Helsmoortel C, Cho MT, Hoischen A, Vissers LELM, Koemans TS, Wissink-Lindhout W, Eichler EE, Romano C, Van Esch H, Stumpel C, Vreeburg M, Smeets E, Oberndorff K, van Bon BWM, Shaw M, Gecz J, Haan E, Bienek M, Jensen C, Loeys BL, Van Dijck A, Innes AM, Racher H, et al. Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on Wnt signaling. Am J Hum Genet. 2015;97:343–52.
White J, Mazzeu JF, Hoischen A, Jhangiani SN, Gambin T, Alcino MC, Penney S, Saraiva JM, Hove H, Skovby F, Kayserili H, Estrella E, Vulto-van Silfhout AT, Steehouwer M, Muzny DM, Sutton VR, Gibbs RA, Baylor-Hopkins Center for Mendelian Genomics, Lupski JR, Brunner HG, van Bon BWM, Carvalho CMB. DVL1 frameshift mutations clustering in the penultimate exon cause autosomal-dominant Robinow syndrome. Am J Hum Genet. 2015;96:612–22.
Bunn KJ, Daniel P, Rösken HS, O'Neill AC, Cameron-Christie SR, Morgan T, Brunner HG, Lai A, Kunst HPM, Markie DM, Robertson SP. Mutations in DVL1 cause an osteosclerotic form of Robinow syndrome. Am J Hum Genet. 2015;96:623–30.
White JJ, Mazzeu JF, Coban-Akdemir Z, Bayram Y, Bahrambeigi V, Hoischen A, van Bon BWM, Gezdirici A, Gulec EY, Ramond F, Touraine R, Thevenon J, Shinawi M, Beaver E, Heeley J, Hoover-Fong J, Durmaz CD, Karabulut HG, Marzioglu-Ozdemir E, Cayir A, Duz MB, Seven M, Price S, Ferreira BM, Vianna-Morgante AM, Ellard S, Parrish A, Stals K, Flores-Daboub J, Jhangiani SN, et al. WNT signaling perturbations underlie the genetic heterogeneity of Robinow syndrome. Am J Hum Genet. 2018;102:27–43.
Fu F, Li R, Li Y, Nie Z-Q, Lei T-Y, Wang D, Yang X, Han J, Pan M, Zhen L, Ou Y-M, Li J, Li F-T, Jing X-Y, Li D-Z, Liao C. Whole exome sequencing as a diagnostic adjunct to clinical testing in fetuses with structural abnormalities. Ultrasound Obstet Gynecol. 2018;51:493–502.
Yates CL, Monaghan KG, Copenheaver D, Retterer K, Scuffins J, Kucera CR, Friedman B, Richard G, Juusola J. Whole-exome sequencing on deceased fetuses with ultrasound anomalies: expanding our knowledge of genetic disease during fetal development. Genet Med. 2017;19:1171–8.
Daum H, Meiner V, Elpeleg O, Harel T, Collaborating Authors. Fetal exome sequencing: yield and limitations in a single tertiary center. Ultrasound Obstet Gynecol. 2018. https://doi.org/10.1002/uog.19168.
Alamillo CL, Powis Z, Farwell K, Shahmirzadi L, Weltmer EC, Turocy J, Lowe T, Kobelka C, Chen E, Basel D, Ashkinadze E, D'Augelli L, Chao E, Tang S. Exome sequencing positively identified relevant alterations in more than half of cases with an indication of prenatal ultrasound anomalies. Prenat Diagn. 2015;35:1073–8.
Drury S, Williams H, Trump N, Boustred C, GOSGene, Lench N, Scott RH, Chitty LS. Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities. Prenat Diagn. 2015;35:1010–7.
Carss KJ, Hillman SC, Parthiban V, McMullan DJ, Maher ER, Kilby MD, Hurles ME. Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Hum Mol Genet. 2014;23:3269–77.
Vora NL, Powell B, Brandt A, Strande N, Hardisty E, Gilmore K, Foreman AKM, Wilhelmsen K, Bizon C, Reilly J, Owen P, Powell CM, Skinner D, Rini C, Lyerly AD, Boggess KA, Weck K, Berg JS, Evans JP. Prenatal exome sequencing in anomalous fetuses: new opportunities and challenges. Genet Med. 2017;19:1207–16.
Shamseldin HE, Kurdi W, Almusafri F, Alnemer M, Alkaff A, Babay Z, Alhashem A, Tulbah M, Alsahan N, Khan R, Sallout B, Mardawi Al E, Seidahmed MZ, Meriki N, Alsaber Y, Qari A, Khalifa O, Eyaid W, Rahbeeni Z, Kurdi A, Hashem M, Alshidi T, Al-Obeid E, Abdulwahab F, Ibrahim N, Ewida N, El-Akouri K, Mulla Al M, Ben-Omran T, Pergande M, et al. Molecular autopsy in maternal-fetal medicine. Genet Med. 2018;20:420–7.
Rasmussen M, Sunde L, Nielsen ML, Ramsing M, Petersen A, Hjortshøj TD, Olsen TE, Tabor A, Hertz JM, Johnsen I, Sperling L, Petersen OB, Jensen UB, Møller FG, Petersen MB, Lildballe DL. Targeted gene sequencing and whole-exome sequencing in autopsied fetuses with prenatally diagnosed kidney anomalies. Clin Genet. 2018;93:860–9.
Stals KL, Wakeling M, Baptista J, Caswell R, Parrish A, Rankin J, Tysoe C, Jones G, Gunning AC, Lango Allen H, Bradley L, Brady AF, Carley H, Carmichael J, Castle B, Cilliers D, Cox H, Deshpande C, Dixit A, Eason J, Elmslie F, Fry AE, Fryer A, Holder M, Homfray T, Kivuva E, McKay V, Newbury-Ecob R, Parker M, Savarirayan R, et al. Diagnosis of lethal or prenatal-onset autosomal recessive disorders by parental exome sequencing. Prenat Diagn. 2017;38:33–43.
Lei T-Y, Fu F, Li R, Wang D, Wang R-Y, Jing X-Y, Deng Q, Li Z-Z, Liu Z-Q, Yang X, Li D-Z, Liao C. Whole-exome sequencing for prenatal diagnosis of fetuses with congenital anomalies of the kidney and urinary tract. Nephrol Dial Transp. 2017;32:1665–75.
Boissel S, Fallet-Bianco C, Chitayat D, Kremer V, Nassif C, Rypens F, Delrue M-A, Dal Soglio D, Oligny LL, Patey N, Flori E, Cloutier M, Dyment D, Campeau P, Karalis A, Nizard S, Fraser WD, Audibert F, Lemyre E, Rouleau GA, Hamdan FF, Kibar Z, Michaud JL. Genomic study of severe fetal anomalies and discovery of GREB1L mutations in renal agenesis. Genet Med. 2017. https://doi.org/10.1038/gim.2017.173.
Rehder CW, David KL, Hirsch B, Toriello HV, Wilson CM, Kearney HM. American College of Medical Genetics and Genomics: standards and guidelines for documenting suspected consanguinity as an incidental finding of genomic testing. Genet Med. 2013;15:150–2.
Charng W-L, Karaca E, Coban-Akdemir Z, Gambin T, Atik MM, Gu S, Posey JE, Jhangiani SN, Muzny DM, Doddapaneni H, Hu J, Boerwinkle E, Gibbs RA, Rosenfeld JA, Cui H, Xia F, Manickam K, Yang Y, Faqeih EA, Asmari Al A, Saleh MAM, El-Hattab AW, Lupski JR. Exome sequencing in mostly consanguineous Arab families with neurologic disease provides a high potential molecular diagnosis rate. BMC Med Genet. 2016;9:42.
Karaca E, Posey JE, Akdemir ZC, Pehlivan D, Harel T, Jhangiani SN, Bayram Y, Song X, Bahrambeigi V, Yuregir OO, Bozdogan S, Yesil G, Isikay S, Muzny D, Gibbs RA, Lupski JR. Phenotypic expansion illuminates multilocus pathogenic variation. Genet Med. 2018. https://doi.org/10.1038/gim.2018.33.
Westerfield LE, Stover SR, Mathur VS, Nassef SA, Carter TG, Yang Y, Eng CM, Van den Veyver IB. Reproductive genetic counseling challenges associated with diagnostic exome sequencing in a large academic private reproductive genetic counseling practice. Prenat Diagn. 2015;35:1022–9.
Acknowledgements
The authors would like to thank James R. Lupski, MD, PhD; Anh Dang, BS; Irene Miloslavskaya, BS; Wenmiao Zhu, MS; Sandra A. Darilek, MS, CGC; and Sarah Huguenard, MS, CGC for their respective contributions to manuscript review, data generation and review, and clinical care described herein.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files. Our raw data cannot be submitted to publicly available databases because the patient families were not consented for sharing their raw data, which can potentially identify the individuals. Our study, which is the review of aggregate clinical data, were approved by Baylor College of Medicine institutional review board with the waiver of informed consented granted.
Author information
Authors and Affiliations
Contributions
EAN drafted the manuscript. EAN, AB, MW, IBVdV, CME, and YY designed the studies and participated in the writing of the manuscript. AB made substantial contributions to data analysis and interpretation. SN acquired, analyzed, and interpreted data and helped draft the manuscript. PAW, FV, WH, VP, CQ, LW, SS, AVD, RG, DM, HD, LM, XW, RX, PL, WB, and FX acquired, analyzed, and interpreted the exome data. YY supervised the studies. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
De-identified reporting of demographic and molecular data from this laboratory was approved by the Institutional Review Board at Baylor College of Medicine. For clinical testing, all exome tests involving a fetal sample required informed consent, which was obtained from parents. This research conformed with the principles of the Declaration of Helsinki.
Consent for publication
Not applicable
Competing interests
IBVdV is a member of the Baylor Genetics scientific advisory board, but receives no direct compensation for this role.
YY is on the Scientific Advisory Board of Veritas Genetics China. YY and XW founded AiLife Diagnostics, Inc. FV, WH, VP, CQ, AVD are/were employees of Baylor Genetics.
The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from genetic testing offered at Baylor Genetics. The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Comparison of proband versus trio exome workflows. Figure S2. Fetal sample types received and culturing time prior to prenatal exome sequencing. Figure S3. Referring practices. Table S1. Quality metrics of exome sequencing data of the fetal and parental samples. Table S2. Excluded samples without a final report. Table S3. Incidental findings reported for prenatal exome tests. Table S4. Reported fetal phenotypes. Table S5. Pairwise statistical analysis of diagnostic rate based on number of affected organ systems, corrected for multiple comparisons. Table S6. Regions of absence of heterozygosity (AOH) in cases with homozygous variants underlying the molecular diagnosis. Table S7. Pregnancy outcomes for locally referred cases. (PDF 8142 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Normand, E.A., Braxton, A., Nassef, S. et al. Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected Mendelian disorder. Genome Med 10, 74 (2018). https://doi.org/10.1186/s13073-018-0582-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-018-0582-x