
Abstract
Epigenetic marks do not change the sequence of DNA but affect gene expression in a cell-type specific manner by altering the activities of regulatory elements. Development of new molecular biology assays, sequencing technologies, and computational approaches enables us to profile the human epigenome in three-dimensional structure genome-wide. Here we describe various molecular biology techniques and bioinformatic tools that have been developed to measure the activities of regulatory elements and their chromatin interactions. Moreover, we list currently available three-dimensional epigenomic data sets that are generated in various human cell types and tissues to assist in the design and analysis of research projects.
Similar content being viewed by others


Background
Nearly every cell in the human body has the same DNA. However, each cell has a distinct gene expression profile. The cell-type specific gene expression patterns come from differences in the epigenome (Fig. 1a). The epigenome is a collection of sequence-independent regulatory modifications to DNA or protein, which include, but are not limited to histone modifications, DNA methylation, and chromatin organization [1]. Histones are proteins that tightly wrap and pack DNA into nucleosomes, and their modifications are associated with the chromatin states [2, 3]. Chromatin states are largely divided into two states: inactive chromatin and active chromatin. Heterochromatin is a form of chromatin that is densely packed and transcriptionally inactive. Heterochromatin regions are marked by histone modification H3K9me3. Inactive chromatin regions also include cis-regulatory elements (e.g., promoters, enhancers, insulators) that are silenced and repressed. These repressed regions are marked by histone modification H3K27me3. DNA methylation, the addition of a methyl group to the cytosine of CpG, is often found in inactive regulatory elements, where their target genes are repressed [4] (Fig. 1b, top). On the other hand, euchromatin is the transcriptionally active form of chromatin. Active regions of chromatin include regulatory elements that are open and accessible for proteins to bind. Regulatory elements bound by transcription factors (TFs) control the rate of transcription [5]. A promoter is located near the transcriptional start site (TSS) of a target gene, and an active promoter is unmethylated and marked by histone modification H3K4me3 [6]. An enhancer, marked by histone modification H3K4me1 for poised and H3K27ac for active status, is located distal to the TSS of a target gene [7]. Enhancers interact with the promoter of a target gene to increase the rate of transcription. An insulator, which is marked by CTCF (CCCTC-binding factor), can either decrease the rate of transcription by interfering with the promoter-enhancer interaction or increase the transcription by acting as a barrier to stop the spread of heterochromatin [8] (Fig. 1b, bottom). The most likely model that has been suggested for explaining the mechanisms by which regulatory elements influence gene expression is a looping model. In a looping model, TFs bring regulatory elements into proximity by forming a loop [9]. For example, forming promoter and enhancer loops increases the expression of a target gene [10]. Insulators also form a loop, often preventing an enhancer located between insulators from interacting with the promoter of a non-target gene [11].

Overview of epigenome change in the human genome. a Human cells within an individual are genetically identical across all cell types, but distinct epigenome profiles are detected between cell types. b Epigenome changes when normal cells become diseased cells, and vice versa. DNA methylation, histone marks H3K9me3 (heterochromatin region) and H3K27me3 (repressed region) are usually associated with inactive, closed chromatin, while unmethylated DNA, histone marks H3K4me3 (active promoter) and H3K27ac (active enhancer), and transcription factor (TF) binding are found in active, open chromatin. An insulator marked by CTCF can act as a barrier to prevent enhancer-promoter interaction and decrease the rate of transcription or stop the spreading of heterochromatin to increase rate of transcription. c DNA methylation arrays and bisulfite sequencing are used to measure DNA methylation levels. ChIP-seq, CUT & RUN, and CUT & TAG are used to identify regulatory elements using histone mark and TF enriched regions. 3C, 4C, 5C, Capture-C, Hi-C, DNase Hi-C, ChIA-PET, and HiChIP are used to map chromatin interactions. PolII: RNA Polymerase II, GTF: General transcription factor
Chromatin states and interactions not only change among cell types but also change between inactive and active status when normal cells become diseased cells, and vice versa (Fig. 1b). Dysregulation of the human epigenome can result in cancer, autoimmune diseases, psychiatric diseases, and many more [12,13,14]. For example, it is reported that changes in DNA methylation of CTCF binding sites result in the loss of insulators and promote chromatin interactions between enhancers and oncogenes in tumors [15]. Profiling and characterizing three-dimensional (3D) epigenomes is crucial to understanding of underlying molecular mechanisms and promoting future development of treatments.
The development of molecular biology techniques coupled with next generation sequencing now enables us to map epigenomes genome-wide. For example, ChIP-seq (Chromatin immunoprecipitation sequencing), CUT & RUN (Cleavage under targets and release using nuclease) sequencing, and CUT & TAG (Cleavage under targets and tagmentation) sequencing are used to profile histone modification and TF enrichment. DNase-seq (Deoxyribonuclease I hypersensitive sites sequencing), MNase-seq (Micrococcal nuclease digestion with sequencing), FAIRE-seq (Formaldehyde-Assisted Isolation of Regulatory Elements sequencing), ATAC-seq (Assay of Transpose Accessible Chromatin sequencing), and NOMe-seq (Nucleosome Occupancy and Methylome sequencing) are used to assess chromatin accessibility and nucleosome positioning. DNA methylation arrays and bisulfite sequencing are used to measure global DNA methylation levels. Chromatin interactions are mapped using 3C (chromatin conformation capture), 4C, 5C, Capture-C, Hi-C, DNase Hi-C, Micro-C, ChIA-PET (chromatin interaction analysis by paired-end tag) and HiChIP (Fig. 1c).
In this paper, we aim to introduce methods that are commonly used to map regulatory elements, chromatin accessibility, and chromatin interactions genome-wide. To facilitate researchers who are new in either molecular or computational biology, we describe wet lab as well as dry lab protocols for each of the methods. In particular, we detail chromatin conformation interaction methods and analysis tools, which are relatively new. We also discuss the advantages and limitations of each method and introduce recently developed single-cell based methods. Furthermore, we list currently available 3D epigenomic data sets that are generated in various human cell types and tissues. Introduction of epigenomic methods and resources described here will assist many researchers in the design and analysis of their research projects.
Main text
Methods to map regulatory elements
The advancement of molecular biology techniques and next generation sequencing has led to the development of methods to identify regulatory elements throughout the entire genome by analyzing protein-DNA interaction, histone modification, chromatin accessibility, and DNA methylation. The enrichment of specific DNA-binding protein and histone modification is used to identify regulatory elements. Chromatin accessibility analysis reveals open and closed chromatin regions and nucleosome positioning. DNA methylation studies identify the location of methylated CpG sites, which is used to infer chromatin states of regulatory elements and their influences in gene expression [16]. As different factors (protein-DNA interaction, histone modification, chromatin accessibility, and DNA methylation) are assayed, various size of regulatory elements can be identified depending on the methods [17]. Here we introduce commonly used methods to map regulatory elements.
Assays for protein-DNA interaction and histone modification
ChIP-seq [18] is one of the popular methods to analyze protein-DNA binding or histone modifications (Fig. 2a). Because regulatory elements are marked by specific proteins that bind to DNA and histone modifications, ChIP-seq has been utilized to profile the activities of regulatory elements [19]. ChIP-seq wet lab protocol includes following steps. First, to identify the regions occupied by TFs or marked by histone modifications, cells can be fixed using crosslinking reagents such as formaldehyde. To localize histone modifications and nucleosome positioning, native ChIP can be done without crosslinking [20]. Second, nuclei are isolated from cells using lysis buffer. Third, the DNA is sonicated or enzymatically fragmented to produce sheared chromatin and quantified for the next step to capture specific regions of interest. Fourth, the sheared chromatin is immunoprecipitated with an antibody specific to the protein or histone modification of interest. Next, the DNA–protein complex is separated by reverse crosslinking as needed. Finally, the pulled down DNA is purified to generate a library by adding adapters for sequencing. The library is sequenced to determine the global genomic regions bound by the protein or marked by the histone modification [21].

Methods to map regulatory elements. a Simplified protocols of methods to identify regulatory elements using histone modifications are shown. ChIP-seq is performed in lysed cells, while CUT & RUN and CUT & TAG are performed in intact nuclei. b Simplified protocols of methods to map chromatin accessibility are shown. Cells are lysed, and the DNA is either fragmented with enzymes or through sonication. c Simplified protocols of methods to measure global DNA methylation levels are shown. Bisulfite sequencing uses bisulfite conversion followed by sequencing. NOMe-seq simultaneously detects endogenous DNA methylation levels (CpG) and chromatin accessibility (GpC). Bisulfite treatment converts unmethylated C into U, which is converted to T during PCR amplification. pA-MN: Protein A and micrococcal nuclease. pA-Tn5: Protein A and Tn5 transposase, M.CviPl: GpC Methyltransferase
ChIP-seq bioinformatic pipeline includes (1) mapping of sequenced reads to the genome, (2) quality check (QC) of sequenced data sets, (3) calling peaks to identify TF binding sites or histone mark enriched regions, and (4) downstream analysis steps to characterize TF binding sites or identify regulatory elements. First, the sequenced reads (e.g., fastq files) are aligned to the reference genome (e.g., human genome assembly 38 (GRCh38) a.k.a. hg38) using mapping software such as BWA [22] or Bowtie2 [23]. Second, the quality of ChIP-seq data sets is checked. To remove poorly sequenced reads, PCR duplicated reads, and unaligned reads, sequenced reads are filtered using programs such as FastQC [24], Picard [25], bedtools [26], or Samtools [27]. The quality of ChIP-seq data sets is further checked by calculating quality metrics such as PCR Bottleneck Coefficient (PBC), Non-Redundant Fraction (NRF), Normalized Strand Cross-correlation coefficient (NSC), and Relative Strand Cross-correlation coefficient (RSC) [28]. These quality control and filtering processes are necessary to determine whether the ChIP-seq data sets are of high quality with library complexity (high fraction of DNA fragments that are non-redundant and mapped to genome) and highly enriched signals [29]. Third, using aligned reads from the ChIP sample and input sample, which indicates background signals, significantly enriched genomic regions are called with peak calling software programs [30,31,32] such as SPP [33] and MACS2 [34]. To reduce technical variation and identify reproducible peaks, it is recommended to perform ChIP using at least two biological replicates. To measure consistency between replicates, metrics such as Irreproducible Discovery Rate (IDR), which identifies reproducible peaks by generating pseudo replicates from true replicates to call and compare peaks, can be also calculated [35]. Finally, for downstream analysis, genomic distributions of called peaks are analyzed to characterize TF binding sites or identify regulatory elements using programs such as HOMER [36] and ChIPseeker [37]. Differential enrichment of ChIP-seq signals between conditions can be evaluated using programs [38, 39] such as DiffBind [40] and MAnorm [41]. Furthermore, enriched TF motifs at identified peaks and regulatory elements can be determined using motif-search programs such as MEME Suite [42], Transfac [43], Jaspar [44], and HOMER [36]. Identified TF motifs by TF ChIP-seq data are archived in databases such as Factorbook [45].
As the traditional ChIP-seq protocol uses sonication to fragment DNA, the resolution of data is not high. Therefore, ChIP-exo, which is a modified ChIP-seq method that uses exonuclease digestion after ChIP, has been developed [46]. ChIP-exo can identify binding locations at single nucleotide resolution with less background signal [46]. To analyze ChIP-exo data sets, the ChIP-seq bioinformatic pipeline can be used. Specialized bioinformatic tools such as MACE and ChExMix have been developed to analyze ChIP-exo data sets [47, 48]. ChIP-seq requires a relatively large number of cells and has a high background noise. Therefore, methods like CUT & RUN sequencing [49] and CUT & TAG sequencing [50] have been developed to compensate for such limitations. Unlike traditional ChIP that uses fixed cells, CUT & RUN and CUT & TAG methods use unfixed permeabilized cells to facilitate the entry of an antibody into the nuclei, where it binds to TF or histone modification. Unlike ChIP that shears DNA and pulls down enriched regions using an antibody, CUT & RUN uses an antibody and pA-MN (protein A and micrococcal nuclease (MNase) fusion protein) to isolate specific protein-DNA complexes. Calcium ion is added to activate pA-MN, which cleaves the DNA on either side of the binding site of the targeted protein or histone modification. The fragmented DNA that diffuses out of the nuclei is extracted and sequenced after making a DNA library [49]. CUT & TAG is similar to CUT & RUN, except it uses pA-Tn5 transposase instead of pA-MN. pA-Tn5 transposase gets activated by magnesium and ligates an adapter sequence during the cleavage process [50]. Advantages of both CUT & TAG and CUT & RUN are low background noise and lower cell input requirement, since only the DNA that binds to the protein of interest is extracted and sequenced [49, 50]. To analyze CUT & RUN and CUT & TAG sequencing data sets, software programs used for ChIP-seq bioinformatic pipeline can be used. Recently, specialized tools such as SEACR [51], CUT&RUNTools [52], and CUT&TAG pipeline [53] have been developed as well.
Assays for chromatin accessibility and DNA methylation
Chromatin accessibility can be measured to identify active regulatory elements and nucleosome depleted regions (NDRs), where TFs bind (Fig. 2b). Commonly used methods to measure chromatin accessibility include DNase-seq [54], MNase-seq [55], FAIRE-seq [56], ATAC-seq [57], and NOMe-seq [58]. Unlike histone mark ChIP-seq, CUT & RUN, and CUT & TAG methods that identify regulatory elements which are several kb in size, methods to measure chromatin accessibility can identify smaller-sized NDRs [17]. Moreover, nucleosome and TF footprints can be examined using these methods. These methods do not require an antibody, since they do not target specific proteins or histone marks, so the analysis is not confined to specific TFs or histone modifications [59]. This is advantageous especially when antibodies of the proteins of interest that work for immunoprecipitation and ChIP are not available.
DNase-seq utilizes the Deoxyribonuclease I (DNase I) enzyme that digests accessible DNA regions. Therefore, DNase I hypersensitivity sites (DHS) identified by DNase-seq include open chromatin regulatory regions, where TFs bind [60]. DNase-seq wet lab protocol includes following steps [54]. First, nuclei are isolated from cells using lysis buffer in a similar fashion as ChIP-seq protocol. Second, nuclei are digested using DNase I. DNA fragment sizes are measured to identify optimal digestion using gel electrophoresis. Third, biotinylated linkers are ligated to the ends of digested DNA after polishing to make blunt ends, and the DNA is isolated. Fourth, the DNA with biotinylated linker is digested by restriction endonuclease MmeI and captured by streptavidin-coated Dynabeads to generate short tags to which the second sequencing adaptor can be ligated. Finally, a second linker is ligated and amplified to generate a library for sequencing [54]. Protocols of DNase I digestion and size selection steps may vary by research groups [54, 61, 62]. DNase-seq bioinformatic pipeline is similar to that of ChIP-seq. First, sequenced reads are aligned to reference genome with BWA [22] or Bowtie2 [23] Second, quality of DNase-seq data sets are checked. Poorly sequenced reads, PCR duplicated reads, and unaligned reads are filtered using programs such as FastQC [24], Samtools [27], or Picard [25]. Signal Portion of Tags (SPOT) is used to measure signal-to-noise levels in the genome [63]. Third, the aligned reads are used to call DHS peaks against input sample (background signal) with programs like Hotspot2 [63] or MACS2 [34]. With high-depth sequencing, DNase I cleavage sites can be revealed at base-pair resolution, revealing the presence of TF protected DNA sequences as footprints [64]. CENTIPEDE [65] and DNase2TF [66] are examples of programs that detect these footprints. While DNase-seq shows a greater sensitivity for regulatory sites, especially promoters [67], DNase-seq suffers from sequence specific cutting bias of DNase I that can complicate genomic footprinting [68].
MNase-seq determines chromatin accessibility with micrococcal nuclease (MNase) that preferentially digests nucleosome-free, protein-unbound DNA regions [55, 69]. MNase-seq wet lab protocol includes following steps [70]. First, nuclei are isolated from either native or crosslinked chromatin similar to ChIP-seq protocol. Second, nuclei are digested using MNase with titration. Usually, three to five test digestions with a broad range of total units of MNase is added for a single experiment to help identify the amount of MNase needed for optimal digestion. Third, the uncut DNA is purified and mononucleosome bands are isolated and excised through gel electrophoresis. Finally, the isolated DNA is amplified by adding adapters to generate a library, and sequenced [55]. MNase-seq primarily sequences regions of DNA bound by histones or other proteins [71]. Therefore, it indirectly determines which regions of DNA are accessible by directly determining which regions are bound to nucleosomes or proteins [70]. It is noted that MNase prefers to cut AT-rich sequences in limiting enzyme concentrations [72,73,74], so careful enzymatic titrations are required for generating accurate and reproducible MNase-seq data sets. While MNase-seq follows most of the software used by DNase-seq for the bioinformatic pipeline (mapping, QC, calling peaks, and downstream analysis), DANPOS2 [75, 76] is reported to be optimized to identify NDRs and dynamic nucleosomes from MNase-seq data sets. Computational analysis with MNase-seq has been also used to predict chromatin interaction and structure [77, 78].
FAIRE-seq is a method, which simply isolates NDRs from chromatin, not using an antibody to target histone mark or TF [56]. FAIRE-seq wet lab protocol includes following steps [79]. First, cells are fixed using formaldehyde so that TFs and histones are crosslinked to interacting DNA like ChIP-seq protocol. Second, crosslinked chromatin is sheared by sonication that generates protein-free DNA and protein-crosslinked DNA fragments. Third, protein-free DNA is isolated using a phenol–chloroform extraction; DNA crosslinked with protein stays in organic phase, while protein-free DNA stays in aqueous phase. Finally, the purified DNA, which includes NDRs, is amplified using adapters to generate a library and then sequenced [56]. The FAIRE-seq bioinformatic pipeline is similar to the DNase-seq pipeline (mapping, QC, calling peaks, and downstream analysis). FAIRE-seq peaks are often called using software such as F-Seq [80], ChIPOTle [81], Mixer [82], or MACS2 [34]. Because FAIRE-seq does not require single-cell suspension or nuclear isolation, it is more adaptable for tissue samples [56]. FAIRE-seq is relatively free from the sequence-specific cleavage bias that is seen in DNase-seq or MNase-seq [59]. However, FAIRE-seq has a higher background level and a lower signal-to-noise ratio, compared to other chromatin accessibility assays, which can limit identifying all open chromatin regions in a given cell [83, 84]. It is reported that FAIRE-seq has lower resolution in identifying open chromatin regions at promoters but captures more distal regulatory elements, compared to DNase-seq [79, 84, 85].
DNase-seq, MNase-seq, and FAIRE-seq require a relatively large number of cells and have high background noise level. Therefore, ATAC-seq was developed to supplement. ATAC-seq uses hyperactive Tn5 transposase that preferentially cuts accessible chromatin regions and simultaneously inserts adapters to the fragmented region [57]. ATAC-seq wet lab protocol includes following steps [57]. First, nuclei are isolated from cells using lysis buffer. Second, Tn5 transposase is added to nuclei, and often cases, detergents such as digitonin, NP40, and Tween-20 are added together in this step to improve cell permeabilization and remove mitochondria from the transposition reaction [86]. Third, DNA is isolated and purified. Finally, fragmented and tagged DNA by Tn5 transposase is purified and then amplified to generate a library and sequenced for analysis. The first step of bioinformatic pipeline of ATAC-seq is adapter trimming. Because of adapter sequences that are added during Tn5 transposase activity, programs like Cutadapt [87] and Trimmomatic [88] are used to remove adapter sequences before alignment. Second, the sequenced reads are mapped to the genome after trimming, similar to other methods. Third, the quality of the data sets is evaluated like ChIP-seq and DNase-seq data sets (see above). Finally, ATAC-seq peaks are called using MACS2 [34] or HMMRATAC, which is a peak calling program specific to ATAC-seq that uses a Hidden Markov model to learn the chromatin structure and predict accessible regions [89]. As in DNase-seq, high-depth ATAC-seq data can be used for genomic footprinting, using HINT-ATAC [90] or CENTIPEDE [65]. The advantage of ATAC-seq is that it is relatively fast and requires a low amount of sample inputs compared to other assays, while maintaining similar specificity [57]. However, ATAC-seq data may be contaminated with a high percentage of mitochondrial DNA [91], so it may require some extra procedures to reduce mitochondrial DNA contamination [92]. Omni-ATAC is one of methods that improve mitochondrial DNA contamination by pretreating DNA with DNase I to remove free-floating and to digest DNA from dead cells [86]. Omni-ATAC is also reported to work using archival frozen tissue samples and 50-μm sections, generating fewer sequencing reads that map to mitochondrial DNA.
NOMe-seq is a method to identify NDRs with M.CviPI methyltransferase that methylates cytosine in GpC dinucleotides not protected by nucleosomes or other proteins (Fig. 2c) [58]. Unlike CmpG, GpCm in the human genome does not occur naturally in most cell types [93,94,95]. Therefore, GpCm levels at open chromatin regions can be compared to background signals and determine NDRs. NOMe-seq wet lab protocol includes following steps [17]. First, nuclei are isolated from cells using lysis buffer. Second, nuclei are treated with M.CviPI and S-adenosylhomocysteine (SAM) to methylate accessible GpC sites. Third, M.CviPI treated DNA is sheared using a sonicator, so that DNA fragments can be sequenced in the later step. Fourth, the DNA is treated with bisulfite, which converts unmethylated cytosine to uracil using sodium bisulfite, while methylated cytosine is unaffected. Finally, library is generated using adapters and sequenced. Since NOMe-seq uses bisulfite treatment, besides GpC methylation, endogenous CpG methylation is also measured [17]. Open chromatin is expected to have high levels of GpCm but low levels of CmpG. Therefore, NOMe-seq identifies NDRs using the two separate methylation analyses that serve as independent (but opposite) measures, providing matched chromatin designations for each regulatory element [17]. Bioinformatic pipeline of NOMe-seq includes following steps. First, the sequenced reads are aligned to a bisulfite-converted genome using mapping programs such as BSMAP [96], BWA-METH [97], Bismark [98], BS-SEEKER [99], or Biscuit [100]. Second, Picard [25], Samtools [27], and BamToElementEnrichment script from ECWorkflows [101] are used for QC and post-alignment processing to identify high quality and mapped reads. Third, the methylation status of CpG sites and GpC sites are identified using Bis-SNP [102] or Biscuit [100] programs. Finally, NDRs from NOMe-seq are identified with aaRon R package [103], and plots are generated using programs such as Bis-tools [104]. Unlike other assays, NOMe-seq can determine NDRs at single molecular resolution, and it has no bias toward open chromatin regions, since there is no sonication or digestion with enzyme in the step that identifies open chromatin regions [17]; sonication is done after identifying open chromatin regions to fragment DNA for sequencing purpose. However, it is noted that sequencing cost of NOMe-seq, which is based on whole genome sequencing, is more expensive than other assays such as ATAC-seq.
Quantification of DNA methylation level in regulatory elements also helps us to understand the activities of regulatory elements (Fig. 2c) [4]. Active regulatory elements have relatively low levels of CmpG, because proteins bound at open chromatin regions block the DNA methyltransferase (DNMT) complex, needed to methylate cytosine in the regions [105]. On the other hand, DNA methylation in regulatory elements such as CpG island promoters leads to gene silencing [106]. The most common method to assess DNA methylation level is to use bisulfite treatment. Depending on the coverage of profiling, reduced representation bisulfite sequencing (RRBS) [107], DNA methylation arrays [108], and whole genome bisulfite sequencing (WGBS) [93] are used. RRBS uses restriction enzyme digestion to produce sequence-specific fragmentation, and it is the method of choice to study specific regions of interest [107]. For genome-wide analyses, most commonly used methods are using Illumina DNA methylation arrays that can target 27,000 (Human Methylation (HM) 27 K BeadChIP) [109], 450,000 (HM 450 K BeadChIP) [110], and 850,000 (Epic BeadChIP) [111] methylation sites across the genome. Unlike arrays that are restricted to probes, WGBS can assess the DNA methylation status of the entire genome, because whole genome sequencing is used after bisulfite conversion [93]. Similar to NOMe-seq, RRBS and WGBS sequenced data are analyzed by bisulfite mapping programs such as BWA-METH [97], BSMAP [96], Bismark [98] and BS-SEEKER [99]. Quality of DNA methylation data sets are checked with Picard [25] and Samtools [27], and methylated regions are identified using programs like MOABS [112] and methylKit [113]. Illumina DNA methylation array data can be analyzed using software such as Illumina GenomeStudio Software, minfi [114], sesame [115], and DMRCate [116].
The processed sequencing data can be visualized in genome browsers like UCSC Genome Browser [117], Integrative Genomics Viewer (IGV) [118], Integrated Genome Browser (IGB) [119], Ensembl Genome Browser [120], or WashU Epigenome Browser [121]. Commonly used file formats for these genome browsers are bam, bigwig, and bedgraph, which show aligned reads and signal intensity of data sets. Files with bed extensions can be also loaded to the genome browsers to visualize peaks. Some genome browsers like UCSC Genome Browser and Ensembl Genome Browser can only be used as the web-based applications, while IGV and IGB can be used from the local desktop. IGV is also now available as web-based application as well. The web-based genome browsers are generally better at importing and exporting sessions, as data sets can be visualized without downloading data to the local desktop and shared between users.
Recently, advanced techniques using single cell sequencing have been developed to better understand heterogeneity of individual cells. For example, single-cell ATAC-seq, which improves the low input requirement of ATAC-seq further by capturing and assaying cells using a programmable microfluidics platform, has been developed [122]. The specificity of single-cell ATAC-seq identifies chromatin accessibility variance among cell populations, and it is useful to identify sets of TFs associated with specific subgroups [123]. Single-cell NOMe-seq has also been developed using fluorescence-activated cell sorting, and it is reported that it can directly estimate the fraction of accessible regions of individual cells [124]. Single-cell WGBS is also performed. For example, single-cell WGBS on human oocytes revealed distinct DNA methylation patterns in three oocyte maturation stages [125]. Currently, a small number of single-cell ATAC-seq, NOMe-seq, and WGBS data sets have been generated, while thousands of data sets have been generated using a population of cells.
Data sets that mapped regulatory elements
Large consortia such as ENCODE (Encyclopedia of DNA Elements) [126] and REMC (Roadmap Epigenomics Mapping Consortium) [127] profiled global regulatory elements using over one hundred different cell types. The ENCODE consortium is a project that aims to assemble comprehensive lists of functional elements in the human and mouse genome (https://www.encodeproject.org/). From the phase III of the ENCODE project, a registry of 926,535 human and 339,815 mouse candidate regulatory elements is developed [126]. Data sets generated by the ENCODE project include, but are not limited to, histone mark and TF ChIP-seq, ATAC-seq, DNase-seq, FAIRE-seq, eCLIP-seq, RRBS, DNA methylation array, and WGBS. For example, 2039 ChIP-seq data sets that annotate regulatory elements (promoters, enhancers, and insulators), and 2066 open chromatin and DNA methylation data sets from various cell and tissue types have been generated as of October 2020. As part of the ENCODE project, the functional genomics database that stores thousands of experimental data sets is established. The distinguishing feature of the ENCODE database compared to other databases is its filtering capabilities. Its user-friendly interface allows one to filter experimental data according to assay, target of assay, organism, cell and tissue type, and even developmental stage. Moreover, some data sets can be visualized using its own genome browser and other genome browsers such as UCSC and Ensembl. REMC is a consortium that aims to produce data sets of the human epigenomes that include ChIP-seq of histone modifications, chromatin accessibility, DNA methylation, and gene expression data sets for hundreds of human cell types and tissues (http://www.roadmapepigenomics.org/). Unlike ENCODE, REMC only profiles the human epigenomes, and it does not produce TF ChIP-seq data sets including CTCF ChIP-seq data that mark insulators. The REMC database has searching tools and a matrix, which allows a user to search data sets based on experiment, cell-, and/or tissue-type. Moreover, it has options to visualize data sets in the UCSC genome browser.
There are additional consortia that profile the human epigenomes focusing on specific tissues or diseases. For example, PsychENCODE has profiled the epigenomes of brain cells and tissues obtained from patients who suffer from psychiatric diseases [128]. On the other hand, Blueprint project focuses on hematopoietic epigenomes [129], and The Cancer Genome Atlas (TCGA) specializes in cancer (https://www.cancer.gov/tcga). PsychENCODE has generated ChIP-seq, ATAC-seq, and DNA methylation data sets of more than 750 samples (http://www.psychencode.org/). Blueprint project includes histone modification ChIP-seq, DNase-seq and WGBS data sets (https://www.blueprint-epigenome.eu/), while TCGA mainly contains ATAC-seq and DNA methylation arrays to profile the epigenomes of tumors. Over 400 chromatin accessibility data sets and over 12,300 methylation data sets generated by TCGA are available in GDC data portal (https://portal.gdc.cancer.gov/).
With the increased amount of epigenome data sets generated by researchers, multiple epigenome databases have been developed and maintained. The most commonly used database for regulatory elements data sets is GEO (Gene Expression Omnibus) (https://www.ncbi.nlm.nih.gov/geo/). GEO is a public functional genomics database that archives and freely distributes numerous genomic data sets as part of the National Center for Biotechnology Information (NCBI) [126]. While GEO [130] allows some searching and filtering based on organism and sample type, its query and search mechanism is not as intuitive as that of ENCODE or REMC. However, GEO stores by far the largest amount of epigenome data sets that profile regulatory elements as any researchers can deposit data sets. In addition, European Nucleotide Archive (ENA) [131] led by the European Molecular Biology Laboratory—European Bioinformatics Institute (EMBL-EBI) archives functional genomic data sets resulting from biomedical research projects (https://www.ebi.ac.uk/ena/). Moreover, the International Human Epigenome Consortium (IHEC) coordinates the production of epigenomes from healthy and diseased human cells [132] (http://ihec-epigenomes.org/). Currently, IHEC data portal archives thousands of epigenome data sets generated from ENCODE, REMC, Blueprint, Canadian Epigenetics, Environment and Health Research Consortium (CEEHRC), Japan Agency of Medical Research and Development & Core Research for Evolutional Science and Technology (AMED-CREST), Korean National Institute of Health (KINH), and Deutsches Epigenom Programm (DEEP). Individual research groups also maintain databases by collecting and processing epigenome data sets generated and reported by the research community. For example, Cistrome Data Browser [133] (http://cistrome.org/) encompasses TF, histone ChIP-seq and chromatin accessibility data from GEO, ENCODE, and REMC. ReMap2020 database [134] (http://remap.univ-amu.fr/) collects data sets specialized in transcriptional regulators of DNA-binding experiments in Homo sapiens and Arabidopsis thaliana.
Methods to map chromatin interactions
The human genome is tightly packed into the nucleus, because the stretched DNA cannot be contained within the cell size. Increasing evidence suggests that chromatin organization and interaction of regulatory elements influence gene regulation and expression. Local chromatin conformation change can also result in human diseases [13, 14, 135,136,137]. For example, chromatin conformations affect promoter-enhancer interactions. An enhancer that is located hundreds of kb away from the promoter of a target gene can activate or inactivate the target gene by changing chromatin interaction and organization. Moreover, studies on relationship with polycomb repressive complexes (i.e., PRC1, PRC2) [138,139,140] and cohesin complex (e.g., CTCF, RAD21) that is enriched at chromatin loop anchors [141, 142] support the importance of chromatin organization in epigenome changes. Here we describe commonly used techniques to profile global chromatin interactions.
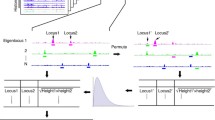
Chromatin conformation capture (3C) based techniques are one of useful methods to study the chromatin interactions and the spatial organization of the human genome (Fig. 3). The standard 3C protocol includes following steps. First, cells are crosslinked to fix chromatin segments connected by a protein complex. Second, nuclei are isolated from fixed cells, and then chromatin segments are fragmented using a restriction enzyme. Third, the chromatin fragments, which are in spatially proximity, are ligated together. Next, crosslinking is reversed to isolate ligated DNA. Finally, the purified ligation product (3C template) is quantified with PCR, using primers designed for two chromatin segments looped (one vs one) [143]. 3C is not coupled with next generation sequencing, so 3C cannot assess chromatin interactions genome-wide. Therefore, many derivatives of 3C-based methods (e.g., 4C, 5C, Hi-C) to measure chromatin interactions in many to all loci are developed.

Methods to map chromatin interactions. Simplified protocols of methods to map chromatin interactions are shown. DNA is first crosslinked, and fragmented with restriction enzymes (3C, 4C, 5C, Capture-C, Hi-C), DNaseI (DNase Hi-C), or MNase (Micro-C). After fragmentation, biotin is added for all methods except for 3C, 4C, 5C or Capture-C. The DNA then goes through proximity ligation, and reverse-crosslinked. Purification and amplification steps are followed. ChIA-PET and HiChIP use an antibody specific to TF or histone modification to map chromatin interactions associated with the specific TF or regulatory elements. IP: Immunoprecipitation
Circular Chromosome Conformation Capture (4C) identifies all possible interactions between a locus of interest with other DNA sequences (one vs all) [144]. 4C wet lab protocol includes additional steps after performing 3C. In 4C, the 3C template is digested again with second restriction enzyme. Then, the product is circularized using ligation. Next, an inverse PCR is performed with primers binding outward on the genomic region of interest to identify and quantify fragments that are ligated to the genomic region of interest. Finally, the amplicons are analyzed using microarray or sequencing to capture all interactions of the genomic region of interest [145]. In 4C-seq (circular chromosome conformation capture, coupled to high throughput sequencing), inverse PCR is performed with a primer that hybridizes to second restriction enzyme fragment and has overhang sequences that corresponds to adapter sequence used in sequencing [145, 146]. 4C-seq bioinformatic pipeline includes following steps. First, the sequenced reads that include the genomic region of interests are kept by demultiplexing and trimmed to extract the sequence including restriction enzyme motifs. Second, data are mapped to reference genome using Bowtie [147] or Novoalign [148]. Third, reads that are mapped to restriction fragment ends with captured regions are quantified using in silico digested reference genome [145]. Finally, read counts are normalized and smoothened, and analyses are performed to identify statistically significant chromatin interactions. Programs like peakC [149], 4C-ker [150], fourSig [151] and FourCSeq [145] are commonly used to identify chromatin interactions from 4C.
Carbon Copy Chromosome Conformation Capture (5C) detects interactions between all restriction fragments within given regions (many vs many) [152]. 5C wet lab protocol includes additional steps after generating the 3C template. To make a 5C library, the 3C template is first converted using multiplex ligation-mediated amplification (LMA), which detects and amplifies specific genomic regions of interest using primer pairs that anneal next to each other on the same DNA strand; In 5C, two sets of primers (5C forward and 5C reverse primers) are annealed to the specific target sequences, and only sequences with both primers attached to the same DNA strand are ligated. The generated 5C library is then followed by microarray or sequencing. For sequencing, universal PCR primers that anneal to tails of 5C primers are used to amplify 5C library for sequencing [152]. The 5C bioinformatic pipeline is similar to 4C. First, paired-end reads are aligned to a pseudo-genome that include all 5C primer sequences using Bowtie [147] or Novoalign [148]. Next, 5C interactions are counted when both paired-end reads are uniquely mapped to the 5C primer pseudo-genome. During this step, invalid interactions that include reads with the same primer or primers of the same type were removed or flagged. Finally, interaction contact matrices are generated using valid interaction counts and normalized for distance and background signals using statistical methods such as quantile normalization [153, 154]. Software such as HiFive [155] and my5C [156] have been developed for 5C data analysis. HiFive is capable of mapping, filtering, normalizing, and visualizing 5C as well as Hi-C data sets, allowing users to analyze the data with a single program [155].
Assays to map chromatin interactions genome-wide
Unlike 3C, 4C, and 5C, Hi-C can map all possible chromatin interactions across the genome (all vs all) [135,136,137, 157,158,159]. Hi-C wet lab protocol includes following steps. First two steps are similar to 3C protocol. First, cells are crosslinked like 3C. Second, nuclei are isolated and then chromatin segments are fragmented by a restriction enzyme. Third, after DNA fragmentation, biotin-labeled nucleotides are added to mark the end. Fourth, segments in proximity are ligated using a DNA ligase. Biotin-label allows enrichment of crosslinked ligation products across the genome. Fifth, the ligated products are reverse-crosslinked. Next, ligation products are fragmented using a sonicator and then pulled down using biotin to generate the biotinylated DNA suitable for sequencing. Finally, by adding adapters needed for sequencing, DNA is amplified and purified. The Hi-C library is then sequenced using paired-end sequencing. By mapping the pair of sequences cut by restriction enzymes and ligated, individually, all possible pairwise interactions between fragments are identified [157].
With the increasing popularity of Hi-C experiments, numerous Hi-C analysis bioinformatic tools have been recently developed. Hi-C bioinformatic pipelines include (1) matrix generation, (2) topologically associating domains (TAD) calling, (3) loop calling, and (4) reproducibility and differential analysis steps (Table 1). Once Hi-C data is generated, the resulting sequencing FASTQ files are first processed to generate a matrix that includes chromatin contact frequencies throughout the entire genome. Examples of matrix generation software include HiC-Pro [160], Juicer [161], Hiclib [162], and Distiller [163] (Table 1). In the first step of matrix generation, read-pairs are aligned to the human genome. During this process, programs account for chimeric reads that span the ligation junction and restriction enzymes that were used. After alignment, the reads are filtered to remove technical artifacts such as PCR duplicates or low-quality alignment reads. Invalid pairs, which are generated due to invalid ligation like dangling end or self-circle circulation, are also filtered. Next, the reads are then mapped through ‘binning’, in which the genome is partitioned into fixed size called ‘bin’, and the number of contacts between bins are assessed and normalized.
Hi-C contact matrices often contain systemic biases that can affect the consistency and analysis of the data sets. Therefore, after Hi-C data sets are mapped, the contact matrices are normalized to remove biases such as GC content, mappability, copy number variations, and fragment length (Table 2). The normalization method can be divided into two categories: implicit and explicit. The explicit normalization assumes specific sources of biases and utilizes additional information like fragment length, mappability score, and GC content to correct biases [164]. Examples of software that normalize using the explicit methods include Hicpipe [165] and HiCNorm [164]. On the other hand, the implicit normalization assumes no known source of bias and assumes that all loci have equal representation when there is no bias. Examples of implicit normalization method include ICE (Iterative Correction and Eigenvector Decomposition) [162] and SCN (Sequential Component normalization) [166]. ICE collectively normalizes bias affecting experimental visibility through iterative correction, while SCN normalizes circulation biases. Moreover, there are additional normalization software for other biases such as calCB [167] that normalizes genomic DNA copy number bias in tumor cells and multiHiCcompare that normalizes across multiple data sets [168].
Mapping genome-wide chromatin interactions by Hi-C and other 3C-derived methods revealed that the human genome consists of compartments and smaller sub-parts. A normalized Hi-C matrix at 1 Mb resolution revealed a plaid pattern, suggesting that chromosome is decomposed into two compartments: compartment A and compartment B [157]. The sequences in compartment A are more closely related with open, accessible, and actively transcribed chromatin, while the sequences in compartment B are more related with closed, inactive chromatin. Compartment A and B partition are cell type-specific, and can be further broken down into sub-compartments, such as A1–A2 and B1–B3 [141]. High-resolution chromatin contact maps revealed highly self-interacting regions that preferentially interact within the domain, and they were referred to as topologically associating domains (TADs) [154, 169, 170]. TADs are suggested to be fundamental components of genome organization as TADs are reported to be conserved across cell types and tissues [171, 172] although recently developed higher resolution of chromatin contact maps revealed that smaller-size TADs (sub-TADs) can vary among cell types [13, 173]. Programs and software such as DI [169], TopDom [174], HiCseg [175], CaTCH [176], and arrowhead [141] have been developed to identify and analyze TADs (Table 1). A previous study has shown that each TAD calling software comes with its own advantages and disadvantages due to their difference in algorithms [177]. Additionally, it is reported that one program can identify TADs that are different in sizes when the bin size of the matrix used to call TADs is changed [177].
Hi-C data sets can be further used to identify chromatin loops [141]. The chromatin loops that have significantly higher contact frequencies, compared with their neighbors were identified as peaks in the Hi-C contact matrix. Examining chromatin loops at higher resolution enables us to study the looping of regulatory elements such as promoter-enhancer loops. Examples of loop calling software include HiCCUPS [141], GOTHiC [178], FitHiC [179], FitHiC2 [180], SIP [181], and Mustache [182] (Table 1). Interaction frequency is compared to the local or global background to determine its significance. Programs like GOTHiC [178], FitHiC [179], and FitHiC2 [180] utilize global background to identify loops, while programs like HiCCUPS [141], SIP [181], and Mustache [182] utilize local background to detect loops. Global background methods can detect interdomain interactions better than local background methods, while the local background methods can detect more significant loops than global background methods [141, 180, 182].
To compare Hi-C data sets, it is crucial to first measure the reproducibility of the generated data sets. However, common statistical methods like Pearson, Spearman or irreproducible discovery rate are not suitable for Hi-C data sets due to their dimensional nature. Therefore, slightly modified methods compatible for Hi-C experiments such as IDR2D [183] and HiCRep [184] are developed (Table 1). IDR2D expands from one-dimensionality of IDR and analyze interactions in two dimensions by a pair of genome coordinates. HiCRep utilizes stratum-adjusted correlation coefficient, a weighted version of Pearson correlation coefficient. Another program called HiC-spector utilizes spectral decomposition to quantify reproducibility of contact maps [185]. After measuring reproducibility of data sets, Hi-C data sets generated in different biological conditions can be further compared to identify regions differentially interacting using programs such as HiCCompare [186], FIND [187], and Selfish [188].
Visualization of Hi-C data sets facilitates data analysis and interpretation. Chromatin contact maps are often represented as a heatmap. In a heatmap, the x-axis and y-axis represent each position along a given chromosome, and each ‘contact’ is represented by a bin, with more frequently interacting contact having stronger color such as dark red, while less frequently interacting contact having weaker color such as white in the white to red color scale. Example software that generate heatmaps include Juicebox [189], HiGlass [190], HiCPlotter [191], HiTC [192], and 3D Genome Browser [193] (Table 3). Heatmaps are sometimes represented as a triangle to facilitate the comparison of Hi-C data sets with other next generation sequencing data sets like ChIP-seq and DNase-seq. Chromatin interactions can be also visualized as loops in genome browsers such as UCSC genome browser [117]
Hi-C that uses 6-cutter restriction enzyme fragmentation yields ~ 4 kb fragment size, and even 4-cutter restriction enzyme and multiple restriction enzyme fragmentation results in ~ 1 kb resolution at the best [194]. Therefore, to improve resolution, variations of Hi-C such as DNase Hi-C [195] and Micro-C [194] that use different enzymes to fragment DNA have been recently developed. Unlike Hi-C that uses restriction enzyme to digest crosslinked DNA, DNase Hi-C uses DNase I and Micro-C uses MNase. After digestion, DNase Hi-C includes a step to mark chromatin fragments with biotinylated adapters that contain BamHI restriction enzyme cut sites, instead of simple addition of biotin-marked nucleotides. These sequences are used later to check the DNase Hi-C library size. Unlike Hi-C, where proximity ligation is performed in solution, proximity ligation step for DNase Hi-C is done in gel to reduce random inter-molecular collisions of small-sized DNase-digested fragments [196]. After proximity ligation and reverse-crosslinking steps, DNA can be optionally sonicated for DNase Hi-C if the size of chromatin fragments is too large for sequencing. Micro-C does not require sonication, because MNase already digest DNA to a size less than 500 bp. DNase Hi-C has shown slightly improved resolution over Hi-C, while Micro-C has provided resolution up to ~ 200 bp [194, 195]. While Hi-C bioinformatic pipeline can be used to analyze DNase Hi-C and Micro-C data, difference in fragmentating enzyme needs to be accounted during the steps of mapping and identifying valid pairs to create contact matrices since most Hi-C bioinformatic pipelines utilize restriction enzyme information. To increase the coverage, targeted sequencing methods such as Capture-C [197] and Capture Hi-C [198] that uses oligonucleotide capture technology to enrich the regions of interest are also developed. These targeted sequencing methods can provide enough resolution to identify chromatin contact maps between selected regions of interest while requiring less sequencing depth.
3C methods can be combined with chromatin immunoprecipitation to identify interactions of loci associated with proteins. Chromatin interaction analysis with paired-end tag sequencing (ChIA-PET) [199] combines ChIP and 3C method to detect genome-wide interactions associated with a particular protein. ChIA-PET wet lab protocol includes additional steps after performing ChIP. After performing first four steps of ChIP (crosslinking, nuclei isolation, chromatin fragmentation, immunoprecipitation), biotinylated oligonucleotide half-linkers containing flanking MmeI restriction enzyme sites are added at the ends of DNA. Next, DNA fragments in proximity are ligated like 3C. Furthermore, MmeI restriction enzyme is used to digest to generate a paired end tag (PET) construct, which includes a pair of tags and a linker between the tag pair. Finally, the PET sequences are purified and then PCR amplified for sequencing [199, 200]. MmeI restriction digestion and amplification steps can be alternatively done using Tn5 transposome digestion, in which sequencing adapters are added to the DNA ends simultaneously [201]. ChIA-PET data can be analyzed using bioinformatic tools such as MANGO [202] and ChIA-PET Tool [200] that are specifically designed to process ChIA-PET data sets by filtering linker sequences and mapping to genome to classify PET. Another method called HiChIP [203] is developed to detect interactions associated with proteins of interest. In HiChIP, the restriction enzyme-mediated fragmented DNA goes through in situ proximity ligation like Hi-C, and then immunoprecipitated with a specific antibody of the protein of interest like ChIP. HiChIP is reported to require fewer cells, compared to ChIA-PET [203]. For HiChIP data processing, Hi-C bioinformatic pipeline can be used. HiChIP specialized bioinformatic tools such as hichipper [204] and FitHiChIP [205] can be also used to identify significant chromatin loops.
Recently, single-cell Hi-C is developed to analyze genome organization and variability in individual cells. The first single-cell Hi-C relied on physical separation of cells and resulted in low throughput [206]. However, it is reported that combinatorial cellular indexing to single-cell Hi-C led to significant improvement in genome coverage and throughput [207]. Moreover, SPRITE (Split-pool recognition of interactions by tag extension) method, which measures high-order interactions within an individual nucleus but does not use proximity ligation, is developed to identify chromatin interactions [208]. SPRITE is reported to able to detect interactions that occur at larger spatial distances than the interactions found in Hi-C. Besides these, DNA fluorescence in situ hybridization (DNA FISH) that utilizes imaging method allows for the study of chromosomal organization [209].
Data sets that mapped chromatin interactions
Because chromatin interaction method is a relatively new technique, there are currently few studies that have generated genome-wide chromatin contact maps in human cells. The 4D Nucleome (4DN) consortium [210] aims to develop experimental and computational approaches to study spatial organization of the genome and its effect on gene regulation and other biological functions (https://www.4dnucleome.org/). Currently, 4DN Data Portal encompasses hundreds of experimental data sets, including Hi-C, Micro-C and DNA FISH data. ENCODE [126] has also generated Hi-C, ChIA-PET, 5C, and SPRITE data, but relatively few data sets compared to other data types. Most of the Hi-C data sets that are currently available are from cell lines and cancer cells. Only few studies have focused on tissues from organs [211, 212], and most of them have a small number of read pairs, which cannot identify all chromatin loops but only identifies large TADs (Table 4). Higher genome coverage is recommended to perform comparison analyses between Hi-C data sets and call chromatin loops for regulatory elements [177, 213]. Therefore, additional higher resolution data sets using Hi-C or 3C-derived methods are greatly needed.
Conclusions
There have been striking improvements in both molecular and computational methods to analyze regulatory elements over the last decade. Chromatin immunoprecipitation, chromatin accessibility, and DNA methylation assays have annotated regulatory elements and revealed interactions between TFs and regulatory elements. Recently developed 3C-based methods have shown how these regulatory elements interact with each other genome-wide. Moreover, new methods enable further research of regulatory elements and their interactions in single cell and single molecule resolution. Although thousands of epigenomic data sets have been generated up until now, profiling of regulatory elements and chromatin structures in additional normal and diseased cells is in great demand, because 3D epigenetic signatures are distinct among cell types and cell populations. Further identification and characterization of regulatory elements that control transcription in a cell-type specific manner will enlighten novel molecular mechanisms of gene regulation and diseases.
Availability of data and materials
Not applicable.
Abbreviations
- 3C:
-
Chromatin conformation capture
- 4C:
-
Circular chromosome conformation capture
- 5C:
-
Carbon copy chromosome conformation capture
- 4DN:
-
The 4D nucleome
- AMED-CREST:
-
Japan agency of medical research and development & core research for evolutional science and technology
- ATAC-seq:
-
Assay of transpose accessible chromatin sequencing
- CEEHRC:
-
Canadian epigenetics, environment and health research consortium
- ChIA-PET:
-
Chromatin interaction analysis by paired-end tag
- ChIP-seq:
-
Chromatin immunoprecipitation sequencing
- CTCF:
-
CCCTC-binding factor
- CUT & RUN:
-
Cleavage under targets and release using nuclease
- CUT & TAG:
-
Cleavage under targets and tagmentation
- DEEP:
-
Deutsches epigenom program
- DNA FISH:
-
DNA fluorescence in situ hybridization
- DNase I:
-
Deoxyribonuclease I
- DHS:
-
DNase hypersensitivity sites
- DNase-seq:
-
Deoxyribonuclease I hypersensitive sites sequencing
- DNMT:
-
DNA methyltransferase
- EMBL-EBI:
-
European molecular biology laboratory–European bioinformatics institute
- ENA:
-
European nucleotide archive
- ENCODE:
-
Encyclopedia of DNA elements
- FAIRE-seq:
-
Formaldehyde-assisted isolation of regulatory elements sequencing
- GEO:
-
Gene expression omnibus
- HM:
-
Human methylation
- ICE:
-
Iterative correction and eigenvector decomposition
- IDR:
-
Irreproducible discovery rate
- IGB:
-
Integrated genome browser
- IGV:
-
Integrative genomics viewer
- IHEC:
-
The international human epigenome consortium
- KINH:
-
Korean national institute of health
- MNase:
-
Micrococcal nuclease
- MNase-seq:
-
Micrococcal nuclease digestion with sequencing
- NCBI:
-
The national center for biotechnology information
- NDRs:
-
Nucleosome depleted regions
- NOMe-seq:
-
Nucleosome occupancy and methylome sequencing
- NRF:
-
Non-redundant fraction
- NSC:
-
Normalized strand cross-correlation coefficient
- pA-MN:
-
Protein A and micrococcal nuclease
- pA-Tn5:
-
Protein A and Tn5 transposase
- PBC:
-
PCR bottleneck coefficient
- QC:
-
Quality check
- REMC:
-
Roadmap epigenomics mapping consortium
- RRBS:
-
Reduced representation bisulfite sequencing
- SCN:
-
Sequential component normalization
- SPRITE:
-
Split-pool recognition of interactions by tag extension
- SPOT:
-
Signal portion of tags
- TADs:
-
Topologically associating domains
- TCGA:
-
The cancer genome atlas
- TFs:
-
Transcription factors
- TSS:
-
Transcription start sites
- WGBS:
-
Whole genome bisulfite sequencing
References
Deichmann U. Epigenetics: The origins and evolution of a fashionable topic. Dev Biol. 2016;416:249–54.
Mariño-Ramírez L, Kann MG, Shoemaker BA, Landsman D. Histone structure and nucleosome stability. Expert Rev Proteomics. 2014;2:719–29.
Andrew JBTK. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–95.
Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610.
Maston GA, Evans SK, Green MR. Transcriptional Regulatory Elements in the Human Genome. Annu Rev Genomics Hum Genet. 2006;7:29–59.
Roy AL, Singer DS. Core promoters in transcription: old problem, new insights. Trends Biochem Sci. 2015;40:165–71.
Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15:272–86.
Gaszner M, Felsenfeld G. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet. 2006;7:703–13.
Holwerda S, de Laat W. Chromatin loops, gene positioning, and gene expression. Front Genet. 2012;3:217.
Iourl Chepelev Gang Wei Dara Wangsa Qingsong Tang Keji Z: Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res. 2012; 22:490–503.
West AG. Insulators: many functions, many mechanisms. Genes Dev. 2002;16:271–88.
Zoghbi HY, Beaudet AL. Epigenetics and Human Disease. Cold Spring Harbor Perspect Biol. 2016;8:a019497.
Rhie SK, Perez AA, Lay FD, Schreiner S, Shi J, Polin J, Farnham PJ. A high-resolution 3D epigenomic map reveals insights into the creation of the prostate cancer transcriptome. Nat Commun. 2019;10:4154.
Rhie SK, Schreiner S, Witt H, Armoskus C, Lay FD, Camarena A, Spitsyna VN, Guo Y, Berman BP, Evgrafov OV, et al. Using 3D epigenomic maps of primary olfactory neuronal cells from living individuals to understand gene regulation. Science advances. 2018;4:eaav8550.
Flavahan WA, Drier Y, Johnstone SE, Hemming ML, Tarjan DR, Hegazi E, Shareef SJ, Javed NM, Raut CP, Eschle BK, et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature. 2019;575:229–33.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92.
Rhie SK, Schreiner S, Farnham PJ. Defining regulatory elements in the human genome using nucleosome occupancy and methylome sequencing (NOMe-Seq). CpG Islands. 2018;1766:209–29.
Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–502.
Farnham PJ. Insights from genomic profiling of transcription factors. Nat Rev Genet. 2009;10:605–16.
Liu ET, Pott S, Huss M. Q&A: ChIP-seq technologies and the study of gene regulation. BMC Biol. 2010;8:56.
Park PJ. ChIP–seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–80.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Comput Appl Biosci. 2009;25:1754–60.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
FastQC: A Quality Control Tool for High Throughput Sequence Data [https://www.bioinformatics.babraham.ac.uk/projects/fastqc/]
Picard Tools [https://github.com/broadinstitute/picard]
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Comput Appl Biosci. 2010;26:841–2.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Comput Appl Biosci. 2009;25:2078–9.
Bailey T, Krajewski P, Ladunga I, Lefebvre C, Li Q, Liu T, Madrigal P, Taslim C, Zhang J. Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data. PLoS Comput Biol. 2013;9:e1003326.
Johnston MJ, Nikolic A, Ninkovic N, Guilhamon P, Cavalli FMG, Seaman S, Zemp FJ, Lee J, Abdelkareem A, Ellestad K, et al. High-resolution structural genomics reveals new therapeutic vulnerabilities in glioblastoma. Genome Res. 2019;29:1211–22.
Chitpin JG, Awdeh A, Perkins TJ. RECAP reveals the true statistical significance of ChIP-seq peak calls. Bioinformatics. 2019;35:3592–8.
Wilbanks EG, Facciotti MT. Evaluation of algorithm performance in ChIP-seq peak detection. PLoS ONE. 2010;5:e11471.
Thomas R, Thomas S, Holloway AK, Pollard KS. Features that define the best ChIP-seq peak calling algorithms. Brief Bioinform. 2017;18:441–50.
Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol. 2008;26:1351–9.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137.
Li Q, Brown JB, Huang H, Bickel PJ. Measuring reproducibility of high-throughput experiments. Ann Appl Stat. 2011;5:1752–79.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B Cell identities. Mol Cell. 2010;38:576–89.
Yu G, Wang L-G, He Q-Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Comput Appl Biosci. 2015;31:2382–3.
Steinhauser S, Kurzawa N, Eils R, Herrmann C. A comprehensive comparison of tools for differential ChIP-seq analysis. Brief Bioinform. 2016;17:953–66.
Lun ATL, Smyth GK. csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 2016;44:e45.
Ross-Innes CS, Stark R, Ali S, Chin S-F, Palmieri C, Caldas C, Carroll JS, Teschendorff AE, Holmes KA, Raza Ali H, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–93.
Shao Z, Zhang Y, Yuan G-C, Orkin SH, Waxman DJ. MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 2012;13:R16.
Bailey TL, Johnson J, Grant CE, Noble WS. The MEME suite. Nucleic Acids Res. 2015;43:W39–49.
Matys V. TRANSFAC(R) and its module TRANSCompel(R): transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–10.
Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranašić D, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2019;48:D87–92.
Wang J, Zhuang J, Iyer S, Lin XY, Greven MC, Kim BH, Moore J, Pierce BG, Dong X, Virgil D, et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2012;41:171–6.
Rhee HS, Pugh BF. ChIP-exo: A method to identify genomic location of DNA-binding proteins at near single nucleotide accuracy. Current Protocol Mol Biol. 2012;21:21–4.
Yamada N, Lai WKM, Farrell N, Pugh BF, Mahony S. Characterizing protein–DNA binding event subtypes in ChIP-exo data. Comput Appl Biosci. 2019;35:903–13.
Wang L, Chen J, Wang C, Uusküla-Reimand L, Chen K, Medina-Rivera A, Young EJ, Zimmermann MT, Yan H, Sun Z, et al. MACE: model based analysis of ChIP-exo. Nucleic Acids Res. 2014;42:156.
Skene PJ, Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife. 2017;6:e21856.
Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, Henikoff S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 1930;2019:10.
Meers MP, Tenenbaum D, Henikoff S. Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenet Chromatin. 2019;12:42.
Zhu Q, Liu N, Orkin SH, Yuan G-C. CUT&RUNTools: a flexible pipeline for CUT&RUN processing and footprint analysis. Genome Biol. 2019;20:192.
CUT&Tag Data Processing and Analysis Tutorial [https://www.protocols.io/view/cut-amp-tag-data-processing-and-analysis-tutorial-bjk2kkye]
Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc. 2010;2010:pdb.prot5384.
Schones DE, Cui K, Cuddapah S, Roh T-Y, Barski A, Wang Z, Wei G, Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–98.
Simon JM, Giresi PG, Davis IJ, Lieb JD. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat Protoc. 2012;7:256–67.
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for multimodal regulatory analysis and personal epigenomics. Nat Methods. 2013;10:1213–8.
Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones PA. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012;22:2497–506.
Tsompana M, Buck MJ. Chromatin accessibility: a window into the genome. Epigenet Chromatin. 2014;7:33.
Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–22.
Koohy H, Down TA, Hubbard TJ. Chromatin accessibility data sets show bias due to sequence specificity of the DNase I enzyme. PLoS ONE. 2013;8:e69853.
John S, Sabo PJ, Canfield TK, Lee K, Vong S, Weaver M, Wang H, Vierstra J, Reynolds AP, Thurman RE, Stamatoyannopoulos JA. Genome-scale mapping of DNase I hypersensitivity. Curr Protocols Mol Biol. 2013;103:21–7.
John S, Sabo PJ, Thurman RE, Sung M-H, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43:264–8.
Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6:283–9.
Pique-Regi R, Degner JF, Pai AA, Gaffney DJ, Gilad Y, Pritchard JK. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 2010;21:447–55.
Sung M-H, Guertin Michael J, Baek S, Hager Gordon L. DNase footprint signatures are dictated by factor dynamics and DNA sequence. Mol Cell. 2014;56:275–85.
Kumar V, Muratani M, Rayan NA, Kraus P, Lufkin T, Ng HH, Prabhakar S. Uniform, optimal signal processing of mapped deep-sequencing data. Nat Biotechnol. 2013;31:615–22.
He HH, Meyer CA, Hu SeS, Chen M-W, Zang C, Liu Y, Rao PK, Fei T, Xu H, Long H, et al. Analysis of optimized DNase-seq reveals intrinsic bias in transcription factor footprint identification. Nat Methods. 2014;11:73–8.
Kuan PF, Huebert D, Gasch A, Keles S. A non-homogeneous hidden-state model on first order differences for automatic detection of nucleosome positions. Statist Applicat Genet Mol Biol. 2009;8:29–45.
Cui K, Zhao K. Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq. Chromatin remodeling. 2012;833:413–9.
Zentner GE, Henikoff S. Surveying the epigenomic landscape, one base at a time. Genome Biol. 2012;13:250.
Hörz W, Altenburger W. Sequence specific cleavage of DNA by micrococcal nuclease. Nucleic Acids Res. 1981;9:2643–58.
Dingwall C, Lomonossoff GP, Laskey RA. High sequence specificity of micrococcal nuclease. Nucleic Acids Res. 1981;9:2659–74.
Cockell M, Rhodes D, Klug A. Location of the primary sites of micrococcal nuclease cleavage on the nucleosome core. J Mol Biol. 1983;170:423–46.
Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, Li W. DANPOS: Dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2012;23:341–51.
Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, Rodriguez B, Xi Y, Xia Z, Chen X, et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet. 2015;47:1149–57.
Zhang H, Li F, Jia Y, Xu B, Zhang Y, Li X, Zhang Z. Characteristic arrangement of nucleosomes is predictive of chromatin interactions at kilobase resolution. Nucleic Acids Res. 2017;45:12739–51.
Schwartz U, Németh A, Diermeier S, Exler JH, Hansch S, Maldonado R, Heizinger L, Merkl R, Längst G. Characterizing the nuclease accessibility of DNA in human cells to map higher order structures of chromatin. Nucleic Acids Res. 2019;47:1239–54.
Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17:877–85.
Boyle AP, Guinney J, Crawford GE, Furey TS. F-Seq: a feature density estimator for high-throughput sequence tags. Comput Appl Biosci. 2008;24:2537–8.
Buck MJ, Nobel AB, Lieb JD. ChIPOTle: a user-friendly tool for the analysis of ChIP-chip data. Genome Biol. 2005;6:R97.
Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3:1101–8.
Barua S, Kuizon S, Chadman KK, Flory MJ, Brown WT, Junaid MA. Single-base resolution of mouse offspring brain methylome reveals epigenome modifications caused by gestational folic acid. Epigenet Chromatin. 2014;7:3.
Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Graf S, Huss M, Keefe D, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21:1757–67.
Giresi PG, Lieb JD. Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements). Methods. 2009;48:233–9.
Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14:959–62.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;2011(17):10–2. https://doi.org/10.14806/ej171200.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Comput Appl Biosci. 2014;30:2114–20.
Tarbell ED, Liu T. HMMRATAC: a hidden markov ModeleR for ATAC-seq. Nucleic Acids Res. 2019;47:e91.
Li Z, Schulz MH, Look T, Begemann M, Zenke M, Costa IG. Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 2019;20:45.
Wu J, Huang B, Chen H, Yin Q, Liu Y, Xiang Y, Zhang B, Liu B, Wang Q, Xia W, et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature. 2016;534:652–7.
Montefiori L, Hernandez L, Zhang Z, Gilad Y, Ober C, Crawford G, Nobrega M, Jo Sakabe N. Reducing mitochondrial reads in ATAC-seq using CRISPR/Cas9. Sci Rep. 2017;7:2451–9.
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo Q-M, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22.
Darst RP, Nabilsi NH, Pardo CE, Riva A, Kladde MP. DNA Methyltransferase accessibility protocol for individual templates by deep sequencing. Methods Enzymol. 2012;513:185–204.
Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013;23:555–67.
Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformat. 2009;10:232.
Fast and accurate alignment of long bisulfite-seq reads [https://github.com/brentp/bwa-meth]
Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Comput Appl Biosci. 2011;27:1571–2.
Chen P-Y, Cokus SJ, Pellegrini M. BS Seeker: precise mapping for bisulfite sequencing. BMC Bioinformat. 2010;11:203.
Biscuit [https://github.com/zhou-lab/biscuit]
ECWorkflows [https://github.com/uec/ECWorkflows]
Liu Y, Siegmund KD, Laird PW, Berman BP. Bis-SNP: Combined DNA methylation and SNP calling for Bisulfite-seq data. Genome Biol. 2012;13:R61.
aaRon R package [https://github.com/astatham/aaRon]
Bis-tools [https://github.com/dnaase/Bis-tools]
Han L, Lin IG, Hsieh CL. Protein binding protects sites on stable episomes and in the chromosome from De Novo methylation. Mol Cell Biol. 2001;21:3416–24.
Jin B, Li Y, Robertson KD. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes & cancer. 2011;2:607–17.
Meissner A. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–77.
Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203.
Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, Gunderson KL. Genome-wide DNA methylation profiling using Infinium ® assay. Epigenomics. 2009;1:177–200.
Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2014;6:692–702.
Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 2016;8:389–99.
Sun D, Xi Y, Rodriguez B, Park H, Tong P, Meong M, Goodell MA, Li W. MOABS: model based analysis of bisulfite sequencing data. Genome Biol. 2014;15:R38.
Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Comput Appl Biosci. 2014;30:1363–9.
Zhou W, Triche TJ, Laird PW, Shen H. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 2018;46:e123.
Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, V Lord R, Clark SJ, Molloy PL. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6.
Karolchik D, Hinrichs AS, Kent WJ. The UCSC genome browser. Curr Proto Bioinformat. 2009;71:18–26.
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6.
Nicol JW, Helt GA, Blanchard SG, Raja A, Loraine AE. The integrated genome browser: free software for distribution and exploration of genome-scale datasets. Comput Appl Biosci. 2009;25:2730–1.
Birney E. An overview of ensembl. Genome Res. 2004;14:925–8.
Zhou X, Wang T. Using the Wash U Epigenome browser to examine genome-wide sequencing data. Curr Prot Bioinformat. 2012;40:10.
Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–90.
Jia G, Preussner J, Chen X, Guenther S, Yuan X, Yekelchyk M, Kuenne C, Looso M, Zhou Y, Teichmann S, Braun T. Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat Commun. 2018;9:4877.
Pott S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. eLife. 2017;6:e23203.
Yu B, Dong X, Gravina S, Kartal Ö, Schimmel T, Cohen J, Tortoriello D, Zody R, Hawkins RD, Vijg J. Genome-wide, single-cell DNA methylomics reveals increased non-cpg methylation during human oocyte maturation. Stem Cell Rep. 2017;9:397–407.
Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, Kaul R, et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699–710.
Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30.
Psych EC, The Psych EC. Revealing the brain’s molecular architecture. Science. 2018;362:1262–3.
The, editorial t, Cell editorial t: a cornucopia of advances in human epigenomics. Cell 2016; 167:1139.
Clough E, Barrett T. The gene expression omnibus database. Statistical Genomics. 2016;1418:93–110.
Leinonen R, Akhtar R, Birney E, Bower L, Cerdeno-Tarraga A, Cheng Y, Cleland I, Faruque N, Goodgame N, Gibson R, et al. The European nucleotide archive. Nucleic Acids Res. 2010;39:D28–31.
Bujold D, Morais DAdL, Gauthier C, Côté C, Caron M, Kwan T, Chen KC, Laperle J, Markovits AN, Pastinen T, et al. The international human epigenome consortium data portal. Cell systems. 2016;3:496–9.
Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, Chen C-H, Brown M, Zhang X, Meyer CA, Liu XS. Cistrome data browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019;47:D729–35.
Chèneby J, Ménétrier Z, Mestdagh M, Rosnet T, Douida A, Rhalloussi W, Bergon A, Lopez F, Ballester B. ReMap 2020: a database of regulatory regions from an integrative analysis of human and Arabidopsis DNA-binding sequencing experiments. Nucleic Acids Res. 2019;48:D180–8.
Sati S, Cavalli G. Chromosome conformation capture technologies and their impact in understanding genome function. Chromosoma. 2016;126:33–44.
Misteli T. Higher-order genome organization in human disease. Cold Spring Harbor Perspect Biol. 2010;2:000794.
Yu M, Ren B. The three-dimensional organization of mammalian genomes. Annu Rev Cell Dev Biol. 2017;33:265–89.
Eagen KP, Aiden EL, Kornberg RD. Polycomb-mediated chromatin loops revealed by a subkilobase-resolution chromatin interaction map. Proceed Nat Acad Sci PNAS. 2017;114:8764–9.
Ngan CY, Wong CH, Tjong H, Wang W, Goldfeder RL, Choi C, He H, Gong L, Lin J, Urban B, et al. Chromatin interaction analyses elucidate the roles of PRC2-bound silencers in mouse development. Nat Genet. 2020;52:264–72.
Rhodes JDP, Feldmann A, Hernández-Rodríguez B, Díaz N, Brown JM, Fursova NA, Blackledge NP, Prathapan P, Dobrinic P, Huseyin MK, et al. Cohesin disrupts polycomb-dependent chromosome interactions in embryonic stem cells. Cell reports. 2020;30:820–35.
Rao Suhas SP, Huntley Miriam H, Durand Neva C, Stamenova Elena K, Bochkov Ivan D, Robinson James T, Sanborn Adrian L, Machol I, Omer Arina D, Lander Eric S, Aiden Erez L. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–80.
Cavalli G, Misteli T. Functional implications of genome topology. Nat Struct Mol Biol. 2013;20:290–9.
Dekker J. Capturing chromosome conformation. Science. 2002;295:1306–11.
Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nat Genet. 2006;38:1348–54.
Klein FA, Pakozdi T, Anders S, Ghavi-Helm Y, Furlong EEM, Huber W. FourCSeq: analysis of 4C sequencing data. Bioinformatics. 2015;31:3085–91.
Krijger PHL, Geeven G, Bianchi V, Hilvering CRE, de Laat W. 4C-seq from beginning to end: a detailed protocol for sample preparation and data analysis. Methods. 2019;170:17–32.
Langmead B. Aligning Short Sequencing Reads with Bowtie. Curr Protocols Bioinformat. 2010;32:11–7.
Novoalign [http://www.novocraft.com/]
Geeven G, Teunissen H, de Laat W, de Wit E. peakC: a flexible, non-parametric peak calling package for 4C and Capture-C data. Nucleic Acids Res. 2018;46:e91.
Raviram R, Rocha PP, Müller CL, Miraldi ER, Badri S, Fu Y, Swanzey E, Proudhon C, Snetkova V, Bonneau R, Skok JA. 4C-ker: a method to reproducibly identify genome-wide interactions captured by 4C-Seq experiments. PLoS Computat Biol. 2016;12:e1004780.
Williams RL, Starmer J, Mugford JW, Calabrese JM, Mieczkowski P, Yee D, Magnuson T. fourSig: a method for determining chromosomal interactions in 4C-Seq data. Nucleic Acids Res. 2014;42:e68.
Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–309.
Beagan Jonathan A, Gilgenast Thomas G, Kim J, Plona Z, Norton Heidi K, Hu G, Hsu Sarah C, Shields Emily J, Lyu X, Apostolou E, et al. Local genome topology can exhibit an incompletely rewired 3D-folding state during somatic cell reprogramming. Cell Stem Cell. 2016;18:611–24.
Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–5.
Sauria MEG, Phillips-Cremins JE, Corces VG, Taylor J. HiFive: a tool suite for easy and efficient HiC and 5C data analysis. Genome Biol. 2015;16:237.
Sanyal A, Dekker J, van Berkum NL, Lajoie BR. My5C: web tools for chromosome conformation capture studies. Nat Methods. 2009;6:690–1.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93.
Pombo A, Dillon N. Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol. 2015;16:245–57.
Bonev B, Cavalli G. Organization and function of the 3D genome. Nat Rev Genet. 2016;17:772.
Servant N, Varoquaux N, Lajoie BR, Viara E, Chen C-J, Vert J-P, Heard E, Dekker J, Barillot E. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259.
Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, Aiden EL. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016;3:95–8.
Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, Mirny LA. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods. 2012;9:999–1003.
Distiller (https://github.com/open2c/distiller-nf)
Hu M, Deng K, Selvaraj S, Qin Z, Ren B, Liu JS. HiCNorm: removing biases in Hi-C data via poisson regression. Bioinformatics. 2012;28:3131–3.
Yaffe E, Tanay A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat Genet. 2011;43:1059–65.
Cournac A, Marie-Nelly H, Marbouty M, Koszul R, Mozziconacci J. Normalization of a chromosomal contact map. BMC Genomics. 2012;13:436.
Wu H-J, Michor F. A computational strategy to adjust for copy number in tumor Hi-C data. Comput Appl Biosci. 2016;32:3695–701.
Stansfield JC, Cresswell KG, Dozmorov MG. multiHiCcompare: joint normalization and comparative analysis of complex Hi-C experiments. Comput Appl Biosci. 2019;35:2916–23.
Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–80.
Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. Three-dimensional folding and functional organization principles of the drosophila genome. Cell. 2012;148:458–72.
Rowley MJ, Nichols MH, Lyu X, Ando-Kuri M, Rivera ISM, Hermetz K, Wang P, Ruan Y, Corces VG. Evolutionarily conserved principles predict 3D chromatin organization. Mol Cell. 2017;67:837–52.
Harmston N, Ing-Simmons E, Tan G, Perry M, Merkenschlager M, Lenhard B. Topologically associating domains are ancient features that coincide with Metazoan clusters of extreme noncoding conservation. Nat Commun. 2017;8:441.
Phillips-Cremins Jennifer E, Sauria Michael EG, Sanyal A, Gerasimova Tatiana I, Lajoie Bryan R, Bell Joshua SK, Ong C-T, Hookway Tracy A, Guo C, Sun Y, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–95.
Shin HJ, Shi Y, Dai C, Tjong H, Gong K, Alber F, Zhou XJ. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2016;44:e70.
Levy-Leduc C, Delattre M, Mary-Huard T, Robin S. Two-dimensional segmentation for analyzing Hi-C data. Comput Appl Biosci. 2014;30:i386–92.
Zhan Y, Mariani L, Barozzi I, Schulz EG, Blüthgen N, Stadler M, Tiana G, Giorgetti L. Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes. Genome Res. 2017;27:479–90.
Zufferey M, Tavernari D, Oricchio E, Ciriello G. Comparison of computational methods for the identification of topologically associating domains. Genome Biol. 2018;19:217.
Mifsud B, Martincorena I, Darbo E, Sugar R, Schoenfelder S, Fraser P, Luscombe NM. GOTHiC, a probabilistic model to resolve complex biases and to identify real interactions in Hi-C data. PLoS ONE. 2017;12:e0174744.
Ay F, Bailey TL, Noble WS. Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. Genome Res. 2014;24:999–1011.
Kaul A, Bhattacharyya S, Ay F. Identifying statistically significant chromatin contacts from Hi-C data with FitHiC2. Nat Protoc. 2020;15:991–1012.
Rowley MJ, Poulet A, Nichols MH, Bixler BJ, Sanborn AL, Brouhard EA, Hermetz K, Linsenbaum H, Csankovszki G, Lieberman Aiden E, Corces VG. Analysis of Hi-C data using SIP effectively identifies loops in organisms from C. elegans to mammals. Genome Res. 2020;30:447–58.
Roayaei Ardakany A, Gezer HT, Lonardi S, Ay F. Mustache: multi-scale detection of chromatin loops from Hi-C and Micro-C maps using scale-space representation. Genome Biol. 2020;21:1–256.
Krismer K, Guo Y, Gifford DK. IDR2D identifies reproducible genomic interactions. Nucleic Acids Res. 2020;48:e31.
Yang T, Zhang F, Yardımcı GG, Song F, Hardison RC, Noble WS, Yue F, Li Q. HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res. 2017;27:1939–49.
Yan K-K, Yardımcı GG, Yan C, Noble WS, Gerstein M. HiC-spector: a matrix library for spectral and reproducibility analysis of Hi-C contact maps. Comput Appl Biosci. 2017;33:2199–201.
Stansfield JC, Cresswell KG, Vladimirov VI, Dozmorov MG. HiCcompare: an R-package for joint normalization and comparison of HI-C datasets. BMC Bioinformat. 2018;19:279.
Djekidel MN, Chen Y, Zhang MQ. FIND: difFerential chromatin INteractions Detection using a spatial Poisson process. Genome Res. 2018;28:412–22.
Ardakany AR, Ay F, Lonardi S. Selfish: discovery of differential chromatin interactions via a self-similarity measure. Comput Appl Biosci. 2019;35:i145–53.
Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016;3:99–101.
Kerpedjiev P, Abdennur N, Lekschas F, McCallum C, Dinkla K, Strobelt H, Luber JM, Ouellette SB, Azhir A, Kumar N, et al. HiGlass: web-based visual exploration and analysis of genome interaction maps. Genome Biol. 2018;19:125.
Akdemir KC, Chin L. HiCPlotter integrates genomic data with interaction matrices. Genome Biol. 2015;16:198.
Servant N, Lajoie BR, Nora EP, Giorgetti L, Chen C-J, Heard E, Dekker J, Barillot E. HiTC: exploration of high-throughput ‘C’ experiments. Comput Appl Biosci. 2012;28:2843–4.
Wang Y, Song F, Zhang B, Zhang L, Xu J, Kuang D, Li D, Choudhary MNK, Li Y, Hu M, et al. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 2018;19:151.
Han HT, Weiner A, Lajoie B, Dekker J, Friedman N, Rando Oliver J. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell. 2015;162:108–19.
Ramani V, Cusanovich DA, Hause RJ, Ma W, Qiu R, Deng X, Blau CA, Disteche CM, Noble WS, Shendure J, Duan Z. Mapping 3D genome architecture through in situ DNase Hi-C. Nat Protoc. 2016;11:2104–21.
Ma W, Ay F, Lee C, Gulsoy G, Deng X, Cook S, Hesson J, Cavanaugh C, Ware CB, Krumm A, et al. Fine-scale chromatin interaction maps reveal the cis-regulatory landscape of human lincRNA genes. Nat Methods. 2015;12:71–8.
Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, De Gobbi M, Taylor S, Gibbons R, Higgs DR. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat Genet. 2014;46:205–12.
Dryden NH, Broome LR, Dudbridge F, Johnson N, Orr N, Schoenfelder S, Nagano T, Andrews S, Wingett S, Kozarewa I, et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res. 2014;24:1854–68.
Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. An oestrogen-receptor-α-bound human chromatin interactome. Nature. 2009;462:58–64.
Li G, Fullwood MJ, Xu H, Mulawadi FH, Velkov S, Vega V, Ariyaratne PN, Mohamed YB, Ooi H-S, Tennakoon C, et al. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010;11:R22.
ChIA-PET Protocol Standards for ENCODE4 [https://www.encodeproject.org/documents/480f9184-07a5-4e57-bc33-ca6f675a1f97/@@download/attachment/ChIA-PET%20protocol%20for%20ENCODE4_Final.pdf]
Phanstiel DH, Boyle AP, Heidari N, Snyder MP. Mango: a bias-correcting ChIA-PET analysis pipeline. Bioinformatics. 2015;31:3092–8.
Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, Chang HY. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods. 2016;13:919–22.
Lareau CA, Aryee MJ. hichipper: a preprocessing pipeline for calling DNA loops from HiChIP data. Nat Methods. 2018;15:155–6.
Bhattacharyya S, Chandra V, Vijayanand P, Ay F. Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat Commun. 2019;10:4221.
Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64.
Ramani V, Deng X, Qiu R, Gunderson KL, Steemers FJ, Disteche CM, Noble WS, Duan Z, Shendure J. Massively multiplex single-cell Hi-C. Nat Methods. 2017;14:263–6.
Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, et al. Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell. 2018;174:744–57.
Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348:aaa6090.
Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, Lomvardas S, Mirny LA, O’Shea CC, Park PJ, Ren B, et al. The 4D nucleome project. Nature. 2017;549:219–26.
Greenwald WW, Li H, Benaglio P, Jakubosky D, Matsui H, Schmitt A, Selvaraj S, D’Antonio M, D’Antonio-Chronowska A, Smith EN, Frazer KA. Subtle changes in chromatin loop contact propensity are associated with differential gene regulation and expression. Nat Commun. 2019;10:1054.
Leung D, Jung I, Rajagopal N, Schmitt A, Selvaraj S, Lee AY, Yen C-A, Lin S, Lin Y, Qiu Y, et al. Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature. 2015;518:350–4.
Cameron CJF, Dostie J, Blanchette M. HIFI: estimating DNA-DNA interaction frequency from Hi-C data at restriction-fragment resolution. Genome Biol. 2020;21:11.
Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, Manke T. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun. 2018;9:189.
Wingett SW, Ewels P, Furlan-Magaril M, Nagano T, Schoenfelder S, Fraser P, Andrews S. HiCUP: pipeline for mapping and processing Hi-C data. F1000 Research. 2015;4:1310.
Serra F, Baù D, Goodstadt M, Castillo D, Filion GJ, Marti-Renom MA. Automatic analysis and 3D-modelling of Hi-C data using TADbit reveals structural features of the fly chromatin colors. PLoS Computat Biol. 2017;13:e1005665.
Chen F, Li G, Zhang MQ, Chen Y. HiCDB: a sensitive and robust method for detecting contact domain boundaries. Nucleic Acids Res. 2018;46:11239–50.
Cresswell KG, Dozmorov MG. TADCompare: an R package for differential and temporal analysis of topologically associated domains. Front Genet. 2020;11:158.
Soler-Vila P, Cuscó P, Farabella I, Di Stefano M, Marti-Renom Marc A. Hierarchical chromatin organization detected by TADpole. Nucleic Acids Res. 2020;48:e39.
Weinreb C, Raphael BJ. Identification of hierarchical chromatin domains. Comput Appl Biosci. 2016;32:1601–9.
Norton HK, Emerson DJ, Huang H, Kim J, Titus KR, Gu S, Bassett DS, Phillips-Cremins JE. Detecting hierarchical genome folding with network modularity. Nat Methods. 2018;15:119–22.
Carty M, Zamparo L, Sahin M, González A, Pelossof R, Elemento O, Leslie CS. An integrated model for detecting significant chromatin interactions from high-resolution Hi-C data. Nat Commun. 2017;8:15454.
FIREcaller [https://github.com/yycunc/FIREcaller]
Vian L, Pękowska A, Rao SSP, Kieffer-Kwon K-R, Jung S, Baranello L, Huang S-C, El Khattabi L, Dose M, Pruett N, et al. The energetics and physiological impact of cohesin extrusion. Cell. 2018;173:1165–78.
Spill YG, Castillo D, Vidal E, Marti-Renom MA. Binless normalization of Hi-C data provides significant interaction and difference detection independent of resolution. Nat Commun. 1938;2019:10.
Lu L, Liu X, Huang W-K, Giusti-Rodríguez P, Cui J, Zhang S, Xu W, Wen Z, Ma S, Rosen JD, et al. Robust Hi-C maps of enhancer-promoter interactions reveal the function of non-coding genome in neural development and diseases. Mol Cell. 2020;79:521–34.
Vidal E, le Dily F, Quilez J, Stadhouders R, Cuartero Y, Graf T, Marti-Renom MA, Beato M, Filion GJ. OneD: increasing reproducibility of Hi-C samples with abnormal karyotypes. Nucleic Acids Res. 2018;46:e49.
Ramirez RN, Bedirian K, Gray SM, Diallo A. DNA Rchitect: an R based visualizer for network analysis of chromatin interaction data. Bioinformatics. 2020;36:644–6.
Kumar R, Sobhy H, Stenberg P, Lizana L. Genome contact map explorer: a platform for the comparison, interactive visualization and analysis of genome contact maps. Nucl Acids Res. 2017;45:e152.
Selvaraj S, R Dixon J, Bansal V, Ren B. Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nat Biotechnol. 2013;31:1111–8.
Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen C-A, Schmitt AD, Espinoza CA, Ren B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290–4.
Le Dily F, Baù D, Pohl A, Vicent GP, Serra F, Soronellas D, Castellano G, Wright RHG, Ballare C, Filion G, et al. Distinct structural transitions of chromatin topological domains correlate with coordinated hormone-induced gene regulation. Genes Dev. 2014;28:2151–62.
Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–6.
Grubert F, Zaugg Judith B, Kasowski M, Ursu O, Spacek Damek V, Martin Alicia R, Greenside P, Srivas R, Phanstiel Doug H, Pekowska A, et al. Genetic control of chromatin states in humans involves local and distal chromosomal interactions. Cell. 2015;162:1051–65.
Adrian LS, Suhas SPR, Su-Chen H, Neva CD, Miriam HH, Andrew IJ, Ivan DB, Dharmaraj C, Ashok C, Jian L, et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proceed Nat Acad Sci PNAS. 2015;112:E6456.
Schmitt Anthony D, Hu M, Jung I, Xu Z, Qiu Y, Tan Catherine L, Li Y, Lin S, Lin Y, Barr Cathy L, Ren B. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell reports. 2016;17:2042–59.
Taberlay PC, Achinger-Kawecka J, Lun ATL, Buske FA, Sabir K, Gould CM, Zotenko E, Bert SA, Giles KA, Bauer DC, et al. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res. 2016;26:719–31.
Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 2016;538:523–7.
Fritz AJ, Ghule PN, Boyd JR, Tye CE, Page NA, Hong D, Shirley DJ, Weinheimer AS, Barutcu AR, Gerrard DL, et al. Intranuclear and higher-order chromatin organization of the major histone gene cluster in breast cancer. J Cell Physiol. 2018;233:1278–90.
Haarhuis JHI, van der Weide RH, Blomen VA, Yáñez-Cuna JO, Amendola M, van Ruiten MS, Krijger PHL, Teunissen H, Medema RH, van Steensel B, et al. The cohesin release factor WAPL restricts chromatin loop extension. Cell. 2017;169:693–707.
Phanstiel DH, Van Bortle K, Spacek D, Hess GT, Shamim MS, Machol I, Love MI, Aiden EL, Bassik MC, Snyder MP. Static and dynamic DNA loops form AP-1-bound activation hubs during macrophage development. Mol Cell. 2017;67:1037–48.
Rao SSP, Huang S-C, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon K-R, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. Cohesin Loss Eliminates All Loop Domains. Cell. 2017;171:305–20.
Rubin AJ, Barajas BC, Furlan-Magaril M, Lopez-Pajares V, Mumbach MR, Howard I, Kim DS, Boxer LD, Cairns J, Spivakov M, et al. Lineage-specific dynamic and pre-established enhancer–promoter contacts cooperate in terminal differentiation. Nat Genet. 2017;49:1522–8.
Li Y, He Y, Liang Z, Wang Y, Chen F, Djekidel MN, Li G, Zhang X, Xiang S, Wang Z, et al. Alterations of specific chromatin conformation affect ATRA-induced leukemia cell differentiation. Cell Death Dis. 2018;9:200–15.
Lin D, Hong P, Zhang S, Xu W, Jamal M, Yan K, Lei Y, Li L, Ruan Y, Fu ZF, et al. Digestion-ligation-only Hi-C is an efficient and cost-effective method for chromosome conformation capture. Nat Genet. 2018;50:754–63.
Abramo K, Valton A-L, Venev SV, Ozadam H, Fox AN, Dekker J. A chromosome folding intermediate at the condensin-to-cohesin transition during telophase. Nat Cell Biol. 2019;21:1393–402.
Gorkin DU, Qiu Y, Hu M, Fletez-Brant K, Liu T, Schmitt AD, Noor A, Chiou J, Gaulton KJ, Sebat J, et al. Common DNA sequence variation influences 3-dimensional conformation of the human genome. Genome Biol. 2019;20:255.
Ray J, Munn PR, Vihervaara A, Lewis JJ, Ozer A, Danko CG, Lis JT. Chromatin conformation remains stable upon extensive transcriptional changes driven by heat shock. Proceed Nat Acad Sci PNAS. 2019;116:19431–9.
Zhang Y, Li T, Preissl S, Amaral ML, Grinstein JD, Farah EN, Destici E, Qiu Y, Hu R, Lee AY, et al. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat Genet. 2019;51:1380–8.
Akdemir KC, Le VT, Chandran S, Li Y, Verhaak RG, Beroukhim R, Campbell PJ, Chin L, Dixon JR, Futreal PA. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat Genet. 2020;52:294–305.
Acknowledgements
We thank the lab members for helpful discussions.
Funding
This work was supported by the following grants from the National Institutes of Health (K01CA229995, R21HG011506), the Wright Foundation, and the University of Southern California Norris Comprehensive Cancer Center (Genomics and Epigenomics Regulation Grant) to SKR.
Author information
Authors and Affiliations
Contributions
BL and SKR wrote the article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lee, B.H., Rhie, S.K. Molecular and computational approaches to map regulatory elements in 3D chromatin structure. Epigenetics & Chromatin 14, 14 (2021). https://doi.org/10.1186/s13072-021-00390-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13072-021-00390-y