
Abstract
Background
Toxoplasma gondii infection can cause adverse pregnancy outcomes, such as recurrent abortion, fetal growth restriction and infants with malformations, among others. Decidual myeloid-derived suppressor cells (dMDSCs) are a novel immunosuppressive cell type at the fetal-maternal interface which play an important role in sustaining normal pregnancy that is related to their high expression of the inhibitory molecule leukocyte immunoglobulin-like receptor B4 (LILRB4). It has been reported that the expression of LILRB4 is downregulated on decidual macrophages after T. gondii infection, but it remains unknown whether T. gondii infection can induce dMDSC dysfunction resulting from the change in LILRB4 expression.
Methods
LILRB4-deficient (LILRB4−/−) pregnant mice infected with T. gondii with associated adverse pregnancy outcomes, and anti-LILRB4 neutralized antibodies-treated infected human dMDSCs were used in vivo and in vitro experiments, respectively. The aim was to investigate the effect of LILRB4 expression on dMDSC dysfunction induced by T. gondii infection.
Results
Toxoplasma gondii infection was observed to reduce STAT3 phosphorylation, resulting in decreased LILRB4 expression on dMDSCs. The levels of the main functional molecules (arginase-1 [Arg-1], interleukin-10 [IL-10]) and main signaling molecules (phosphorylated Src-homology 2 domain-containing protein tyrosine phosphatase [p-SHP2], phosphorylated signal transducer and activator of transcription 6 [p-STAT6]) in dMDSCs were all significantly reduced in human and mouse dMDSCs due to the decrease of LILRB4 expression induced by T. gondii infection. SHP-2 was found to directly bind to STAT6 and STAT6 to bind to the promoter of the Arg-1 and IL-10 genes during T. gondii infection.
Conclusions
The downregulation of LILRB4 expression on dMDSCs induced by T. gondii infection could regulate the expression of Arg-1 and IL-10 via the SHP-2/STAT6 pathway, resulting in the dysfunction of dMDSCs, which might contribute to adverse outcomes during pregnancy by T. gondii infection.
Graphical abstract

Similar content being viewed by others


Background
Toxoplasma gondii is an obligate intracellular protozoan parasite that can infect almost all warm-blooded animals. It is estimated that about 30% of the human population is affected by this parasite worldwide [1]. Toxoplasmosis is a life-threatening infection in immunosuppressed individuals; in this context, infected pregnant women often experience recurrent abortion, preterm birth, stillbirth, fetal growth restriction and infants with malformations [2,3,4,5]. Several studies have shown that T. gondii infection during pregnancy leads to the dysfunction of decidual immune cells at the maternal–fetal interface, which causes the disruption of the immune microenvironment [6,7,8,9]. The immune microenvironment comprises various decidual immune cells, such as decidual natural killer (dNK) cells, decidual macrophages (dMφ), decidual dendritic cells (dDCs) and decidual myeloid-derived suppressor cells (dMDSCs), all of which play an important role in sustaining normal pregnancy [10]. MDSCs are a heterogeneous population of cells consisting of myeloid progenitor cells and immature myeloid cells [11, 12]. In humans, MDSCs are defined as cells of the CD33+ human leukocyte antigen DR (HLA-DR)neg/l°w phenotype [13]. In mice, MDSCs are characterized by the co-expression of CD11b+ Gr-1+ [14]. Healthy pregnant women have more MDSCs in their peripheral blood than nonpregnant women, and women who have had a miscarriage have fewer MDSCs in their blood and endometrium than healthy pregnant women [15]. In addition, the accumulation of MDSCs on the maternal–fetal interface during gestation has been directly implicated in the maintenance of maternal–fetal tolerance [16, 17]. When compared to healthy pregnant mice, mice that respond to abnormal delivery have fewer MDSCs in their placenta and uteri [18, 19]. All of these findings suggest that MDSCs are closely related to a successful pregnancy and are essential for maternal–fetal immune tolerance. More recently, MDSCs were observed in human gestation, and they seemed to play an important role in suppressing CD4+ and CD8+ T-cell proliferation [20]. Studies have shown that the immunosuppressive capacity of MDSCs is adjusted by the inhibitory molecule leukocyte immunoglobulin-like receptor B4 (LILRB4) in patients with non-small-cell lung cancer and in mice with melanoma occurring as subcutaneous tumors [21, 22]. LILRB4 belongs to the leukocyte immunoglobulin G (Ig)-like receptor superfamily and is expressed in a wide variety of immune cell types, including DCs, Mφ and MDSCs [22, 23]. This molecule consists of an extracellular domain with two Ig-like domains, a transmembrane segment and a cytoplasmic domain with three immunoreceptor tyrosine-based inhibitory motifs (ITIMs) [24, 25]. LILRB4 expressed on MDSCs has been shown to regulate the immunological function of MDSCs and facilitate tumor immune tolerance [26]. Our previous studies have shown that the downregulation of LILRB4 expressed on dMφ and dDCs may contribute to the adverse outcomes during pregnancy caused by T. gondii infection [9, 27]. In addition, the results of one study implied that the expression of LILRB4 was mediated by the phosphorylation of signal transducer and activator of transcription 3 (STAT3) in human vascular endothelial cells [28]. However, further research is required to determine whether T. gondii infection can affect LILRB4 expression in dMDSCs and characterize the molecular mechanism of regulation.
Some studies have reported that two intracellular ITIMs of LILRB4 interact with Src-homology 2 domain-containing protein tyrosine phosphatase (SHP-2) and activate the phosphorylation of SHP-2 [29, 30], and that the SHP-2 domain recruits STAT6 and stimulates the phosphorylation of signal transducer and activator of transcription 6 (STAT6) followed by its dimerization [31]. The phosphorylated STAT6 (p-STAT6) accumulates in the nucleus of macrophages and regulates the expression of arginase-1 (Arg-1) [32]. According to several studies, STAT6 activation (p-STAT6) in M2 macrophages of patients with acute liver failure and in the cerebrospinal fluid of those with primary central nervous system lymphoma significantly correlates with the presence of interleukin -10 (IL-10) [33, 34]. Results from our previous study showed that the downregulation of LILRB4 on dMφ induced by T. gondii infection could result in the decrease of Arg-1 and IL-10 [27]. However, more research is necessary to determine whether the change in LILRB4 expression level during T. gondii infection influences the expression of Arg-1 and IL-10 on dMDSCs via the SHP-2/STAT6 pathway.
In the present study, human dMDSCs and LILRB4−/− pregnant mice were utilized to investigate the change in LILRB4 expression on dMDSCs during T. gondii infection and explain the molecular mechanism of the change in LILRB4 expression, leading to the immunosuppressive dysfunction of dMDSCs.
Methods
Toxoplasma gondii, RH strain
The tachyzoites of the T. gondii RH strain were harvested from human foreskin fibroblast (HFF) cells grown in 10% fetal bovine serum (FBS) and 100 IU/ml penicillin/streptomycin. After co-culture of HFF cells and toxoplasma tachyzoites for 48 h, the cultures were collected, the cells removed by centrifugation at 433 g for 5 min and the tachyzoites remaining in the supernatant purified by centrifugation at 2810 g for 7 min.
Experimental animals and T. gondii infection
Wild-type (WT) C57BL/6 mice (8- to 10 week-old females and males) were purchased from Pengyue Laboratory Animal Technology Co., Ltd. (Jinan, China). LILRB4-deficient (LILRB4−/−) C57BL/6 mice were obtained from Bioray Laboratories Inc. (Shanghai, China). All mice were maintained in a specific pathogen-free facility at 23 °C ± 2 °C, 55% ± 5% relative humidity and a 12/12-h (light/dark) cycle, with access to abundant sterilized water and food (Jiangsu Biological Engineering Co., Ltd., China). The females with a vaginal plug were designated as gestational day (Gd) 0 of pregnancy following overnight cohabitation with males. WT pregnant C57/BL6 mice were randomly divided into three groups: (i) an uninfected group; (ii) an infected group; and (iii) an infected group treated with the STAT3 inhibitor cucurbitacin I (synonym: JSI-124; MCE Technologies, Suzhou, China). LILRB4−/− pregnant mice were utilized as the infected LILRB4−/− group. Each group included 10 mice.
To explore the molecular mechanism of LILRB4 regulating the function of decidual MDSCs in adverse pregnancy outcomes caused by T. gondii infection early in the first trimester (from 0 to 7 day [35]), pregnant mice in the infected group received an intraperitoneal (i.p.) injection of 200 tachyzoites in 200 μl sterile phosphate-buffered saline (PBS) on Gd 7. The uninfected group received an i.p. injection of 200 μl sterile PBS only. Fetuses, uteri and placentas were harvested on Gd 13 for analysis, and the number of dMDSCs [36] and the expression level of LILRB4 [9, 37] were determined during pregnancy.
For JSI-124 treatment, WT pregnant mice were infected with T. gondii as described above and then i.p. injected with 1 mg/kg body weight of JSI-124 3 times a week (Gd 7, Gd 9 and Gd 11). The infected WT pregnant mice received an i.p. injection with 200 μl sterile PBS only.
The experimental protocols used in all experiments adhered to all relevant ethical regulations for animal testing and research and were approved by the Institutional Animal Care and Research Advisory Committee of Binzhou Medical University, China (permit number: 2017-009-09). The use of animals in our study was in accordance with the guidelines issued by the Chinese Council on Animal Care.
Genotyping of LILRB4−/− mice
Genomic DNA was extracted from mouse tails, and complementary DNA (cDNA) was synthesized by PCR, as described previously [27]. After initial denaturation (30 s at 95 °C), PCR was performed with 40 amplifcation cycles of denaturation for 10 s at 95 °C, annealing for 30 s at 65 °C, and extension for 5 s at 72 °C, followed by a final extension for 3 min at 72 °C. The PCR products were run on gels and stained with GelStain (Transgene S.A., Illkirch-Graffenstaden, France) to visualize DNA. Primers for PCR amplification were LILRB4 P1 (5′-ACCGGTGGATGTGGAATGTGTG-3′), LILRB4 P2 (5′-GTCCTGGGTTCCAGAATAAGAC-3′) and LILRB4 P3 (5′-TCTGCTCTTAGGAAATTACAGAA-3′). The expected PCR product sizes were 260 bp (mutant), 371 and 260 bp (heterozygote) and 371 bp (wild-type).
Cell preparation of mice
The experimental protocol was performed as previously described [17] with modifications. The uteri and placentas of the mice were carefully separated and washed several times with cold PBS, following which the tissues were cut into small pieces and digested with 0.1% collagenase type IV (Sigma-Aldrich, St. Louis, MO, USA) and 25 IU/ml DNase I (Sigma-Aldrich) for 1 h at 37 °C with shaking. The digested pieces were filtered through 48-μm sterile mesh. Mononuclear cells were collected from the white film layer after Ficoll density gradient centrifugation in mouse lymphocyte separation medium (TBD Science, Tianjin, China). As a last step, the cells were collected, resuspended in cold PBS and utilized for subsequent flow cytometry analysis.
Collection of human samples
Clinical samples of decidual tissues were provided by the Department of Obstetrics and Gynecology of Yantai Affiliated Hospital associated with Binzhou Medical University and by the Yantai Zhifu District Maternal and Child Health Hospital. Aborted tissues from pregnant women who had undergone healthy voluntary abortions during the first trimester of pregnancy (gestational age: 6–10 weeks) were aseptically collected and washed 5 to 8 times with PBS under sterile conditions. Decidual tissues were delivered to our laboratory within 2 h of the abortion and kept in Dulbecco’s Modified Eagle Medium (DMEM)/high-glucose medium (HyClone, GLogan, UT, USA) with 100 IU/ml penicillin and 100 μg/ml streptomycin (Sigma-Aldrich).
Prior to beginning the study, written informed consent was obtained from all the participants or their legal guardians. The sample collection procedure was approved by the Ethics Committee of Binzhou Medical University (Approval Number: 2017-016-01).
Isolation and purification of human dMDSCs
The decidual tissues were washed with cold PBS, cut into small fragments using ophthalmic scissors and digested with 0.1% collagenase IV (Sigma-Aldrich) and 25 IU/ml DNase I (Sigma-Aldrich) in an incubator at 37 °C for 60 min. The resulting suspension was filtered through 48-µm mesh and washed twice in cold PBS. The decidual mononuclear cells used for flow cytometry were collected from the white film layer after Ficoll density gradient centrifugation using human lymphocyte separation medium (TBD Science). To remove decidual macrophages and stromal cells, mononuclear cells were cultured at 37 °C for 1 h, and cells were collected in the supernatant. These cells were used for flow cytometry staining and analysis. The dMDSCs were purified using a human CD33-positive isolation kit and HLA-DR-negative selection kit (Stemcell, Canada) according to the manufacturer’s instructions. The purified dMDSCs were analyzed using Western blot, quantitative reverse transcription PCR (qRT–PCR), co-immunoprecipitation (Co-IP) and chromatin-immunoprecipitation (ChIP) assays. The specific culture and treatment conditions of the purified dMDSCs are explained for each experiment.
Human decidual mononuclear cell culture and treatment
The mononuclear cells were divided equally into two groups: uninfected and infected groups. Toxoplasma gondii tachyzoites were added to the cells of the infected group at a ratio of 1:5 (T. gondii tachyzoites:cells). All cells were cultured in RPMI 1640 medium supplemented with 10% FBS (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), 100 IU/ml streptomycin and 100 IU/ml penicillin for 24 h at 37 °C in a humidified 5% CO2 incubator. All of the cells were prepared for flow cytometry staining and analysis.
Flow cytometry staining and analysis
The prepared human and murine decidual mononuclear cell (1 × 106 cells of each group) suspensions were first stained with membrane molecules and then with intracellular molecules, such as Arg-1, IL-10, phosphorylated SHP2 (p-SHP2) or p-STAT6, after the cells were treated with a membrane rupture kit according to the manufacturer’s instructions (eBioscience, San Diego, CA, USA). The experiments were replicated at least 8 times. Cells were analyzed using a BD FACSCanto™ TM II Flow Cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and using FlowJo analysis software (FlowJo LLC, Ashland, OR, USA). The gating strategies are provided in Additional file 1: Figure S1.
Western blot analysis
Purified human dMDSCs were infected with T. gondii tachyzoites at a 1:5 ratio (T. gondiitachyzoites: cells) with or without 10 μM Static (STAT3 inhibitor in vitro [STAT3i]), 10 μg/ml anti-LILRB4 (αLILRB4) neutralizing antibody (Invitrogen, Thermo Fisher Scientific), 500 ng/ml recombinant human apolipoprotein E (APOE), 10 μM SHP099 (SHP-2 inhibitor [SHP-2i]) or 100 nM AS1517499 (STAT6 inhibitor [STAT6i]). About 2 × 107 dMDSCs of each group were cultured in RPMI 1640 medium supplemented with 10% FBS (Gibco, Thermo Fisher Scientific) and 100 IU/ml penicillin/streptomycin (Sigma-Aldrich) for 24 h at 37 °C in a humidified 5% CO2 incubator. All of the cultured human dMDSCs were harvested, lysed with RIPA lysis buffer (Beyotime Biotechnology, Nantong, China) and then centrifuged at 13,400 g for 20 min at 4 °C. After the concentration was measured, equal amounts of protein were loaded into 10% or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) gels and the products were then transferred to polyvinylidene fluoride (PVDF) membranes (MilliporeSigma, Burlington, MA, USA). The membranes were blocked at room temperature for 2 h in 5% skim milk in TBS-T buffer and then incubated overnight at 4 °C with primary antibodies on a shaker. Next, the membranes were incubated with horseradish peroxidase (HRP)-labeled secondary antibodies (Abmart Inc., Berkeley Heights, NJ, USA) at 37 °C for 1 h. The hybridization signal bands were visualized using an enhanced chemiluminescence (ECL) detection kit (Yeasen Biotechnology, Shanghai, China). Protein expression levels were determined using Image J software. The experiments were replicated 5 times.
Co-immunoprecipitation
To investigate the interaction between SHP-2 and STAT6, purified human dMDSCs were infected with T. gondii tachyzoites at a ratio of 1:5 (T. gondii tachyzoites:cells) for 24 h with or without 500 ng/ml APOE. About 2 × 107 cells of each group were collected and washed twice with PBS. All cells were lysed with IP Binding Buffer (Solarbio Life Science, Beijing, China) supplemented with phenylmethylsulfonyl fluoride (PMSF). After 20 min on ice, the supernatant was centrifuged at 14,000 g at 4 °C, following which the protein was extracted and quantified: 15% of the whole-cell lysis was prepared for input with 5× loading buffer boiling for 10 min at 100 °C. After being preabsorbed with protein, A/G agarose beads (Solarbio Life Science) together with anti-STAT6 antibody (Abcam, Oxford, UK) for 2 h at 4 °C with shaking, the remaining extracts were incubated with antibody-conjugated protein A/G magnetic beads overnight with shaking at 4 °C. Isotype IgG was used as a negative control. Beads were washed 4 times with immunoprecipitation washing buffer (Solarbio Life Science) and denatured at 100 °C for 10 min. Protein samples were used for Western blot analyses. Equal amounts of extracts were separated by SDS–PAGE, and the products were transferred onto nitrocellulose membranes and blotted with specific antibodies.
Reagents and antibodies
The reagents and antibodies used in the study are listed in Additional file 2: Table S1.
Quantitative RT‒PCR
Total RNA from human dMDSCs was extracted with TRIzol reagent (Invitrogen, Thermo Fisher Scientific), and cDNA was synthesized using a SuperRT cDNA Synthesis Kit (CoWin BioSciences, Cambridge, MA, USA) according to the manufacturer’s recommended protocol. All messenger RNA (mRNA) expression levels were analyzed by qRT‒PCR using an UltraSYBR One Step RT‒qPCR Kit (CoWin BioSciences) in a Bio-Rad iQ5 multicolor RT‒PCR system (Bio-Rad Laboratories, Hercules, CA, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used for the normalization of mRNA expression. The experiments were replicated 6 times. The primers used for the RT-qPCR are shown in Additional file 2: Table S1. The relative change in the expression of LILRB4 was estimated using the 2−ΔΔCT method.
ChIP assay
The STAT6 binding regions on the promoters of the genes for Arg-1 and IL-10 (ARG1 and IL10) were predicted with the NCBI JASPAR database online. The primers used for ChIP-qPCR were synthesized by Sangon Biotech and are listed in Additional file 2: Table S1.
The ChIP experiment was carried out using a SimpleChIP® Enzymatic Chromatin IP Kit (Cell Signaling, Danvers, MA, USA) according to the manufacturer’s instructions. For cross-linking of proteins to DNA, about 2 × 107 cells from the uninfected group and the infected group were treated with 1% paraformaldehyde at room temperature for 10 min; then glycine was added to a final concentration of 0.125 M, and the sample was incubated for 5 min at room temperature to stop cross-linking. After washing twice with ice-cold PBS, the cells were digested to an optimal DNA length of approximately 150–900 bp by using a lysis buffer and sonication (sonication at 20% power: 5 s ON/10 s OFF for 5 min on ice). For immunoprecipitation, protein–DNA complexes were incubated overnight with an antibody with shaking at 4 °C, following which protein G agarose beads were added, and the samples were incubated for 2 h at 4 °C with shaking. Protein G agarose beads were washed 4 times using a low-salt washing buffer and 2 times using a high-salt washing buffer at 4 °C for 5 min with shaking. DNA was eluted in ChIP elution buffer, reversed cross-linked by incubation in 5 M NaCl and proteinase K for 2 h at 65 °C, purified via DNA wash buffer and purification collection tube and subjected to qPCR. The DNA was analyzed by qPCR using an UltraSYBR One-Step RT-qPCR Kit (CoWin BioSciences) in a Bio-Rad iQ5 multicolor RT‒PCR system (Bio-Rad Laboratories). The experiments were replicated 3 times.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8 software (GraphPad Software, La Jolla, CA, USA). Data are presented as the mean ± standard error (SE). Unpaired and paired t-tests were used to identify the differences. One-way analysis of variance was applied with 95% confidence intervals. Significance was defined as p < 0.05.
Results
LILRB4 is decreased in dMDSCs after T. gondii infection
To explore the change in the expression of LILRB4 on dMDSCs during T. gondii infection, we detected LILRB4 expression levels on human dMDSC using Western blot, qPCR and flow cytometry analyses. We also detected LILRB4 expression levels on mouse dMDSCs by using flow cytometry. The results showed that the expression levels of both LILRBA protein and LILRBA mRNA were significantly decreased in the infected group compared with those in the uninfected group (Fig. 1a, b). The representative flow cytometry results showed that the expression of LILRB4 by the infected dMDSCs was always lower than that by the uninfected dMDSCs both in vitro and in vivo (Fig. 1c, d).

The expression level of LILRB4 in dMDSCs was decreased after Toxoplasma gondii infection. The expression level of LILRB4 on human decidual myeloid-derived suppressor cells (dMDSCs) was analyzed in the uninfected and infected groups by Western blot (a), qRT–PCR (b) and flow cytometry (c). d The levels of LILRB4 on mouse dMDSCs from the WT and infected mice were analyzed by flow cytometry. The human data were identified by a paired t-test; the mouse data were identified by the unpaired t-test. Asterisks indicate significant difference at *p < 0.05, ** p < 0.01. FMO, fluorescence minus one; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; INF, infected; LILRBA, leukocyte immunoglobulin-like receptor B4; mRNA, messenger RNA; NC, negative control; WT, wild type.
LILRB4 expression on dMDSCs was regulated by STAT3 during T. gondii infection
The expression of LILRB4 has been reported previously to be mediated by STAT3 [28]. In the present study, the p-STAT3 expression level in human dMDSCs was detected by Western blotting. The results showed that the level of p-STAT3 expression in human dMDSCs was lower in the infected group than in the uninfected group (Fig. 2a). Also, the expression level of p-STAT3 in dMDSCs of the infected mice was downregulated after T. gondii infection in vivo (Fig. 2b). In order to investigate whether the LILRB4 expression level on dMDSCs is regulated by STAT3, we used Static (STAT3i) and JSI-124 (STAT3i cucurbitacin I) to inhibit STAT3 phosphorylation during T. gondii infection both in vitro and in vivo. Human dMDSCs were analyzed by Western blotting, and mouse dMDSCs were analyzed by flow cytometry. The expressions of both p-STAT3 and LILRB4 were found to be downregulated in the infected human dMDSCs treated with STAT3i (Fig. 2c) and in the infected mouse dMDSCs treated with JSI-124 (Fig. 2d, e). These results suggested that the change in LILRB4 expression on dMDSCs after T. gondii infection is regulated by p-STAT3.

LILRB4 expression level on dMDSCs was regulated by p-STAT-3 during T. gondii infection. a Analysis of the expression of STAT3 and p-STAT3S727 in human dMDSCs with or without T. gondii infection. b Results of p-STAT3 expression in mouse dMDSCs of the uninfected and infected WT mice were detected by flow cytometry. c Expression levels of STAT3, p-STAT3S727 and LILRB4 in human dMDSCs treated with or without STAT3i during T. gondii infection were detected by Western blot. d, e Results of p-STAT3 and LILRB4 expression in mouse dMDSCs treated with or without JSI-124 during T. gondii infection were detected by flow cytometry. The data on human dMDSCs were identified by a paired t-test. The data on mouse dMDSCs were identified by the unpaired t-test. Asterisks indicate a significant difference at *p < 0.05 and **p < 0.01. FMO, fluorescence minus one; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; INF, infected; JSI-124, the inhibitor of STAT3 in vivo; LILRB4, leukocyte immunoglobulin-like receptor B4; mRNA, messenger RNA; NC, negative control; p-STAT3, phosphorylated STAT3; STAT3, signal transducer and activator of transcription 3; STAT3i, the inhibitor of STAT3 in vitro; WT, wild type.
The decrease in LILRB4 induced by T. gondii infection could further regulate the expression level of p-SHP2, p-STAT6, Arg-1 and IL-10 in dMDSCs in vitro and in vivo
As a ligand, APOE can combine directly with LILRB4 and activate it [38]. A number of authors have reported that there are two intracellular ITIMs of LILRB4 interacting with SHP-2 and activating the phosphorylation of SHP-2 [29, 30], with the SHP-2 domain recruiting STAT6 and stimulating the phosphorylation of STAT6 followed by its dimerization [31]. To explain whether the downregulation of LILRB4 on dMDSCs after T. gondii infection could affect the production of Arg-1 and IL-10 through the SHP-2/STAT6 pathway, we used a specific neutralizing antibody of LILRB4 or APOE to block or activate the function of LILRB4 in vitro, and the infected LILRB4−/− mice with unfavorable abnormal pregnancies were established in vivo. The results showed that the phosphorylation levels of the pathway molecules (p-SHP2 and p-STAT6) and functional molecules (Arg-1 and IL-10) were decreased in the anti-LILRB4-neutralized (α-LILRB4) human dMDSCs and increased in the APOE-stimulated human dMDSCs compared with those in the infected dMDSCs (Fig. 3a). Similarly, the phosphorylation levels of pathway molecules (p-SHP2 and p-STAT6) and Arg-1 were all decreased in the infected LILRB4−/− mice compared to the infected WT mice (Fig. 3B–D). Overall, our findings indicated that the change in LILRB4 induced by T. gondii infection might regulate the expression levels of Arg-1 and IL-10 through the SHP-2/STAT6 pathway.

Blocking or activating LILRB4 could downregulate or upregulate the levels of p-SHP2, p-STAT6, Arg-1 and IL-10 in dMDSCs during T. gondii infection. a The expression levels of SHP-2, p-SHP2, STAT6, p-STAT6, Arg-1 and IL-10 in human dMDSCs treated with or without α-LILRB4 and APOE after infection with T. gondii were detected by Western blot. b–d The expression levels of p-SHP2, p-STAT6 and Arg-1 in mouse dMDSCs from the WT and LILRB4−/− infected mice were detected by flow cytometry. The data on human dMDSCs were identified by a paired t-test. The data on mouse dMDSCs were identified by the unpaired t-test. Asterisks indicate a significant difference at *p < 0.05 and **p < 0.01. APOE, apolipoprotein E; Arg-1, arginase-1; FMO, fluorescence minus one; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IL-10, interleukin-10; INF, infected; LILRB4-/-, LILRB4-deficient; p-SHP2, phosphorylated SHP-2; p-STAT6, phosphorylated STAT6; SHP-2, src-homology 2 domain-containing protein tyrosine phosphatase; STAT6, signal transducer and activator of transcription 6; WT, wild type; αLILRB4: anti-LILRB4-neutralized.
LILRB4 modulates Arg-1 and IL-10 expression via the SHP-2/STAT6 pathway in dMDSCs during T. gondii infection
To investigate whether SHP-2 can stimulate STAT6 activation, SHP099 (a selective SHP-2 inhibitor [SHP-2i]) was utilized in human dMDSCs after treatment with or without APOE during T. gondii infection. The results showed that p-SHP2 expression was inhibited by SHP-2i and that the phosphorylation of STAT6 was also reduced in human dMDSCs, while reduced expression levels of Arg-1 and IL-10 were observed after T. gondii infection (Fig. 4a). To further explore the role of STAT6 activation in regulating the expression levels of Arg-1 and IL-10 after T. gondii infection, human dMDSCs were treated with the STAT6 inhibitor AS1517499 (STAT6i) or APOE. The results suggested that STAT6i inhibited the activation of STAT6, whereas the production of Arg-1 and IL-10 was decreased in human dMDSCs during T. gondii infection (Fig. 4b). These results demonstrated that the expression levels of Arg-1 and IL-10 in dMDSCs resulting from the downregulation of LILRB4 induced by T. gondii infection were regulated through the SHP-2/STAT6 pathway. The Co-IP assay was carried out to further verify the interaction between SHP-2 and STAT6 in human dMDSCs treated with or without APOE after T. gondii infection. The SHP-2 antibody was used as a bait protein. The results showed that SHP-2 directly interacted with STAT6 in human dMDSCs during T. gondii infection (Fig. 4c).

LILRB4 regulates Arg-1 and IL-10 expression levels via the SHP-2/STAT6 pathway in dMDSCs during T. gondii infection. a Expression levels of SHP-2, p-SHP2, STAT6, p-STAT6, Arg-1 and IL-10 in human dMDSCs treated with or without SHP-2i and APOE after T. gondii infection were detected by Western blot. b Expression levels of STAT6, p-STAT6, Arg-1 and IL-10 in human dMDSCs treated with or without STAT6i and APOE during T. gondii infection were detected by Western blot. c Interaction between SHP-2 and STAT6 in human dMDSCs treated with or without APOE during T. gondii infection was analyzed by Co-IP. d The schematic diagram shows the possible binding sites of STAT6 on the ARG1 and IL10 promoter regions. e–f ChIP-PCR of STAT6 binding regions on the promoter of ARG1 (e) and IL10 (f). ChIP assays were performed using anti-STAT6 and then qRT–PCR. The data of human dMDSCs were identified by a paired t-test. Asterisks indicate a significant difference at *p < 0.05 and **p < 0.01. APOE, apolipoprotein E; Arg-1, arginase-1; ARG1, the gene for Arg-1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IgG, immunoglobulin G; IL-10, interleukin-10; IL10, the gene for IL-10; INF, infected; IP, immunoprecipitation; NC, negative control; P1-P5, primer 1 to primer 5; p-SHP2, phosphorylated SHP-2; p-STAT6, phosphorylated STAT6; SHP-2, src-homology 2 domain-containing protein tyrosine phosphatase; SHP-2i, the inhibitor of SHP-2; STAT6, signal transducer and activator of transcription 6; STAT6i, the inhibitor of STAT6.
STAT6 has been reported to be a transcription factor regulating the expression levels of Arg-1 and IL-10 [33, 39]. Therefore, the ChIP assay was also used to explore the activity of STAT6 binding to the promoter of ARG1 and IL10 in dMDSCs during T. gondii infection. The results showed that the enrichment of STAT6 was decreased after T. gondii infection in the promoter regions of ARG1, which span from − 1637 to− 1476 bp, from− 1475 to − 1316 bp, from − 1320 to − 1125 bp, from − 1123 to − 969 bp and from − 946 to− 768 bp (Fig. 4d, e). Moreover, the enrichment of STAT6 was reduced after T. gondii infection in the promoter regions of IL10, which span from − 871 to− 792 bp, from− 700 to − 617 bp and from − 615 to − 448 bp (Fig. 4d, f). These results suggest that STAT6 could regulate the expression levels of Arg-1 and IL-10 via binding to their respective promoters.
Discussion
The immune microenvironment at the maternal–fetal interface plays an important role in normal pregnancy [40]. The most critical immune cells within the decidual tissue are dNK cells, Mφ, T cells, DCs and MDSCs [10, 41], and these immune cells are important for a successful pregnancy [42]. Infection by bacteria, viruses, and parasites may impair the maternal–fetal tolerance function, which can lead to fetal death, preterm birth or secondary sequelae [43, 44]. Toxoplasma gondii is one of the TORCH pathogens, and its infection could cause the occurrence of abnormal outcomes during pregnancy, such as spontaneous abortion, congenital retinal disease and fetal intrauterine growth retardation [45]. Our previous studies have shown that T. gondii infection resulted in the dysfunction of several decidual immune cells, such as Mφ [27], NK cells [8] and DCs [46]; however, it remains unknown whether T. gondii infection can result in the dysfunction of dMDSCs. dMDSCs have emerged as one of the novel immuno-modulators for the maintenance of maternal–fetal immune tolerance [47]. One study showed that LILRB4 expressed on MDSCs played a distinct role in inducing immunosuppression in cancer [22]. The authors of a recent study reported that LILRB4 plays a major role in regulating the immunosuppressive function of MDSCs by inhibiting the miR-1 family of microRNAs and facilitating tumor migration and invasion in the tumor microenvironment [26]. However, whether T. gondii infection can affect the expression of LILRB4 in dMDSCs has not been reported yet.
To explore the change in the expression of LILRB4 on dMDSCs after T. gondii infection, human dMDSCs infected with T. gondii in vitro and infected mice with abnormal pregnancy were established in vivo. We found that the expression level of LILRB4 was clearly reduced on human dMDSCs examined by Western blotting and qPCR after infection. Similarly, the downregulation of LILRB4 was observed in infected human and mouse dMDSCs examined by flow cytometry. Thus, T. gondii infection can result in a decrease in the expression of LILRB4 on dMDSCs. However, the molecular mechanism of the downregulation of LILRB4 induced by T. gondii infection needs further exploration. In one study, the expression of LILRB4 was mediated by STAT3 phosphorylation [28], and the results of the present investigation showed that STAT3 phosphorylation in dMDSCs was reduced after T. gondii infection. To explore whether the downregulation of LILRB4 on dMDSCs after T. gondii infection resulted from weak STAT3 phosphorylation, STAT3i was used to inhibit STAT3 phosphorylation in infected human dMDSCs and infected pregnant mice. As anticipated, the expression levels of p-STAT3 and LILRB4 were all reduced when dMDSCs were treated with STAT3i during T. gondii infection. Therefore, the results of the present study demonstrated that the downregulation of LILRB4 in dMDSCs induced by T. gondii infection resulted from the inhibition of STAT3 phosphorylation (Fig. 5).

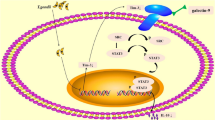
Diagrammatic representation of LILRB4 downregulation induced by T. gondii infection resulting in the dysfunction of dMDSCs. The phosphorylation level of STAT3 was decreased after T. gondii infection, resulting in the downregulation of LILRB4 on dMDSCs. The decreased level of LILRB4 reduced the phosphorylation of SHP-2 and then inhibited the activity of STAT6, which in turn inhibited the expression levels of Arg-1 and IL-10 via binding to their respective promoter during T. gondii infection. Arg-1, arginase-1; ARG1, the gene for Arg-1; dMDSCs, decidual myeloid-derived suppressor cells; Ig, immunoglobulin; IL-10, interleukin-10; IL10, the gene for IL-10; ITIM, immunoreceptor tyrosine-based inhibitory motifs; LILRB4, leukocyte immunoglobulin-like receptor B4; p-SHP2, phosphorylated SHP-2; p-STAT3, phosphorylated STAT3; p-STAT6, phosphorylated STAT6; SHP-2, src-homology 2 domain-containing protein tyrosine phosphatase; STAT3, signal transducer and activator of transcription 3; STAT6, signal transducer and activator of transcription 6.
The immunosuppressive function was the essential feature of MDSCs and was due to the production of Arg-1 and IL-10 [48]. Some studies have found that the functional molecules of Arg-1 and IL-10 in MDSCs play an important role in suppressing the functions of NK and T cells, further contributing to the maternal–fetal tolerance [49, 50]. In our study, the expression of Arg-1 was downregulated in dMDSCs of LILRB4−/− mice with T. gondii infection. Also, both Arg-1 and IL-10 in human dMDSCs were decreased or increased after the function of LILRB4 was blocked by neutralized antibody or activated by APOE under the condition of T. gondii infection, respectively. These results indicate that the reduction of Arg-1 and IL-10 levels in dMDSCs might be related to the downregulation of LILRB4 after T. gondii infection. However, while the level of LILRB4 decreased, it remains to be determined how the production of Arg-1 and IL-10 in MDSCs during T. gondii infection is regulated.
Some studies have suggested that LILRB4 activates the phosphorylation of SHP-2 and then stimulates the phosphorylation of STAT6 [29, 30]. In the present study, the phosphorylation of SHP-2 and STAT6 in human dMDSCs was inhibited or activated after treatment with LILRB4-neutralizing antibody or APOE, respectively, during T. gondii infection. In order to explore whether the reduced LILRB4 expression could affect the activation of SHP-2 and further inhibit STAT6 phosphorylation, SHP-2i was utilized to inhibit SHP-2 phosphorylation in human dMDSCs after T. gondii infection. This experiment revealed that SHP-2i could inhibit the phosphorylation of STAT6 after T. gondii infection, suggesting that STAT6 may be a downstream molecule of SHP-2. In addition, A Co-IP experiment revealed that the antibody of SHP-2 can interact with STAT6 during T. gondii infection. Moreover, the expression levels of Arg-1 and IL-10 were decreased after treatment with STAT6i following T. gondii infection. Studies found that STAT6 could regulate the expression levels of Arg-1 and IL-10 in macrophages [33, 39]. In order to observe whether STAT6 could combine with the promoters of ARG1 and IL10, we performed ChIP-qPCR analyses. Our results showed that STAT6 can bind to the promoters of ARG1 and IL10 in dMDSCs during T. gondii infection.
Conclusions
In summary, our study demonstrated that the downregulation of LILRB4 after T. gondii infection can regulate the expression levels of Arg-1 and IL-10 in dMDSCs through the SHP-2/STAT6 pathway. Ultimately, we revealed a novel mechanism of dMDSC dysfunction after T. gondii infection. This mechanism resulted in a decrease in the expression levels of Arg-1 and IL-10 via the SHP-2/STAT6 pathway, resulting in the reduction of LILRB4 levels, which may contribute to the abnormal outcomes during pregnancy.
Availability of data and materials
All data generated in this study are presented within the published article.
Abbreviations
- APOE:
-
Apolipoprotein E
- Arg-1:
-
Arginase-1
- ChIP:
-
Chromatin immunoprecipitation
- Co-IP:
-
Co-immunoprecipitation
- dDCs:
-
Decidual dendritic cells
- dMφ:
-
Decidual macrophages
- dMDSCs:
-
Decidual myeloid-derived suppressor cells
- dNK:
-
Decidual natural killer cells
- FBS:
-
Foetal bovine serum
- Gd:
-
Gestational day
- HLA-DR:
-
Human leukocyte antigen DR
- IL-10:
-
Interleukin-10
- LILRB4:
-
Leukocyte immunoglobulin-like receptor B4
- p-SHP2:
-
Phosphorylated SHP-2
- p-STAT3:
-
Phosphorylated STAT3
- p-STAT6:
-
Phosphorylated STAT6
- PBS:
-
Phosphate buffered saline
- qRT-PCR:
-
Quantitative reverse transcription PCR
- SHP-2:
-
Src-homology 2 domain-containing protein tyrosine phosphatase
- STAT3:
-
Signal transducer and activator of transcription 3
- STAT6:
-
Signal transducer and activator of transcription 6
References
Tenter AM, Heckeroth AR, Weiss LM. Toxoplasma gondii: from animals to humans. Int J Parasitol. 2000;30:1217–58.
Nayeri T, Sarvi S, Daryani A. Toxoplasma gondii in mollusks and cold-blooded animals: a systematic review. Parasitology. 2021;148:895–903.
Patel KK, Burrows E, Heuer C, Asher GW, Wilson PR, Howe L. Investigation of Toxoplasma gondii and association with early pregnancy and abortion rates in New Zealand farmed red deer (Cervus elaphus). Parasitol Res. 2019;118:2065–77.
Zhou X, Zhang XX, Mahmmod YS, et al. A transcriptome analysis: various reasons of adverse pregnancy outcomes caused by acute Toxoplasma gondii infection. Front Physiol. 2020;11:115.
Conceição AR, Belucik DN, Missio L, Gustavo Brenner L, Henrique Monteiro M, Ribeiro KS, et al. Ocular findings in infants with congenital toxoplasmosis after a toxoplasmosis outbreak. Ophthalmology. 2021;128:1346–55.
Li T, Cui L, Xu X, Zhang H, Jiang Y, Ren L, et al. The role of Tim-3 on dNK cells dysfunction during abnormal pregnancy with Toxoplasma gondii infection. Front Cell Infect Microbiol. 2021;26:587150.
Xie H, Li Z, Zheng G, Yang C, Liu X, Xu X, et al. Tim-3 downregulation by Toxoplasma gondii infection contributes to decidual dendritic cell dysfunction. Parasit Vectors. 2022;15:393.
Xu X, Zheng G, Ren Y, He X, Peng B, Hu X, et al. A novel 2B4 receptor leads to worse pregnancy outcomes by facilitating TNF-α and IFN-γ production in dNK cells during Toxoplasma gondii infection. Parasit Vectors. 2022;15:337.
Zhan S, Zheng J, Zhang H, Zhao M, Liu X, Jiang Y, et al. LILRB4 decrease on uDCs exacerbate abnormal pregnancy outcomes following Toxoplasma gondii infection. Front Microbiol. 2018;28:588.
Ghaebi M, Nouri M, Ghasemzadeh A, Farzadi L, Jadidi-Niaragh F, Ahmadi M, et al. Immune regulatory network in successful pregnancy and reproductive failures. Biomed Pharmacother. 2017;88:61–73.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68.
Bartmann C, Junker M, Segerer SE, Häusler SF, Krockenberger M, Kämmerer U. CD33(+ /HLA-DR(neg) and CD33(+)/HLA-DR(+/-) cells: rare populations in the human decidua with characteristics of MDSC. Am J Reprod Immunol. 2016;75:539–56.
Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–99.
Zhao AM, Xu HJ, Kang XM, Zhao AM, Lu LM. New insights into myeloid-derived suppressor cells and their roles in feto-maternal immune cross-talk. J Reprod Immunol. 2016;113:35–41.
Ostrand-Rosenberg S, Sinha P, Figley C, Long R, Park D, Carter D, et al. Frontline science: myeloid-derived suppressor cells (MDSCs) facilitate maternal-fetal tolerance in mice. J Leukoc Biol. 2017;101:1091–101.
Pan T, Liu Y, Zhong LM, Shi MH, Duan XB, Wu K, et al. Myeloid-derived suppressor cells are essential for maintaining feto-maternal immunotolerance via STAT3 signaling in mice. J Leukoc Biol. 2016;100:499–511.
Fainaru O, Paz G, Hantisteanu S, Hallak M. Myeloid cell alterations in the mouse placenta precede the onset of labor and delivery. Am J Obstet Gynecol. 2014;210:73.e1-7.
Zhao H, Kalish F, Schulz S, Yang Y, Wong RJ, Stevenson DK. Unique roles of infiltrating myeloid cells in the murine uterus during early to midpregnancy. J Immunol. 2015;194:3713–22.
Köstlin N, Kugel H, Spring B, Leiber A, Marmé A, Henes M, et al. Granulocytic myeloid derived suppressor cells expand in human pregnancy and modulate T-cell responses. Eur J Immunol. 2014;44:2582–91.
de Goeje PL, Bezemer K, Heuvers ME, Dingemans AC, Groen HJ, Smit EF, et al. Immunoglobulin-like transcript 3 is expressed by myeloid-derived suppressor cells and correlates with survival in patients with non-small cell lung cancer. Oncoimmunology. 2015;4:e1014242.
Singh L, Muise ES, Bhattacharya A, Grein J, Javaid S, Stivers P, et al. ILT3 (LILRB4) promotes the immunosuppressive function of tumor-educated human monocytic myeloid-derived suppressor cells. Mol Cancer Res. 2021;19:702–16.
Gu X, Laouar A, Wan J, Daheshia M, Lieberman J, Yokoyama WM, et al. The gp49B1 inhibitory receptor regulates the IFN-gamma responses of T cells and NK cells. J Immunol. 2003;170:4095–101.
Cella M, Döhring C, Samaridis J, Dessing M, Brockhaus M, Lanzavecchia A, et al. A novel inhibitory receptor (ILT3) expressed on monocytes, macrophages, and dendritic cells involved in antigen processing. J Exp Med. 1997;185:1743–51.
Li Z, Deng M, Huang F, et al. LILRB4 ITIMs mediate the T cell suppression and infiltration of acute myeloid leukemia cells published correction appears. Cell Mol Immunol. 2020;17:272–82.
Su MT, Kumata S, Endo S, Okada Y, Takai T. LILRB4 promotes tumor metastasis by regulating MDSCs and inhibiting miR-1 family miRNAs. Oncoimmunology. 2022;11:2060907.
Li Z, Zhao M, Li T, Zheng J, Liu X, Jiang Y, et al. Decidual macrophage functional polarization during abnormal pregnancy due to Toxoplasma gondii: role for LILRB4. Front Immunol. 2017;24:1013.
Gleissner CA, Zastrow A, Klingenberg R, Kluger MS, Konstandin M, Celik S, et al. IL-10 inhibits endothelium-dependent T cell costimulation by up-regulation of ILT3/4 in human vascular endothelial cells. Eur J Immunol. 2007;37:177–92.
Zhou H, Li N, Yuan Y, Jin YG, Wu Q, Yan L, et al. Leukocyte immunoglobulin-like receptor B4 protects against cardiac hypertrophy via SHP-2-dependent inhibition of the NF-κB pathway. J Mol Med (Berl). 2020;98:691–705.
Li J, Gao A, Zhang F, Wang S, Wang J, Wang J, et al. ILT3 promotes tumor cell motility and angiogenesis in non-small cell lung cancer. Cancer Lett. 2021;31:263–76.
Waqas SFH, Ampem G, Röszer T. Analysis of IL-4/STAT6 signaling in macrophages. Methods Mol Biol. 2019;1966:211–24.
Tian LX, Tang X, Zhu JY, Zhang W, Tang WQ, Yan J, et al. Cytochrome P450 1A1 enhances Arginase-1 expression, which reduces LPS-induced mouse peritonitis by targeting JAK1/STAT6. Cell Immunol. 2020;349:104047.
Li Y, Sheng Q, Zhang C, Han C, Bai H, Lai P, et al. STAT6 up-regulation amplifies M2 macrophage anti-inflammatory capacity through mesenchymal stem cells. Int Immunopharmacol. 2021;91:107266.
Mondello P, Cuzzocrea S, Arrigo C, Pitini V, Mian M, Bertoni F. STAT6 activation correlates with cerebrospinal fluid IL-4 and IL-10 and poor prognosis in primary central nervous system lymphoma. Hematol Oncol. 2020;38:106–10.
Schmidt M, Lapert F, Brandwein C, Deuschle M, Kasperk C, Grimsley JM, et al. Prenatal stress changes courtship vocalizations and bone mineral density in mice. Psychoneuroendocrinology. 2017;75:203–12.
Chabtini L, Mfarrej B, Mounayar M, Zhu B, Batal I, Dakle PJ, et al. TIM-3 regulates innate immune cells to induce fetomaternal tolerance. J Immunol. 2013;190:88–96.
Matsumoto Y, Wang LL, Yokoyama WM, Aso T. Uterine macrophages express the gp49B inhibitory receptor in midgestation. J Immunol. 2001;166:781–6.
Deng M, Gui X, Kim J, Xie L, Chen W, Li Z, et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature. 2018;562:605–9.
Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol. 2004;172:7565–73.
PrabhuDas M, Bonney E, Caron K, Dey S, Erlebacher A, Fazleabas A, et al. Immune mechanisms at the maternal-fetal interface: perspectives and challenges. Nat Immunol. 2015;16:328–34.
Erlebacher A. Immunology of the maternal-fetal interface. Annu Rev Immunol. 2013;31:387–411.
Yousefzadeh Y, Soltani-Zangbar MS, Hemmatzadeh M, Shomali N, Mahmoodpoor A, Ahmadian Heris J, et al. Fetomaternal immune tolerance: crucial mechanisms of tolerance for successful pregnancy in humans. Immunol Invest. 2022;51:1108–25.
Adams Waldorf KM, McAdams RM. Influence of infection during pregnancy on fetal development. Reproduction. 2013;146:R151–62.
Condrat CE, Filip L, Gherghe M, Cretoiu D, Suciu N. Maternal HPV infection: effects on pregnancy outcome. Viruses. 2021;13:2455.
Jones JL, Dubey JP. Waterborne toxoplasmosis–recent developments. Exp Parasitol. 2010;124:10–25.
Sun X, Xie H, Zhang H, Li Z, Qi H, Yang C, et al. B7–H4 reduction induced by Toxoplasma gondii infection results in dysfunction of decidual dendritic cells by regulating the JAK2/STAT3 pathway. Parasit Vectors. 2022;15:157.
Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21:485–98.
Wang Y, Tian J, Wang S. The potential therapeutic role of myeloid-derived suppressor cells in autoimmune arthritis. Semin Arthritis Rheum. 2016;45:490–5.
Ren J, Zeng W, Tian F, Zhang S, Wu F, Qin X, et al. Myeloid-derived suppressor cells depletion may cause pregnancy loss via upregulating the cytotoxicity of decidual natural killer cells. Am J Reprod Immunol. 2019;81:e13099.
Wang Y, Ding Y, Guo N, Wang S. MDSCs: key criminals of tumor pre-metastatic niche formation. Front Immunol. 2019;7:172.
Acknowledgements
We thank Aiqun Xu, Zhihua Lv and Jing Zheng for their help in collecting clinical specimens.
Funding
This work was supported by funds from the Shandong Natural Science Foundation (No. ZR2020MH304), the National Natural Science Foundation of China (No. 81871680, No. 81672049) and Taishan Scholar Foundation of Shandong province (No. ts201712066).
Author information
Authors and Affiliations
Contributions
YTL, JJG, HXZ and XMH designed the experiments. ZDL, YSR and YZJ contributed to sample collection. YTL, JJG, HXZ and XMH analyzed the data. YTL, HXZ and XMH wrote the manuscript. HXZ and XMH edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki and its later amendments, and was approved by the Ethics Committee of Binzhou Medical University (Approval Number 2016-029). The women who participated in the study were diagnosed by a professional obstetrician and gynecologist as having normal early pregnancies without any complications, were not on any medication, had no history of toxoplasma infection and voluntarily chose to terminate their pregnancies. All samples were taken with the participants' permission, and all participants signed informed consent forms. Animals in this study were anesthetized with pentobarbital sodium to minimize suffering.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig S1.
Representative FACS gating scheme of dMDSC analyses. A gating strategy of human dMDSCs. After lymphocyte cells were gated by FSC-A and SSC-A, CD33+ HLA-DR- cells were gated as dMDSCs for further analyses. The FMO was used to analyze the expression of functional molecules in human dMDSCs. B Gating strategy of mouse dMDSCs. After lymphocyte cells were gated by FCS-A and SSC-A, CD11b+ Gr-1+ cells were gated as dMDSCs for further analysis. The FMO was used to analyze the expression of functional molecules in mouse dMDSCs.
Additional file 2: Table S1.
Reagents and antibodies used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Guo, J., Zhang, H. et al. LILRB4 regulates the function of decidual MDSCs via the SHP-2/STAT6 pathway during Toxoplasma gondii infection. Parasites Vectors 16, 237 (2023). https://doi.org/10.1186/s13071-023-05856-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-023-05856-4