
Abstract
Leishmania infections span a range of clinical syndromes and impact humans from many geographic foci, but primarily the world’s poorest regions. Transmitted by the bite of a female sand fly, Leishmania infections are increasing with human movement (due to international travel and war) as well as with shifts in vector habitat (due to climate change). Accurate diagnosis of the 20 or so species of Leishmania that infect humans can lead to the successful treatment of infections and, importantly, their prevention through modelling and intervention programs. A multitude of laboratory techniques for the detection of Leishmania have been developed over the past few decades, and although many have drawbacks, several of them show promise, particularly molecular methods like polymerase chain reaction. This review provides an overview of the methods available to diagnostic laboratories, from traditional techniques to the now-preferred molecular techniques, with an emphasis on polymerase chain reaction-based detection and typing methods.
Graphical abstract

Similar content being viewed by others


Background
Among parasitic diseases, leishmaniasis is second only to malaria as a cause of human mortality [1]. Human leishmaniasis is considered a neglected tropical disease (NTD) by the World Health Organization (WHO). It is widespread, occurring in all continents except for Australia and Antarctica, but primarily impacts developing nations in the tropics. However, its endemicity in developed nations is changing due to human migration as a result of war, and expansion in sand fly habitats linked to changes in environmental factors that are often associated with climate change [2,3,4,5].
Human leishmaniasis is caused by 20 or more species of the protozoan genus Leishmania (Kinetoplastida: Trypanosomatidae). These are often referred to as New World or Old World species based on their geographic localisation either in the Western Hemisphere (specifically, Mexico, Central and South America) or the Eastern Hemisphere (specifically, southern Europe, Africa, the Middle East and parts of Asia), respectively. New World species include Leishmania infantum, Leishmania braziliensis, Leishmania guyanensis, Leishmania panamensis, Leishmania peruviana, Leishmania lainsoni, Leishmania naiffi, Leishmania mexicana (syn. Leishmania pifanoi) and Leishmania amazonensis (syn. Leishmania garnhami) [6]. Old World species include Leishmania donovani (syn. Leishmania archibaldi), Leishmania infantum, Leishmania tropica (syn. Leishmania killicki), Leishmania major and Leishmania aethiopica [6, 7]. Other species that infect humans include Leishmania shawi, Leishmania lindenbergi, Leishmania venezuelensis, Leishmania martiniquensis, Leishmania waltoni (all in the New World), and Leishmania arabica and the newly described Leishmania orientalis (in the Old World) [7,8,9]. It must be acknowledged, and kept in mind, however, that the classification of Leishmania is problematic largely due to the use of different genetic markers in evolutionary relationship studies [10, 11]. There have been repeated calls for a consensus classification of the genus, but this has yet to be achieved [6, 12].
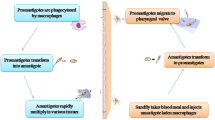
Humans become infected with Leishmania through the bite of female sand flies of the genera Lutzomyia (in the New World) or Phlebotomus (in the Old World) (Fig. 1). Transmission has also been documented through needle sharing, congenital transmission and sexually transmitted infection, albeit rarely [14,15,16]. Clinical presentation of leishmaniasis consists of two main forms: cutaneous leishmaniasis (CL), which includes manifestations such as mucocutaneous leishmaniasis (MCL), diffuse cutaneous leishmaniasis, disseminated leishmaniasis, leishmaniasis recidivans and post-kala-azar dermal leishmaniasis; and kala-azar or visceral leishmaniasis (VL) (Table 1). Each manifestation may be associated with certain species of Leishmania, although there is considerable overlap, exceptions, hybrid species and mixed infections that are recognized [18, 19]. Furthermore, Leishmania has been shown to exhibit mosaic aneuploidy, which influences genetic diversity not only within a given species but within a single isolate [20, 21]. The clinical manifestations of Leishmania infection may also be affected by the presence of sand fly saliva and the host’s genetic makeup and immune status [22, 23].

Life cycle of Leishmania species. The sand flies inject metacyclic promastigote stages of the parasite as they take their blood meal [13]. These parasites transform into asexually reproducing amastigotes within macrophages and can affect different organs and tissues depending, in part, on the parasite species and the species and immune status of the host. The Leishmania species that infect humans are also found in a variety of mammalian reservoir hosts, including canids (particularly domestic dogs), rodents and marsupials [17]
Despite being listed as a NTD in 2007, and being the subject of concerted global efforts for its control, the incidence of leishmaniasis remains significant, and the risk to vulnerable, mostly poor, populations remains lamentably high [24, 25]. In 2018, over 200,000 new cases were reported to the WHO, and it is now estimated that there are 1 billion people at risk of contracting Leishmania [26, 27]. These figures are probably underestimates, as Leishmania is not always a notifiable disease and treatment is not always sought due to financial or geographic constraints [28, 29]. Mortality rates are also under-reported, being confined mainly to official hospital deaths. VL has case fatality rates of between 1.5%, in Bangladesh, to 20%, in South Sudan; based on a global case fatality rate of 10%, these figures represent approximately 20,000–40,000 deaths per year [30]. In recent years (2006–2016), morbidity related to leishmaniasis, in terms of disability-adjusted life years, has risen by 12.5% for CL/MCL but decreased by 61.1% for VL [31]. Other burdens exist, such as psychological morbidity arising from the social stigma surrounding the disfiguring lesions or scarring caused by CL and MCL [17].
The importance of the detection and identification of Leishmania
Worldwide control of leishmaniasis is believed to be achievable, but it does present a multifactorial problem because transmission of Leishmania species takes place in a complex biological interplay involving a human host, a diversity of parasite species, sand fly vectors, and animal reservoirs of infection, and is affected by a number of factors, including climate change, deforestation and war. Asymptomatic infections (and paucisymptomatic infections) add a further challenge, as undiagnosed patients become unobserved reservoirs of the disease, contributing to further transmission and maintenance of leishmaniasis foci [24, 32, 33]. Mild cutaneous infection, such as L. major CL infection, often goes undiagnosed and untreated, especially in resource-limited settings, which increases the risk of spread to the community [34]. Asymptomatic infections have been identified in several screening studies, including those of blood donors, and some studies have found these to be more common than symptomatic infection [35,36,37,38]. This was quantified in an epidemiological survey of peripheral blood by using real-time polymerase chain reaction (PCR), where the authors were able to determine a threshold of five parasitic genomes per millilitre of blood at which asymptomatic disease progressed to symptomatic disease [39].
One particularly confounding factor for any leishmaniasis control program is that Leishmania infection induces a broad spectrum of disease states and the clinical presentation cannot be linked to individual species with certainty; moreover, geographic foci may harbor multiple species. Thus, diagnosis on clinical grounds alone, through physical examination and interrogation of patient travel history, is not sufficient for complete case assessment. Figure 2 shows the recommended diagnostic techniques for the various clinical forms of leishmaniasis given by the WHO, the Centers for Disease Control and Prevention, the Walter Reed Army Institute of Research, the National Reference Centre for Parasitology (Canada) guidelines, and relevant studies. For clinical case management, species-level data provide important information for educated prognosis and therapeutic decision-making. For instance, species-led treatment was found to be imperative for cutaneous leishmaniasis in Peru, as tolerance and susceptibility to antimony was dependent upon the infecting species [40]. At a national and global level, the epidemiological monitoring of individual species and their prevalence and transmission patterns guide public health responses, such as the VL elimination program launched in 2005 in India, Nepal and Bangladesh [41]. Moreover, tracking the presence of exotic species, e.g. L. tropica discovered in a returned traveller to Mexico, allows local health authorities to enact sanitary measures (such as indoor residual spraying) [42]. Consequently, a globally applicable technique that captures both genus- and species-level data has been suggested, to mitigate assay design complications, such as intraspecies heterogeneity and gene target copy number variations, and to detect asymptomatic and multiple-organism infections (including co-infection with multiple Leishmania species) [6]. Seven criteria have been proposed for species-typing tools: discrimination of species, global applicability, sensitivity, specificity, standardisation, applicability for particular settings, and validation [6].

Recommended laboratory-based diagnostic techniques based on clinical presentation. For diffuse cutaneous leishmaniasis, disseminated leishmaniasis and leishmaniasis recidivans, where recommendations are not available from a public health organisation or reference laboratory, guidance is taken from relevant studies [43,44,45,46,47,48,49,50]. The asterisk indicates the rK39 rapid immunochromatographic test kit only. PCR Polymerase chain reaction
Additionally, test availability and timeliness affect opportune treatment. Shortages of diagnostic materials and a long period of time needed to deliver diagnostic results impede patient outcomes [51, 52]. These limitations result in a lack of useful information for medical decision-making. Furthermore, many current diagnostic tests do not have the resolving power required to provide molecular epidemiological data for public health policy makers. Accurate and qualitative detection and identification of a Leishmania infection are, therefore, key to the diagnosis of leishmaniasis, and should be at the heart of any successful control program. This can lead to the achievement of early and improved treatment regimens, implementation of control measures leading to better patient outcomes, and a reduction of sustained reservoirs in the transmission cycle [53]. Additionally, optimised diagnostic tools would be important additions to the One Health approach for the control of leishmaniasis [54]. The comparative strengths and weaknesses of existing diagnostic approaches, with a focus on the widely used and robust PCR-based methods that address many of the requirements for an optimal diagnostic test, are reviewed below.
Methods for the detection and diagnosis of leishmaniasis
Conventional detection methods
Detection via microscopy, histology, culture and serology, and other methods, are commonly utilised by laboratories globally, and especially in endemic, resource-poor nations [55].
For microscopic detection of parasites, direct aspirate smears are used, often with staining; amastigotes appear round in shape and 2–4 μm in diameter, and cultured promastigotes range between 15 and 25 μm in length and are ellipsoid to slender in shape [56,57,58]. Staining methods help to clarify the cells and Giemsa and Leishman stains (both derivatives of Romanowsky stain) are the most widely used for this [59]. Upon staining, Leishmania amastigotes are generally observed within macrophages and have a pale blue cytoplasm, red nucleus and adjacent purple-pink-stained kinetoplasts [60, 61]. Parasitic load may be estimated using the modified Ridley’s parasitic index (Table 2), which quantifies the number of amastigotes [62]. The sensitivity of detection varies (54.0–96.4%), and specificity as low as 46.0% has been reported, depending upon the primary sample taken, the quality of the reagent used for staining and technical expertise [43, 63,64,65,66,67,68]. The stage of infection can also greatly affect sensitivity; in CL, amastigote levels decrease as infection progresses, including in MCL infection, whereas in VL, parasitic load increases as infection becomes chronic [69,70,71]. Generally, in VL, the highest sensitivity when using microscopy is for more invasive specimens, such as those seen in splenic aspirates [70, 72, 73]. Microscopy is of use at the genus level, but cannot be used for species differentiation as all species of Leishmania are morphologically very similar [74]. Recently, machine learning has been incorporated into microscopical examination for leishmaniasis, with sensitivity and specificity of 83% and 35%, respectively, although efficacy and speed are dependent on image quality and the particular algorithm employed [75].
Histological examination for CL cases uses 4- to 5-μm tissue sections, stained with Giemsa or haematoxylin and eosin stains, and fixation to reveal histiocytes containing intracellular amastigotes, often near the epidermis [77]. The marquee sign, where organisms are located around the periphery of the dermal macrophage, is regarded as a typical characteristic of leishmaniasis [78]. Histological examination does, however, depend upon the disease stage, since the number of amastigotes decreases as CL progresses until they are undetectable [62]. Indeed, it is considered the least sensitive diagnostic method, with sensitivities of between 42.0% and 70.0%, although 100% specificity has been reported [79,80,81]. Furthermore, Leishmania cells may be mistakenly identified in histological sections as Toxoplasma gondii, Mycobacterium leprae, fungi, including Histoplasma, or even artifacts, and differential diagnosis requires alternative stains [81,82,83,84]. Histological examination is less used in VL, where clusters of histiocytes, amastigote presence or morphological changes may be observed [85]. Amastigotes are often unevenly distributed, as sections are of varying thickness, which results in lengthy analysis [86].
Culture of Leishmania promastigotes is a useful tool for increasing the sensitivity of downstream detection and identification by microscopy or molecular applications [87]. A variety of semi-solid, liquid or biphasic media are used to culture promastigotes, including sloppy Evans, Novy-MacNeil-Nicole (the reference medium for isolation), Tobie’s, Schneider’s Drosophila medium, Senekjie’s, Medium 199, RPMI 1640, Grace’s insect medium, brain–heart infusion medium, blood agar (including rabbit blood) and chocolate agar [88,89,90]. These media generate growth in differing ways; of note, Tobie’s medium encourages the transformation of amastigotes to promastigotes, and cell density is increased on Grace’s medium [73]. Novel culture methods include the microcapillary culture method, which concentrates the sample in capillary tubes, or liquid (single-phase) media, used to create the microaerophilic conditions that are optimal for amastigote transformation into promastigotes [91]. One study found an improvement from 69.2% sensitivity with traditional culture methods to 92.3% sensitivity with the newer microcapillary method, with a minimal change in specificity (98.9% vs 97.8%, respectively) [92]. It can take days to weeks to produce a result by culture methods, which are also expensive and labour-intensive to set up [86]. Furthermore, the distribution, transport and storage of cultures and culture material, including antibiotics used to prevent contamination from other microorganisms, make it an impractical method in many clinical settings [93, 94].
Serological methods test for Leishmania by detecting antigens or antileishmanial antibodies in the blood or, sometimes, urine or saliva. Antibody detection is primarily used in cases of VL rather than for CL, as the humoral response to the latter is poor [95]. Many antigens have been assessed for antibody detection, and recombinant antigens are preferred over natural antigens, as the latter often cause problems such as cross-reactivity and resultant false-positive results [61, 96]. Some immunoassays, such as enzyme-linked immunosorbent assay (ELISA) and western blot, require relatively sophisticated and expensive equipment and materials, which renders them less useful in endemic regions in poorer countries, despite their good sensitivities and robustness [97, 98]. However, ELISA is widely used as a serological method in countries where leishmaniasis is endemic or non-endemic, and it can provide detailed information on antibody responses [99, 100]. The sensitivity of ELISA depends on the antigen used to capture a specific antibody, with the commonly used crude soluble antigen (CSA), for example, providing sensitivities of between 80 and 100%. However, cross-reactions with trypanosomiasis, tuberculosis and toxoplasmosis have been observed with this method [101]. Flow cytometry for serological Leishmania detection is a recent development and can quantify antibodies rapidly with lower sample input volumes than other serological tests [102]. Despite the range of tests available and the ability of some of these tests to be used in the field, serological assays share the same limitations. The antigen load may not be sufficient in early infection for detection; conversely, because antigen-specific antibodies persist long after cure, active relapsed disease cannot always be discerned [61, 103]. Furthermore, these tests are less accurate for immunocompromised patients, and cross-reactivity is reported with other diseases endemic to Leishmania-affected areas, including Chagas disease [53, 88, 104,105,106,107,108]. Simplified assays have been introduced, such as the rK39 immunochromatographic assay (ICT), the direct agglutination test, the indirect fluorescent antibody test and latex agglutination testing [109]. They include the Food and Drug Administration-approved ICTs, CL Detect, based on the peroxiredoxin antigens, and Kalazar Detect, based on a 39 amino acid repeat recombinant leishmanial antigen, rK39. Although the rk39 ICT is widely used, its sensitivity has been found to be higher in some regions, with studies carried out in Southeast Asian countries and the Indian subcontinent documenting higher sensitivities than those undertaken in East Africa and Brazil [110,111,112]. Furthermore, because antibodies are present in asymptomatic individuals and remain present for several years after cure, antibody tests must be used in conjunction with strictly standardised clinical case definition (i.e., more than 2 weeks of fever, weight loss and splenomegaly) for VL diagnosis [24]. These limitations must be considered in a modern diagnostic setting, hence, serological methods are being reassessed as principal diagnostic options [86, 113].
Other, lesser-used, methods include the leishmanin (or Montenegro) skin test. In CL, this test can detect past and active cases with high sensitivity, whereas in active VL, the test shows negative as patients are anergic, so it is used in screening studies for an indication of past exposure only [57, 114]. It involves intradermal injection of antigen, whereby induration of an area of the skin of 5 mm or greater is considered a positive test result; sensitivity and specificity with this cut-off point have been reported as 97.4% and 93.9%, respectively [15, 115, 116]. When promastigote levels are too low for culture growth, xenodiagnosis can be used to detect Leishmania, through inoculation of the footpad of a hamster with a test sample; however, this approach is time-consuming and involves euthanasia of the hamster [56, 117, 118].
DNA-based detection methods
In DNA-based detection of Leishmania a multitude of genomic targets are used which vary in sensitivity and are influenced largely by the target’s copy number in the genome of the organism. Table 3 summarises the targets that have been investigated for diagnostics. The design of molecular diagnostics is complex, however, and aside from the copy number of a chosen target, which is selected to increase the sensitivity of a given assay (for instance, the 18S rRNA gene), other attributes are sought in molecular assay design [119, 120]. For detection to the subgenus, species complex or species level, selection of gene targets exhibiting increased divergence may necessitate a loss of assay sensitivity by using a target with fewer copy numbers per cell (such as the mini-exon gene) [121]. For instance, polymorphisms, copy number variation and high copy number are attractive features of the kinetoplast DNA (kDNA) minicircle, whereas single copy genes, despite their decreased sensitivity, may be chosen for their stability or to normalise minicircle copy numbers [122, 123]. Moreover, detection of multiple species distinctly and concurrently requires multiplex PCR assays, and designing a single PCR cycling protocol to suit each primer pair can present assay constraints. The use of the novel bisulphite-conversion technique can mitigate such limitations [124].
DNA-based methods represent a new era in clinical Leishmania diagnostics, where the limitations of previous detection methods have been overcome in terms of sensitivity, specificity, rapidity, ease of use and access in endemic settings. Similarly, the prices of molecular methods are decreasing globally; Table 4 illustrates the varying costs associated with Leishmania diagnostics in low income and upper-middle income nations [125,126,127,128]. The costs associated with molecular methods are comparable to those of other detection methods; however, the methodologies used to collect the costing data varied between studies. Some studies compiled comprehensive costings, including that of the healthcare setting that the test may be performed in, to the basic supplier cost of the kit [125, 126]. Despite their differences, these cost analyses were performed at similar times, and their costing conclusions are similar. These advances have also provided new data on frequencies of asymptomatic carriage, and have been used in the monitoring of specific geographic disease burden and to measure the outcomes of intervention programs [109]. Techniques such as real-time PCR can give more information on parasite load and responsiveness to therapy than other methods [122]. It can be used to identify asymptomatic patients who carry the infection, or infected patients before the onset of symptoms, which is important for the development of control measures and for blood donor monitoring [129, 130].
DNA may be acquired from a vast range of clinical specimens, and each approach has varying advantages in terms of diagnostic sensitivity and specificity, ease of collection, transport and storage and invasiveness. For CL, the punch biopsy is the most commonly performed diagnostic procedure, but less invasive sampling can be achieved by using skin scrapings, fine needle aspiration and swabs, although sensitivity is sacrificed with these methods [131,132,133,134]. For post-kala-azar dermal leishmaniasis, split skin smears and skin biopsies are most commonly used [135]. In VL, splenic, bone marrow or lymph aspiration are used, and sensitivity increases with the invasive nature of the sampling method (93–99% for spleen, 53–86% for bone marrow, and 53–65% for lymph [136]). Specimens obtained from less invasive procedures, e.g., the taking of peripheral blood, are being explored for VL testing. Testing of peripheral blood by PCR is associated with a vast range of reported sensitivities, 62—93.2%, depending on the timing of sample collection during the infection process, or the fact that some Leishmania species may circulate at lower levels in the peripheral blood [137, 138]. The use of peripheral blood is also being explored as a diagnostic option for CL, and a limit of detection of 0.1 parasites per reaction for Leishmania (Viannia) spp. parasites has been achieved [139]. Optimal sampling, storage and transport to a receiving laboratory (such as the Centres for Disease Control and Prevention, US, or Fiocruz, Brazil) is critical. Before detection, DNA must first be extracted and purified; this is often performed by using commercial kits, such as the silica-based DNeasy Blood and Tissue Kit (Qiagen, Germany), NucliSENS easyMAG system (BioMerieux, France) or via in-house extraction protocols based on phenol extraction and ethanol precipitation [36, 140, 141]. Some rapid extraction methods have been developed recently, e.g., SpeedXtract (Qiagen, Hilden, Germany), which greatly reduce the time to test result [142]. These methods may be performed manually or through the use of automated systems, as discussed below.
Standardisation of protocols and quality control for assays that are used to detect Leishmania are important, particularly for molecular techniques [143]. Only few studies have compared sampling, extraction, gene target choice and primer design, and some of the findings differ from one report to an other [55]. These inconsistencies can be limited if experiments incorporate controls, which is of particular importance in settings where re-testing is expensive or the number of specimens limited. Poor DNA recovery due to losses during extraction and degradation during storage was determined by the addition of both an external and internal control to a conventional PCR (cPCR) assay for Leishmania [143]; DNA recovery was poor for 15.1% of samples, and a reliable result was not produced for up to 1/6 of the samples [143]. Furthermore, few multi-site studies have been undertaken to validate protocols and examine their reproducibility. An endogenous extraction control used in the measurement of a host sequence can help to account for sample quality and extraction efficacy—two major issues associated with PCR methods—and may also be used to normalise parasite load [122, 143]. For this, an exogenous internal control is spiked into a sample at a known concentration, and sample inhibition is indicated if the control is not detected or is detected at lower levels than expected. This is especially useful for potential inhibitors in samples, such as in peripheral blood; a human β-actin gene was used in a real-time PCR assay to control for this [144]. Other controls that may be included are external positive controls that are used to assess the performance of the PCR, negative template control for PCR contamination, or a negative process control for contamination of the extraction process. Laboratories should also enrol in some form of an external quality control testing program, such as the Pan American Health Organization’s Regional External Quality Assessment Program (developed for microscopic diagnosis), as an indicator of performance [55, 145].
A molecular technique that can be easily used in a resource-limited setting is nucleic acid sequence-based amplification (NASBA), which is based on the isothermal amplification of nucleic acids by enzymatic action. However, NASBA is prone to contamination, as it is not a closed tube system, which potentially leads to false–positive results. It can be used as a quantitative test, targeting RNA in the DNA background, with one method targeting 18S rDNA showing a sensitivity of 79.8% and specificity of 100% [146]. Quantitative NASBA can be combined with electrochemiluminescence, although this is more expensive and time-consuming; however, as the reaction includes a fluorescent beacon, it may be used in a real-time, closed tube format [147]. Loop-mediated isothermal amplification is another alternative to PCR that may be used in an endemic setting, as only basic equipment is required, with no need for a thermal cycler, and a total amplification time of about 40 min [148]. It involves amplification in a water bath and a visible colour change, which can be detected by the naked eye or under blue light, and more recently, in real-time by fluorimetry [149,150,151]. Sensitivity of this technique has been reported as 80–90% with specificities of 94–100% for human Leishmania diagnosis [152]. Additionally, as it is a closed tube test there is no need for post-amplification handling, thus the risk of laboratory contamination is low [153]. Recombinase polymerase amplification (RPA) and recombinase-aided amplification (RAA) are isothermal amplifications wherein recombinase enzymes and proteins avoid the need for temperature cycling as used in PCR methods [154, 155]. In RPA, the recombinase is derived from a phage, whereas in RAA, it is derived from bacteria and/or fungi. RPA was paired with a rapid extraction method to detect L. donovani in two studies [142, 156]; the resultant detection systems were rapid, mobile and avoided the need for refrigerated reagents. The kDNA minicircle target was used in both assays, which had sensitivities and specificities of 100% and 100% [142] and 65.5% and 100% [156], respectively. However, there are challenges in the use of RPA and RAA, as robust design guidelines have yet to be published for either method, and both are time-consuming, labour intensive and expensive.
Polymerase chain reaction-based detection methods
The use of polymerase chain reaction (PCR) as a molecular technique for Leishmania detection, in which purified nucleic acids of the pathogen are amplified, is becoming more widespread. A PCR product may be detected at the end of amplification (cPCR) by gel electrophoresis, amplified further before detection (nested PCR) or detected as amplification occurs (real-time PCR) [157,158,159]. PCR has the best-reported sensitivities and specificities of all the diagnostic methods, and has been suggested as the future gold standard for Leishmania detection [123, 132, 160, 161]. One systematic review and meta-analysis reported sensitivities of up to 100% and specificities up to 100% [136]. Another systematic review assessing assays designed for New World parasitic species regularly found limits of detection of less than one copy [119]. Several commercially available kits based on PCR amplification and detection of Leishmania genes have been developed and are given in Table 5. PCR performance depends on certain factors, such as the nucleic acid extraction method employed, the type of sample, the copy number of the gene target and the design of the primers that target these [53, 109]. Thus, in-house PCR methods, which are developed and used widely, have great differences in the types of DNA targets used, primer and probe design and PCR cycling protocol [129]. Technology transfer from the research and development phase does not always occur, and as these types of PCR assays are far from standardised, data comparison between laboratories is compromised [109, 162]. More recently, PCR has been used to monitor a subject of growing concern in Leishmania infections, particularly in endemic regions: the relapse of disease or resistance to chemotherapy. Both issues present major challenges for the control of leishmaniasis [163]. A PCR-based study that monitored parasite load in VL showed that the presence of 10 parasites/mL of blood after treatment indicated relapse, thus gave useful information for disease prognosis [164]. Four single nucleotide polymorphisms of cysteine protease B gene were identified and indicated resistance to a widely used drug, amphotericin-B; thus, detection of these could be incorporated into a PCR assay [165]. These techniques have yet to be standardised, thus are not widely available in clinical settings, but have great potential to aid public health responses [166].
cPCR: In this method, DNA is amplified using a thermal cycler, amplicons are separated by electrophoresis due to their molecular weight and detected by staining (usually ethidium bromide) and ultraviolet light (via a transilluminator) [167, 168]. This method can be time consuming, requires an array of equipment and, as the PCR tube containing amplicons must be opened for electrophoresis, is associated with a risk of contamination [169]. Clinical sensitivity and specificity of up to 100% each have been achieved for lesion samples, with detection of as low as 0.01–0.1 pg of cultured Leishmania promastigote DNA [170].
Nested PCR: This method is used to overcome poor sensitivity and specificity [171, 172]. Two sequential PCRs are used: first, an outer set of primers is used to amplify the target gene (first round), then the amplicons of this PCR are re-amplified with a set of inner primers (second round) [173]. The disadvantages of this approach are that the use of two PCRs is more time consuming and requires more reagent, and since the amplicon tube is opened for the second PCR, it is an open system, which poses a contamination risk [174]. Encouragingly, however, 100% sensitivity and 100% specificity were reported for a recently developed Leishmania spp.-specific modified version of a nested PCR that was created to reduce carryover and cross-contamination [175].
Real-time PCR: This method, in which fluorescent dyes give a visual indication of amplification as the reaction occurs, is a more recent advance than cPCR [122, 176]. Although limiting in some settings due to the equipment and expertise necessary, real-time PCR is faster than cPCR and is a closed system, so contamination risk is reduced [147, 177]. Real-time PCR is generally used with intercalating dyes, e.g. SYBR green due to its lower cost, but this method is prone to false–positives as it will visualise any amplified double-stranded DNA [178]. Use of probe-based real-time PCR increases specificity, thus avoids this issue, and targets can be multiplexed, whereby multiple assays occur in the same reaction, which saves reagents and time and increases throughput [179]. Real-time PCR was compared to cPCR, and higher sensitivity and specificity, 93.9% and 100%, respectively, for the former was reported compared to two cPCR assays tested (75.6% and 100% for kDNA, and 53.7% and 88.8% for ITS1, respectively) [180]. Higher sensitivity, specificity and reproducibility of real-time PCR have also been reported in other studies [181,182,183,184]. For example, using peripheral buffy coat from cases of VL, 100% sensitivity and 100% specificity were achieved with real-time PCR [103, 129, 185]. The technique can give data on parasite load, which in turn provides information for prognosis and treatment, and is useful for epidemiological studies [186, 187]. Bisulphite modification is a method for reducing the complexity of the genome prior to application of PCR that has been adapted to Leishmania detection in real-time PCR [124, 188]. This assay achieved an analytical sensitivity of 10 genomic copies per real-time PCR reaction, and clinical sensitivity of 97.0% and specificity of 100.0%. Through treatment with sodium bisulphite, cytosine is converted to uracil and ultimately thymine during the first round of PCR. This causes the genomes of subtypes to become more similar to each other, making genus-level primer and probe design possible for highly polymorphic gene targets [189]. The resulting simplified genome enables the design of simplified primers with unique characteristics, and the consequently fewer mismatches result in a similar melting temperature (Tm), allowing for the design of longer oligonucleotides, thereby increasing the specificity of the assay. Additionally, proximate Tms between species-level oligonucleotides can be achieved, making multiplexing more efficient, as a uniform PCR cycling protocol can be easily designated [124].
Droplet digital PCR: The advent of this method has enabled the absolute quantitative measurement of target DNA, negating the need for calibration curves in PCR assays [190]. DNA molecules are partitioned into tens of thousands of replicate PCR reactions, and amplification to endpoint PCR occurs, at which point each “droplet” has template or no template present [191]. Due to the vast numbers of binary (positive or negative) results, the number of target DNA molecules can be calculated precisely. A droplet digital PCR was developed based on 18S rDNA and validated for seven Leishmania species; however, despite the accurate quantification of DNA, the assay was marginally less sensitive and specific than an equivalent real-time PCR assay (84.0% for the former vs 85.0% for the latter) [192]. Moreover, the authors reported that the cost of droplet digital PCR is three times that of real-time PCR, thus is not suitable for the routine diagnosis of Leishmania.
Methods for the identification of species of Leishmania
Differentiation of Leishmania species to discriminate the species, or group of species, that cause disease is important, both clinically and epidemiologically, for disease prognosis, determining therapeutic options and surveillance of populations [122, 193]. As in Leishmania detection, no single differentiation method is considered to be a gold standard, although several techniques have been proposed, including PCR, multilocus enzyme electrophoresis (MLEE) and multilocus sequence typing (MLST) [122, 133, 193,194,195,196,197,198]. The lack of a definitive gold standard for species typing may be attributed to inherent issues related to a lack of standardisation, particularly with regard to the interpretation of the typed result. This pitfall is complicated by the closely related Leishmania species and inter-species hybrids that exist, and the novel species that continue to be discovered [199, 200]. Moreover, methods of interpretation differ between laboratories and, if species assignment is based on centralised programs like BLAST, results can be dependent upon the evaluation of resultant similarity scores [201]. Despite this, multiple robust methods exist and the results obtained with their use continue to strengthen the molecular epidemiological, taxonomic and clinical databases, even if these methods are not yet optimal for routine uses.
Multilocus studies are now used in preference to single locus analysis for population-wide studies as they achieve better resolution [129]. By combining genomic targets in parallel, multilocus schemes capture genetic relationships that may be missed by one genetic locus, which is particularly advantageous in intraspecific variation studies or for species typing within species complexes due to the amount of information gathered [6]. MLEE distinguishes between organisms through electrophoresis of enzymes, and is regarded by some as the gold standard for Leishmania typing and taxonomic studies; information acquired using MLEE led to the development of the first phylogenetic trees of Leishmania [18, 202, 203]. Differences in enzyme mobilities are due to differences in their protein structures, which comprise different amino acids, and lead to the creation of banding patterns from which zymodemes (populations with similar isoenzyme patterns) can be assigned [24]. MLEE is laborious as it requires a large volume of cell culture and can take 1–2 months to produce results, which cannot be compared, with confidence, between laboratories; e.g., different enzyme panels are used in Europe and South America [204, 205]. Furthermore, one zymodeme, MON-1, for L. infantum, the causative agent of most cases of VL in the Mediterranean Basin and South America, was shown to be heterogeneous and polymorphic [129].
DNA-based identification methods
MLST, which involves DNA sequencing sections of defined housekeeping genes (usually seven or more) and the many allelic combinations produced, results in unambiguous characterisation of isolates, giving both inter- and intra-species information on heterogeneity [206, 207]. This method is considered so powerful it has also been proposed as the new gold standard for taxonomic determination of Leishmania [208]. MLST has high reproducibility and can be compared between laboratories; however, it is technically demanding [209].
Like MLST, multilocus microsatellite typing (MLMT) uses co-dominant markers, and because of the relatively high mutation rate of microsatellites, comparison of closely related organisms is possible [209, 210]. It works by the amplification of repeat sequences found in microsatellites, where polymorphisms in the copy number of repeats define the type assigned [211]. For instance, MLMT has been used to analyse L. donovani strains. In one study, the identification of heterogeneous genotypes by MLMT negated the usefulness of MLEE determining genetic relationships in zymodeme MON-37. Not only were the isolates genetically diverse but, geographically, they were spread globally, leading the authors to surmise that the discriminatory power of MLMT adds depth to both diagnostic and population genetic studies [212].
DNA sequencing is based on the classic Sanger sequencing (chain termination) method and, more recently, next generation sequencing (NGS), both of which identify the precise order of nucleotide bases in a targeted DNA locus [213]. DNA sequencing provides important information for genetic, clinical and epidemiological studies. For instance, gene sequence analysis has been used to detect Leishmania hybrids using the cytochrome b gene, to study differences in genetic composition between healed and non-healed patients using the ITS1, 7SL RNA and heat-shock protein 70 regions, and to illuminate the diversity of kDNA minicircle classes [213,214,215]. It is viewed currently as an impractical technique for endemic areas, as it is technically demanding, although simplified techniques such as nanopore sequencing are being developed [198, 216].
NGS involves the extremely high-throughput sequencing of nucleotides in parallel [217]. It provides deep genome sequencing and whole genome sequencing (WGS) of organisms, and provides population-level data in a clinical context, although it is too costly for routine typing [218, 219]. The first complete genome sequence of any Leishmania parasite (i.e., L. major) was completed in 2005 and, since then, the complete genomes of L. mexicana, L. tropica, L. amazonensis, L. donovani, L. infantum, L. panamensis, L. braziliensis, L. guyanensis, L. naiffi, L. peruviana, L. lainsoni, L. martiniquensis and L. orientalis have been sequenced [220, 221]. One type of NGS couples WGS with MLST, and is a methodological advance for the provision of standardised epidemiological, molecular evolution and pathogenicity data [222]. Aneuploidy, which is observed in laboratory cultivated samples, as well as mosaicism, can be challenging in WGS, and thus read depth is critical for correct interpretation of Leishmania NGS data [223,224,225].
PCR-based identification methods
Restriction fragment length polymorphism (RFLP) technology: This methodology is used after amplification by PCR, and is widely applied in species identification [199]. Post-PCR amplicons are digested with a restriction enzyme and the products are detected on a gel. The banding pattern produced can be used to identify a particular species, depending on the presence or absence of a restriction enzyme site in the PCR product derived from the target [226]. It is a relatively inexpensive technique compared to real-time PCR, and a multitude of gene targets can be utilised, including heat-shock protein 70, glycoprotein 63, kDNA, cysteine protease B, mini-exon or NADH dehydrogenase subunit 7 [121, 227,228,229,230,231].
An ITS1-RFLP reported by Schönian et al. [231] was able to differentiate most Leishmania species but was less useful for those species within the L. braziliensis complex [232, 233]. Such findings can be problematic, as the clinical presentation of the MCL tropism is caused by more than one of these species, but the response to treatment differs between them [129]. Improvements in species discrimination using RFLP were achieved recently using the NADH dehydrogenase subunit 7 maxicircle gene target, where it was possible to discriminate L. braziliensis from other species within the L. braziliensis complex (Fig. 3) [231]. RFLP presents multiple difficulties, as it needs a relatively high parasite load, and is thus often paired with cell culture, and multiple restriction enzymes may have to be employed depending on the DNA target. Additionally, RFLP can be difficult to compare between laboratories as banding patterns may be dissimilar due to differing gel size or concentration [234].

Polymerase chain reaction—restriction fragment length polymorphism analysis of Leishmania species targeting the NADH dehydrogenase subunit 7 gene, digested with NIaIII (a) or HpyCH4IV (b) run alongside 50-base pair (bp) molecular weight markers. Digestion of in silico, culture and direct clinical samples enabled differentiation of Leishmania species. Reprinted with permission of Kaufer et al. [231]
Melt curve analysis: This method is used to differentiate species following real-time PCR. Its effectiveness relies on the fact that the temperature at which a sequence of double-stranded DNA dissociates (or “melts”) is a function of the GC/AT ratio and the length of an amplicon [235]. Different species exhibit different melting points, which allows for discrimination [236]. In a study that used Tms to group infecting species, species that caused different clinical presentations (i.e., CL/MCL and diffuse cutaneous leishmaniasis) could be differentiated [234]. Either a standard melt curve or high resolution melt can be use for analysis, the latter being a method that is able to detect more subtle differences in temperature, which potentially gives better species discrimination [237,238,239].
Biosensors
Biosensors lead the emerging field of nanodiagnostics, spanning target detection of DNA/RNA, proteins and even volatile organic compounds from exhaled breath. They are devices that, put simply, convert a biological signal into an electrical signal via a recognition element; they are reported to be low-cost and portable, with high sensitivity and specificity of performance documented [240, 241]. Biosensors use one of several modes of signal generation, like electrochemiluminescence or optical signals, or are based on surface plasmon resonance (SPR). Genosensors, recognising DNA (or RNA), dominate biosensor diagnostics for Leishmania. Other recognition elements may be antibodies, antigens or the newer aptamer-based sensors [242]. Aptamers show great promise in binding biological targets with high affinity; they are short, single-stranded nucleic acids that form unique three-dimensional structures and recognise and bind target molecules in a similar fashion to antibodies, such as those targeting the L. infantum histone H3 or poly(A) binding protein [243, 244]. A DNA-based biosensor using SPR that targeted the kDNA detected L. major and L. tropica [245]. Also using SPR biosensing techniques, Ferreira et al. [245] developed an immunosensor based on circulating antibodies against L. infantum, which achieved antibody detection within 7 min [246]. Another DNA detection biosensor, which uses fluorescent probes based on the kDNA of L. infantum and nanostructured films as sensing platforms, provided sensitive results (1.1 nM of target DNA) even for complex sample types such a human blood [247]. To determine selectivity for the target molecule, the authors measured fluorescence recovery intensity when a target DNA sequence with a single base mismatch was introduced, and observed a reduction of 32% when compared to a fully complementary sequence [247]. Most recently, another genosensor based on the recognition of a single-stranded DNA sequence of L. infantum on cadmium sulfide nanosheets was described; the detection limit for L. infantum DNA was 1.2 ng/uL without reaction with L. major and L. tropica DNA [248]. Biosensor development for Leishmania detection is in its early stages and requires more research to improve efficiencies and standardisation. Despite this, recent publications on their use in NTDs have indicated the potential for their increased performance as well as a reduction in interactions with interfering substances, good stability and the miniaturisation of devices, allowing their portability [249]. Furthermore, this detection method has been widely integrated into smartphone technology, simplifying the interpretation of results and allowing for multiplexing of targets, such as multiple species [250, 251]. Biosensor technology, though in its infancy with regard to leishmaniasis detection, may be a good solution to the challenge of providing a cost-effective, fast and portable detection method [252].
Conclusions
A multitude of diagnostic assays exist for the detection of Leishmania species, but there is no widely accepted gold standard [122, 195, 196]. There have been, however, huge developments in the speed and accuracy of methods with advances in technology, and different approaches may be better suited to different diagnostic health care settings, ranging from primary health care centres (where technical staff perform point-of-care or single-use tests for outpatients) to district hospital laboratories (where limited numbers of staff perform selected routine tests), regional or provincial hospital laboratories (where high numbers of laboratory staff are present to cover many pathology disciplines) and, ultimately, to national reference laboratories (providing highly specialised tests, education and training in research or for teaching hospitals) [253]. All the techniques for the detection and identification of Leishmania, of which there is a vast array, have their own strengths and limitations, but high sensitivity, high specificity, low turnaround times and affordability are the critical features of an ideal test. Also important is discrimination between Leishmania species, which is vital for epidemiological studies, disease prognosis and for the implementation of patient treatment regimens. Figure 4 illustrates typical workflows for the techniques used in diagnostic laboratories, and highlights that, whilst more traditional protocols can be used to identify Leishmania to the genus level relatively speedily when in the hands of trained, experienced professionals, only molecular-based techniques can give species-specific diagnostic information. As global efforts increase to control and eliminate NTDs, there is a need to develop, validate and standardise novel diagnostics for the detection and differentiation of Leishmania spp. With the successful implementation of such methods, the global burden of this disease could be reduced dramatically, with positive outcomes being seen for the people that need them the most.

Example workflows for the detection of Leishmania in clinical diagnostic laboratories at each laboratory tier, as set out in the list of essential in vitro diagnostics (World Health Organization) [253]. qPCR Real-time PCR, ELISA enzyme-linked immunosorbent assay, DAT direct agglutination test
Availability of data and materials
Not applicable.
Abbreviations
- CL:
-
Cutaneous leishmaniasis
- cPCR:
-
Conventional polymerase chain reaction
- ELISA:
-
Enzyme-linked immunosorbent assay
- ICT:
-
Immunochromatography test
- MCL:
-
Mucocutaneous leishmaniasis
- MLEE:
-
Multilocus enzyme electrophoresis
- MLMT:
-
Multilocus microsatellite typing
- MLST:
-
Multilocus sequence typing
- NASBA:
-
Nucleic acid sequence-based amplification
- NGS:
-
Next-generation sequencing
- NTD:
-
Neglected tropical disease
- PCR:
-
Polymerase chain reaction
- RAA:
-
Recombinase-aided amplification
- RFLP:
-
Restriction fragment length polymorphism
- RPA:
-
Recombinase polymerase amplification
- SPR:
-
Surface plasmon resonance
- Tm:
-
Melting temperature
- VL:
-
Visceral leishmaniasis
- WGS:
-
Whole genome sequencing
- WHO:
-
World Health Organization
References
Savoia D. Recent updates and perspectives on leishmaniasis. J Infect Dev Ctries. 2015;9:588–96.
World Health Organization. First WHO report on neglected tropical diseases 2010: working to overcome the global impact of neglected tropical diseases. 2010.
Gonzalez C, Wang O, Strutz SE, Gonzalez-Salazar C, Sanchez-Cordero V, Sarkar S. Climate change and risk of leishmaniasis in North America: predictions from ecological niche models of vector and reservoir species. PLoS Negl Trop Dis. 2010;4:e585.
Alawieh A, Musharrafieh U, Jaber A, Berry A, Ghosn N, Bizri AR. Revisiting leishmaniasis in the time of war: the Syrian conflict and the Lebanese outbreak. Int J Infect Dis. 2014;29:115–9.
Curtin JM, Aronson NE. Leishmaniasis in the United States: emerging issues in a region of low endemicity. Microorganisms. 2021;9:578.
Van der Auwera G, Dujardin JC. Species typing in dermal leishmaniasis. Clin Microbiol Rev. 2015;28:265–94.
Espinosa OA, Serrano MG, Camargo EP, Teixeira MM, Shaw JJ. An appraisal of the taxonomy and nomenclature of trypanosomatids presently classified as Leishmania and Endotrypanum. Parasitology. 2016;145:430–42.
Akhoundi M, Downing T, Votypka J, Kuhls K, Lukes J, Cannet A, et al. Leishmania infections: molecular targets and diagnosis. Mol Aspects Med. 2017;57:1–29.
Sereno D. Leishmania (Mundinia) spp.: from description to emergence as new human and animal Leishmania pathogens. New Microbes New Infect. 2019;30:100540.
Schönian G, Lukeš J, Stark O, Cotton JA. Molecular evolution and phylogeny of Leishmania. In: Ponte-Sucre A, editor. Drug resistance in Leishmania parasites. Cham: Springer; 2018.
Fraga J, Montalvo AM, De Doncker S, Dujardin J-C, Van der Auwera G. Phylogeny of Leishmania species based on the heat-shock protein 70 gene. Infect Genet Evol. 2010;10:238–45.
Schönian G, Mauricio I, Cupolillo E. Is it time to revise the nomenclature of Leishmania? Trends Parasitol. 2010;26:466–9.
Fernandes Shimabukuro PH, de Andrade AJ, Bianchi Galati EA. Checklist of American sand flies (Diptera, Psychodidae, Phlebotominae): genera, species, and their distribution. ZooKeys. 2017;660:67–106.
Haque A, Ekram ARMS, Sharmin LS, Belaluddin M, Salam MA. Congenital visceral leishmaniasis. Pak J Med Sci. 2010;26:485–7.
Magill AJ, Meyers WM, Klassen-Fischer MK, Neafie RC. Visceral leishmaniasis. In: Topics on the pathology of protozoan and invasive arthropod diseases. Edited by Sciences USUotH. Bethesda, MD, USA: Uniformed Services University of the Health Sciences; 2011: 1–11.
Guedes DL, van Henten S, Cnops L, Adriaensen W, van Griensven J. Sexual transmission of visceral leishmaniasis: a neglected story. Trends Parasitol. 2020;36:950–2.
Bern C, Maguire JH, Alvar J. Complexities of assessing the disease burden attributable to leishmaniasis. PLoS Negl Trop Dis. 2008;2:e313.
Bañuls A-L, Hide M, Prugnolle F. Leishmania and the leishmaniases: a parasite genetic update and advances in taxonomy, epidemiology and pathogenicity in humans. Adv Parasitol. 2007;64:1–109.
Raja M, Gonzales Zamora JA. Visceral leishmaniasis with cutaneous involvement caused by Leishmania infantum-chagasi. IDCases. 2018;11:16–7.
Sterkers Y, Lachaud L, Bourgeois N, Crobu L, Bastien P, Pages M. Novel insights into genome plasticity in eukaryotes: mosaic aneuploidy in Leishmania. Mol Microbiol. 2012;86:15–23.
Prieto Barja P, Pescher P, Bussotti G, Dumetz F, Imamura H, Kedra D, et al. Haplotype selection as an adaptive mechanism in the protozoan pathogen Leishmania donovani. Nat Ecol Evol. 2017;1:1961–9.
Pace D. Leishmaniasis. J Infect. 2014;69:S10–8.
Chagas AC, Oliveira F, Debrabant A, Valenzuela JG, Ribeiro JM, Calvo E. Lundep, a sand fly salivary endonuclease increases Leishmania parasite survival in neutrophils and inhibits XIIa contact activation in human plasma. PLoS Pathog. 2014;10:e1003923.
World Health Organization. Control of the leishmaniases. 2010. https://www.who.int/publications/i/item/WHO-TRS-949.
Hotez P, Molyneux D, Fenwick A, Ottesen E, Ehrlich Sachs S, Sachs J. Incorporating a rapid-impact package for neglected tropical diseases with programs for HIV/AIDS, tuberculosis, and malaria. PLoS Med. 2006;3:e102.
World Health Organization. Global leishmaniasis surveillance update, 1998–2016. In: Global leishmaniasis surveillance update, 1998–2016. World Health Organization. 2018.
World Health Organization. Leishmaniasis. https://www.who.int/health-topics/leishmaniasis. 2022. Accessed August 2022.
Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004;27:305–18.
Okwor I, Uzonna J. Social and economic burden of human Leishmaniasis. Am J Trop Med Hyg. 2016;94:489–93.
Wamai RG, Kahn J, McGloin J, Ziaggi G. Visceral leishmaniasis: a global overview. J Glob Health Sci. 2020;2.
Collaborators GDH. Global, regional, and national disability-adjusted life-years (DALYs) for 333 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1260–344.
Ostyn B, Gidwani K, Khanal B, Picado A, Chappuis F, Singh SP, et al. Incidence of symptomatic and asymptomatic Leishmania donovani infections in high-endemic foci in India and Nepal: a prospective study. PLoS Negl Trop Dis. 2011;5:e1284.
Das VNR, Bimal S, Siddiqui NA, Kumar A, Pandey K, Sinha SK, et al. Conversion of asymptomatic infection to symptomatic visceral leishmaniasis: a study of possible immunological markers. PLoS Negl Trop Dis. 2020;14:e0008272.
Kamink S, Abdi A, Kamau C, Ashraf S, Ansari MA, Qureshi NA, et al. Failure of an innovative low-cost, noninvasive thermotherapy device for treating cutaneous leishmaniasis caused by Leishmania tropica in Pakistan. Am J Trop Med Hyg. 2019;101:1373–9.
Foroutan M, Dalvand S, Khademvatan S, Majidiani H, Khalkhali H, Masoumifard S, et al. A systematic review and meta-analysis of the prevalence of Leishmania infection in blood donors. Transfus Apher Sci. 2017;56:544–51.
Aliaga L, Ceballos J, Sampedro A, Cobo F, Lopez-Nevot MA, Merino-Espinosa G, et al. Asymptomatic Leishmania infection in blood donors from the south of Spain. Infection. 2019;47:739–47.
Mannan SB, Elhadad H, Loc TTH, Sadik M, Mohamed MYF, Nam NH, et al. Prevalence and associated factors of asymptomatic leishmaniasis: a systematic review and meta-analysis. Parasitol Int. 2021;81:102229.
Andrade-Narvaez FJ, Loria-Cervera EN, Sosa-Bibiano EI, Van Wynsberghe NR. Asymptomatic infection with American cutaneous leishmaniasis: epidemiological and immunological studies. Mem Inst Oswaldo Cruz. 2016;111:599–604.
Sudarshan M, Sundar S. Parasite load estimation by qPCR differentiates between asymptomatic and symptomatic infection in Indian visceral leishmaniasis. Diagn Microbiol Infect Dis. 2014;80:40–2.
Arevalo J, Ramirez L, Adaui V, Zimic M, Tulliano G, Miranda-Verastegui C, et al. Influence of Leishmania (Viannia) species on the response to antimonial treatment in patients with American tegumentary leishmaniasis. J Infect Dis. 2007;195:1846–51.
Mondal D, Singh SP, Kumar N, Joshi A, Sundar S, Das P, et al. Visceral leishmaniasis elimination programme in India, Bangladesh, and Nepal: reshaping the case finding/case management strategy. PLoS Negl Trop Dis. 2009;3:e355.
Fernandez-Figueroa EA, Sanchez-Montes S, Miranda-Ortiz H, Mendoza-Vargas A, Cervantes-Sarabia R, Cardenas-Ovando RA, et al. Relevance of epidemiological surveillance in travelers: an imported case of Leishmania tropica in Mexico. Rev Inst Med Trop Sao Paulo. 2020;62:e41.
Zijlstra EE, Alvar J. The post-kala-azar dermal leishmaniasis (PKDL) atlas: a manual for health workers. World Health Organization. 2012.
World Health Organization. Post-kala-azar dermal leishmaniasis: a manual for case management and control: report of a WHO consultative meeting. World Health Organization. 2012.
Ganguly S, Saha P, Chatterjee M, Roy S, Ghosh TK, Guha SK, et al. PKDL–a silent parasite pool for transmission of leishmaniasis in kala-azar endemic areas of Malda District, West Bengal, India. PLoS Negl Trop Dis. 2015;9:e0004138.
Hashiguchi Y, Gomez EL, Kato H, Martini LR, Velez LN, Uezato H. Diffuse and disseminated cutaneous leishmaniasis: clinical cases experienced in Ecuador and a brief review. Trop Med Health. 2016;44:2.
Pan American Health Organization. Manual of procedures for leishmaniases surveillance and control in the Americas. Pan American Health Organization. 2019.
Gitari JW, Nzou SM, Wamunyokoli F, Kinyeru E, Fujii Y, Kaneko S, et al. Leishmaniasis recidivans by Leishmania tropica in Central Rift Valley Region in Kenya. Int J Infect Dis. 2018;74:109–16.
Walter Reed Army Institute of Research. Cutaneous leishmaniasis scrapings procedures. Edited by Laboratory LD. Silver Spring, USA: Walter Reed Army Institute of Research; 2022: 1–3.
Centres for Disease Control and Prevention. Diagnosis. https://www.cdc.gov/parasites/leishmaniasis/diagnosis.html. 2022. Accessed 1 July 2022.
Coulborn RM, Gebrehiwot TG, Schneider M, Gerstl S, Adera C, Herrero M, et al. Barriers to access to visceral leishmaniasis diagnosis and care among seasonal mobile workers in western Tigray, northern Ethiopia: a qualitative study. PLoS Negl Trop Dis. 2018;12:e0006778.
van Henten S, Adriaensen W, Fikre H, Akuffo H, Diro E, Hailu A, et al. Cutaneous leishmaniasis due to Leishmania aethiopica. EClinicalMedicine. 2018;6:69–81.
de Paiva-Cavalcanti M, de Morais RC, Pessoa ESR, Trajano-Silva LA, Goncalves-de-Albuquerque Sda C, Tavares Dde H, et al. Leishmaniases diagnosis: an update on the use of immunological and molecular tools. Cell Biosci. 2015;5:31.
Hong A, Zampieri RA, Shaw JJ, Floeter-Winter LM, Laranjeira-Silva MF. One health approach to leishmaniases: understanding the disease dynamics through diagnostic tools. Pathogens. 2020;9:809.
Reithinger R, Dujardin JC. Molecular diagnosis of leishmaniasis: current status and future applications. J Clin Microbiol. 2007;45:21–5.
Sakkas H, Gartzonika C, Levidiotou S. Laboratory diagnosis of human visceral leishmaniasis. J Vector Borne Dis. 2016;53:8–16.
Torres-Guerrero E, Quintanilla-Cedillo M, Ruiz-Esmenjaud J, Arenas R. Leishmaniasis: a review. F1000 Research. 2017;6:1–15.
Ogden G, Melby P. Leishmania. In: MS, editor. Encyclopedia of microbiology, third edn. San Diego: Academic Press; 2009; 663–73.
Barratt JL, Harkness J, Marriott D, Ellis JT, Stark D. Importance of nonenteric protozoan infections in immunocompromised people. Clin Microbiol Rev. 2010;23:795–836.
Barcia J. The Giemsa stain: its history and applications. Int J Surg Pathol. 2007;15:292–6.
Ejazi SA, Ali N. Developments in diagnosis and treatment of visceral leishmaniasis during the last decade and future prospects. Expert Rev Anti Infect Ther. 2016;11:79–98.
Yehia L, Adib-Houreih M, Raslan WF, Kibbi AG, Loya A, Firooz A, et al. Molecular diagnosis of cutaneous leishmaniasis and species identification: analysis of 122 biopsies with varied parasite index. J Cutan Pathol. 2012;39:347–55.
Mukhtar M, Ali SS, Boshara SA, Albertini A, Monnerat S, Bessell P, et al. Sensitive and less invasive confirmatory diagnosis of visceral leishmaniasis in Sudan using loop-mediated isothermal amplification (LAMP). PLoS Negl Trop Dis. 2018;12:e0006264.
Da Silva MRB, Stewart JM, Costa CHN. Sensitivity of bone marrow aspirates in the diagnosis of visceral leishmaniasis. Am J Trop Med Hyg. 2005;72:811–4.
Zijlstra EE, Ali MS, El-Hassan AM, El-Toum IA, Satti M, Ghalib HW, et al. Kala-azar: a comparative study of parasitological methods and the direct agglutination test in diagnosis. Trans R Soc Trop Med Hyg. 1992;86:505–7.
de Goes TC, de Morais RCS, de Melo MG, Rezende AM, Rezende AM, de Paiva-Cavalcanti M. Analysis of the IGS rRNA region and applicability for Leishmania (V.) braziliensis characterization. J Parasitol Res. 2020;2020:8885070.
Abd El-Salam NM, Ayaz S, Ullah R. PCR and microscopic identification of isolated Leishmania tropica from clinical samples of cutaneous leishmaniasis in human population of Kohat region in Khyber Pakhtunkhwa. Biomed Res Int. 2014;2014:861831.
Bensoussan E, Nasereddin A, Jonas F, Schnur LF, Jaffe CL. Comparison of PCR assays for diagnosis of cutaneous leishmaniasis. J Clin Microbiol. 2006;44:1435–9.
Daboul MW. Is the amastigote form of Leishmania the only form found in humans infected with cutaneous leishmaniasis? Lab Med. 2008;39:38–41.
Goto H, Lindoso JA. Current diagnosis and treatment of cutaneous and mucocutaneous leishmaniasis. Expert Rev Anti Infect Ther. 2010;8:419–33.
Rodrigues MM, Verma S, Kumar R, Katara GK, Singh LC, Negi NS, et al. Quantification of parasite load in clinical samples of leishmaniasis patients: IL-10 level correlates with parasite load in visceral leishmaniasis. PLoS ONE. 2010;5:e10107.
Siddig M, Ghalib H, Shillington D, Petersen E, Khidir S. Visceral leishmaniasis in Sudan. Clinical features. Trop Geogr Med. 1990;42:107–12.
Zijlstra E, Siddig Ali M, El-Hassan A, Isam K, El-Tourn A, Satti M, et al. Kala-azar: a comparative study of parasitological methods and the direct agglutination test in diagnosis. Trans R Soc Trop Med Hyg. 1992;86:505–7.
Stark D, van Hal S, Lee R, Marriott D, Harkness J. Leishmaniasis, an emerging imported infection: report of 20 cases from Australia. J Travel Med. 2008;15:351–4.
Zare M, Akbarialiabad H, Parsaei H, Asgari Q, Alinejad A, Bahreini MS, et al. A machine learning-based system for detecting leishmaniasis in microscopic images. BMC Infect Dis. 2022;22:48.
Ridley DS, Ridley MJ. The evolution of the lesion in cutaneous leishmaniasis. Pathology. 1983;141:83–96.
Von Stebut E. Leishmaniasis. J Dtsch Dermatol Ges. 2015;13:191–200
Oetken T, Hiscox B, Orengo I, Rosen T. Cutaneous leishmaniasis mimicking squamous cell carcinoma. Dermatol Online J. 2017;23.
Aronson NE, Joya CA. Cutaneous leishmaniasis: updates in diagnosis and management. Infect Dis Clin North Am. 2019;33:101–17.
Ranawaka R, Abeygunasekara P, Weerakoon H. Correlation of clinical, parasitological and histopathological diagnosis of cutaneous leishmaniasis in an endemic region in Sri Lanka. Ceylon Med J. 2013;57:149–52.
Daneshbod Y, Oryan A, Davarmanesh M, Shirian S, Negahban S, Aledavood A, et al. Clinical, histopathologic, and cytologic diagnosis of mucosal leishmaniasis and literature review. Arch Pathol Lab Med. 2011;135:478–82.
Roiko MS, Schmitt BH, Relich RF, Meyer TL, Zhang S, Davis TE. An unusual presentation of leishmaniasis in a human immunodeficiency virus-positive individual. JMM Case Rep. 2016;3:e005011.
Masia R, Misdraji J. Liver and bile duct infections. In: Kradin R, editor. Diagnostic pathology of infectious disease. Amsterdam: Elsevier; 2018.
Venkataram M, Moosa M, Devi L. Histopathological spectrum in cutaneous leishmaniasis: a study in Oman. Indian J Dermatol Venereol Leprol. 2001;67:294–8.
Hermida MD, de Melo CVB, Lima IDS, Oliveira GGS, Dos-Santos WLC. Histological disorganization of spleen compartments and severe visceral leishmaniasis. Front Cell Infect Microbiol. 2018;8:394.
Sundar S, Rai M. Laboratory diagnosis of visceral leishmaniasis. Clin Diagn Lab Immunol. 2002;9:951–8.
Nasiri V. An overview of the recent findings in the cultivation of Leishmania. Rev Med Microbiol. 2017;28:34–42.
Antinori S, Calattini S, Longhi E, Bestetti G, Piolini R, Magni C, et al. Clinical use of polymerase chain reaction performed on peripheral blood and bone marrow samples for the diagnosis and monitoring of visceral leishmaniasis in HIV-infected and HIV-uninfected patients: a single-center, 8-year experience in Italy and Revi. Clin Infect Dis. 2007;44:1602–10.
Castelli G, Galante A, Lo Verde V, Migliazzo A, Reale S, Lupo T, et al. Evaluation of two modified culture media for Leishmania infantum cultivation versus different culture media. J Parasitol. 2014;100:228–30.
Muniaraj M, Das P. Leishmania donovani promastigotes on “chocolate” agar. Ann Trop Med Parasitol. 2008;102:451–3.
Allahverdiyev A, Bagirova M, Uzun S, Alabaz D, Aksaray N, Kocabas E, et al. The value of a new microculture method for diagnosis of visceral leishmaniasis by using bone marrow and peripheral blood. Am J Trop Med Hyg. 2005;73:276–80.
Aberra L, Abera A, Belay T, Kebede A, Gadisa E, Tasew G. Evaluation of microcapillary culture method for the isolation of Leishmania aethiopica parasites from patients with cutaneous lesions in Ethiopia. Diagn Progn Res. 2019;3:4.
Mäser P, Grether-Bühler Y, Kaminsky R, Brun R. An anti-contamination cocktail for the in vitro isolation and cultivation of parasitic protozoa. Parasitol Res. 2014;88:172–4.
Pratlong F, Balard Y, Lami P, Talignani L, Ravel C, Dereure J, et al. The Montpellier Leishmania Collection, from a laboratory collection to a biological resource center: a 39-year-long story. Biopreserv Biobank. 2016;14:470–9.
de Vries HJ, Reedijk SH, Schallig HD. Cutaneous leishmaniasis: recent developments in diagnosis and management. Am J Clin Dermatol. 2015;16:99–109.
Dias DS, Ribeiro PAF, Salles BCS, Santos TTO, Ramos FF, Lage DP, et al. Serological diagnosis and prognostic of tegumentary and visceral leishmaniasis using a conserved Leishmania hypothetical protein. Parasitol Int. 2018;67:344–50.
Maciel M, Soares MF, Costa SF, Bragato JP, de Freitas JH, Venturin GL, et al. Development of plasmonic ELISA for the detection of anti-Leishmania sp. IgG antibodies. J Immunol Methods. 2019;474:112664.
Srivastava P, Dayama A, Mehrotra S, Sundar S. Diagnosis of visceral leishmaniasis. Trans R Soc Trop Med Hyg. 2011;105:1–6.
Maia Z, Lirio M, Mistro S, Mendes CM, Mehta SR, Badaro R. Comparative study of rK39 Leishmania antigen for serodiagnosis of visceral leishmaniasis: systematic review with meta-analysis. PLoS Negl Trop Dis. 2012;6:e1484.
Abeijon C, Alves F, Monnerat S, Mbui J, Viana AG, Almeida RM, et al. Urine-based antigen detection assay for diagnosis of visceral leishmaniasis using monoclonal antibodies specific for six protein biomarkers of Leishmania infantum/Leishmania donovani. PLoS Negl Trop Dis. 2020;14:e0008246.
Kumar A, Pandey SC, Samant M. A spotlight on the diagnostic methods of a fatal disease visceral leishmaniasis. Parasite Immunol. 2020;42:e12727.
da Silva ED, de Oliveira BC, Pereira AMS, Guedes DL, de Melo Neto OP, Costa CHN, et al. A flow cytometry-based serological assay to detect visceral leishmaniasis in HIV-infected patients. Front Med. 2021;8:553280.
Hossain F, Ghosh P, Khan MAA, Duthie MS, Vallur AC, Picone A, et al. Real-time PCR in detection and quantitation of Leishmania donovani for the diagnosis of visceral leishmaniasis patients and the monitoring of their response to treatment. PLoS ONE. 2017;12:e0185606.
Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Peeling RW, et al. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat Rev Microbiol. 2007;5:873–82.
Ready PD. Epidemiology of visceral leishmaniasis. Clin Epidemiol. 2014;6:147–54.
Oliveira GG, Magalhaes FB, Teixeira MC, Pereira AM, Pinheiro CG, Santos LR, et al. Characterization of novel Leishmania infantum recombinant proteins encoded by genes from five families with distinct capacities for serodiagnosis of canine and human visceral leishmaniasis. Am J Trop Med Hyg. 2011;85:1025–34.
Caballero ZC, Sousa OE, Marques WP, Saez-Alquezar A, Umezawa ES. Evaluation of serological tests to identify Trypanosoma cruzi infection in humans and determine cross-reactivity with Trypanosoma rangeli and Leishmania spp. Clin Vaccine Immunol. 2007;14:1045–9.
Ortalli M, Lorrai D, Gaibani P, Rossini G, Vocale C, Re MC, et al. Serodiagnosis of visceral leishmaniasis in northeastern Italy: evaluation of seven serological tests. Microorganisms. 2020;8:1847.
Sundar S, Singh OP. Molecular diagnosis of visceral leishmaniasis. Mol Diagn Ther. 2018;22:443–57.
Chappuis F, Rijal S, Soto A, Menten J, Boelaert M. A meta-analysis of the diagnostic performance of the direct agglutination test and rK39 dipstick for visceral leishmaniasis. Br Med J. 2006;333:723.
Reimao JQ, Coser EM, Lee MR, Coelho AC. Laboratory diagnosis of cutaneous and visceral leishmaniasis: current and future methods. Microorganisms. 2020;8:1632.
World Health Organization. Visceral leishmaniasis rapid diagnostic test performance. In: Visceral leishmaniasis rapid diagnostic test performance. World Health Organization. 2011
Varani S, Ortalli M, Attard L, Vanino E, Gaibani P, Vocale C, et al. Serological and molecular tools to diagnose visceral leishmaniasis: 2-years’ experience of a single center in northern Italy. PLoS ONE. 2017;12:e0183699.
Carstens-Kass J, Paulini K, Lypaczewski P, Matlashewski G. A review of the leishmanin skin test: a neglected test for a neglected disease. PLoS Negl Trop Dis. 2021;15:e0009531.
Bettaieb J, Toumi A, Ghawar W, Chlif S, Nouira M, Belhaj-Hamida N, et al. A prospective cohort study of cutaneous leishmaniasis due to Leishmania major: dynamics of the leishmanin skin test and its predictive value for protection against infection and disease. PLoS Negl Trop Dis. 2020;14:e0008550.
Skraba CM, de Mello TF, Pedroso RB, Ferreira EC, Demarchi IG, Aristides SM, et al. Evaluation of the reference value for the Montenegro skin test. Rev Soc Bras Med Trop. 2015;48:437–44.
Cardo LJ. Leishmania: risk to the blood supply. Transfusion. 2006;46:1641–5.
Singh OP, Hasker E, Boelaert M, Sacks D, Sundar S. Xenodiagnosis to address key questions in visceral leishmaniasis control and elimination. PLoS Negl Trop Dis. 2020;14:e0008363.
Conter CC, Mota CA, dos Santos BA, de Souza BL, de Souza TM, Navasconi TR, et al. PCR primers designed for New World Leishmania: a systematic review. Exp Parasitol. 2019;207:107773.
Deborggraeve S, Boelaert M, Rijal S, De Doncker S, Dujardin JC, Herdewijn P, et al. Diagnostic accuracy of a new Leishmania PCR for clinical visceral leishmaniasis in Nepal and its role in diagnosis of disease. Trop Med Int Health. 2008;13:1378–83.
Marfurt J, Niederwieser I, Makia N, Beck H, Felger I. Diagnostic genotyping of Old and New World Leishmania species by PCR-RFLP. Diagn Microbiol Infect Dis. 2003;46:115–24.
Galluzzi L, Ceccarelli M, Diotallevi A, Menotta M, Magnani M. Real-time PCR applications for diagnosis of leishmaniasis. Parasit Vectors. 2018;11:273.
Mary C, Faraut F, Lascombe L, Dumon H. Quantification of Leishmania infantum DNA by a real-time PCR assay with high sensitivity. J Clin Microbiol. 2004;42:5249–55.
Gow I, Millar D, Ellis J, Melki J, Stark D. Semi-quantitative, duplexed qPCR assay for the detection of Leishmania spp. using bisulphite conversion technology. Trop Med Infect Dis. 2019;4:135.
Machado de Assis TS, Azeredo-da-Silva AL, Werneck GL, Rabello A. Cost-effectiveness analysis of diagnostic tests for human visceral leishmaniasis in Brazil. Trans R Soc Trop Med Hyg. 2016;110:464–71.
Mohammadi M, Bamorovat M, Fasihi Harandi M, Karimi T, Sharifi I, Aflatoonian M. Comparison of three PCR-based methods for simplicity and cost effectiveness identification of cutaneous leishmaniasis due to Leishmania tropica. Iran J Parasitol. 2017;12:215–23.
Aerts C, Vink M, Pashtoon SJ, Nahzat S, Picado A, Cruz I, et al. Cost effectiveness of new diagnostic tools for cutaneous leishmaniasis in Afghanistan. Appl Health Econ Health Policy. 2019;17:213–30.
Castillo-Rodriguez L, Ovalle-Bracho C, Diaz-Jimenez D, Sanchez-Vanegas G, Muvdi-Arenas S, Castaneda-Orjuela C. Cost-effectiveness analysis of mucosal leishmaniasis diagnosis with PCR-based vs parasitological tests in Colombia. PLoS ONE. 2019;14:e0224351.
Tsokana CN, Athanasiou LV, Valiakos G, Spyrou V, Manolakou K, Billinis C. Molecular diagnosis of leishmaniasis, species identification and phylogenetic analysis. In: Claborn DM, editor. Leishmaniasis- trends in epidemiology diagnosis & treatment. London: In Tech Open. 2014. p. 161–93.
Ortalli M, De Pascali AM, Longo S, Pascarelli N, Porcellini A, Ruggeri D, et al. Asymptomatic Leishmania infantum infection in blood donors living in an endemic area, northeastern Italy. J Infect. 2020;80:116–20.
Hosseinzadeh M, Omidifar N, Lohrasb MH. Use of fine needle aspiration cytology in the diagnosis of cutaneous leishmaniasis: a comparison with the conventional scraping method. Trop Doct. 2012;42:112–3.
Saab M, Hage HE, Charafeddine K, Habib RH, Khalifeh I. Diagnosis of cutaneous leishmaniasis: why punch when you can scrape? Am J Trop Med Hyg. 2015;92:518–22.
Zhang WW, Miranda-Verastegui C, Arevalo J, Ndao M, Ward B, Llanos-Cuentas A, et al. Development of a genetic assay to distinguish between Leishmania viannia species on the basis of isoenzyme differences. Clin Infect Dis. 2006;42:801–9.
Daoui OAKM, Mhaidi I, El Kacem S, Hjiyej Andaloussi L, Akarid K, Lemrani M. The role of sampling by cotton swab in the molecular diagnosis of cutaneous leishmaniasis. Transbound Emerg Dis. 2021;68:2287–94.
Adams ER, Versteeg I, Leeflang MM. Systematic review into diagnostics for post-kala-azar dermal leishmaniasis (PKDL). J Trop Med. 2013;2013:150746.
De Ruiter CM, Van Der Veer C, Leeflang MMG, Deborggraeve S, Lucas C, Adams ER. Molecular tools for diagnosis of visceral leishmaniasis: systematic review and meta-analysis of diagnostic test accuracy. J Clin Microbiol. 2014;52:3147–55.
Eberhardt E, Van den Kerkhof M, Bulte D, Mabille D, Van Bockstal L, Monnerat S, et al. Evaluation of a pan-Leishmania spliced-leader RNA detection method in human blood and experimentally infected Syrian golden hamsters. J Mol Diagn. 2018;20:253–63.
Deborggraeve S, Laurent T, Espinosa D, Van der Auwera G, Mbuchi M, Wasunna M, et al. A simplified and standardized polymerase chain reaction format for the diagnosis of leishmaniasis. J Infect Dis. 2008;198:1565–72.
Saldarriaga OA, Castellanos-Gonzalez A, Porrozzi R, Baldeviano GC, Lescano AG, de Los Santos MB, et al. An innovative field-applicable molecular test to diagnose cutaneous Leishmania viannia spp. Infect PLoS Negl Trop Dis. 2016;10:e0004638.
Suzuki RB, Cabral AD, Tonhosolo R, Marcili A, de Oliveira Campos Camargo Sanches C, Martins LPA, et al. A highly sensitive and specific conventional molecular diagnosis for Leishmania infantum chagasi based on a single copy gene. J Mol Biomark Diagn. 2016;07:1–4.
Beldi N, Mansouri R, Bettaieb J, Yaacoub A, Souguir Omrani H, Saadi Ben Aoun Y, et al. Molecular characterization of Leishmania parasites in Giemsa-stained slides from cases of human cutaneous and visceral leishmaniasis. Eastern Algeria. Vector Borne Zoonotic Dis. 2017;17:416–24.
Gunaratna G, Manamperi A, Bohlken-Fascher S, Wickremasinge R, Gunawardena K, Yapa B, et al. Evaluation of rapid extraction and isothermal amplification techniques for the detection of Leishmania donovani DNA from skin lesions of suspected cases at the point of need in Sri Lanka. Parasit Vectors. 2018;11:665.
da C Gonçalves-de-Albuquerque S, Pessoa-e-Silva R, Trajano-Silva LA, de Morais RC, Brandão-Filho SP, de Paiva-Cavalcanti M. Inclusion of quality controls on leishmaniases molecular tests to increase diagnostic accuracy in research and reference laboratories. Mol Biotechnol. 2015;57:318–24.
Franca AO, Pompilio MA, Pontes E, de Oliveira MP, Pereira LOR, Lima RB, et al. Leishmania infection in blood donors: a new challenge in leishmaniasis transmission? PLoS ONE. 2018;13:e0198199.
Pan American Health Organisation. Leishmaniasis regional health program. In: Communicable diseases and health analysis neglected, tropical, and vector borne diseases panaftosa - Veterinary Public Health; 2017. https://www.paho.org/en/documents/leishmaniasis-regional-program-americas-2010-2017-2017. Accessed 21 Mar 2019.
Basiye FL, Mbuchi M, Magiri C, Kirigi G, Deborggraeve S, Schoone GJ, et al. Sensitivity and specificity of the Leishmania OligoC-TesT and NASBA-oligochromatography for diagnosis of visceral leishmaniasis in Kenya. Trop Med Int Health. 2010;15:806–10.
van der Meide W, Guerra J, Schoone G, Farenhorst M, Coelho L, Faber W, et al. Comparison between quantitative nucleic acid sequence-based amplification, real-time reverse transcriptase PCR, and real-time PCR for quantification of Leishmania parasites. J Clin Microbiol. 2008;46:73–8.
Adams ER, Schoone G, Versteeg I, Gomez M, Diro E, Mori Y, et al. Development and evaluation of a novel loop-mediated isothermal amplification assay for diagnosis of cutaneous and visceral leishmaniasis. J Clin Microbiol. 2018;56:1–8.
Ibarra-Meneses AV, Cruz I, Chicharro C, Sanchez C, Bieler S, Broger T, et al. Evaluation of fluorimetry and direct visualization to interpret results of a loop-mediated isothermal amplification kit to detect Leishmania DNA. Parasit Vectors. 2018;11:250.
Sukphattanaudomchoke C, Siripattanapipong S, Thita T, Leelayoova S, Piyaraj P, Mungthin M, et al. Simplified closed tube loop mediated isothermal amplification (LAMP) assay for visual diagnosis of Leishmania infection. Acta Trop. 2020;212:105651.
Dixit KK, Ramesh V, Gupta R, Negi NS, Singh R, Salotra P. Real-time fluorimetry loop-mediated isothermal amplification for diagnosis of leishmaniasis and as a tool for assessment of cure for post-kala-azar dermal leishmaniasis. Am J Trop Med Hyg. 2021;104:2097–107.
Nzelu CO, Kato H, Peters NC. Loop-mediated isothermal amplification (LAMP): an advanced molecular point-of-care technique for the detection of Leishmania infection. PLoS Negl Trop Dis. 2019;13:e0007698.
Castellanos-Gonzalez A, White AC, Melby P, Travi B. Molecular diagnosis of protozoan parasites by recombinase polymerase amplification. Acta Trop. 2018;182:4–11.
Lin H, Zhao S, Liu YH, Shao L, Ying QJ, Yang K. Development of a fluorescent recombinase-aided isothermal amplification-based nucleic acid assay for detection of Leishmania. Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi. 2021;33:452–6.
Khan MAA, Faisal K, Chowdhury R, Ghosh P, Hossain F, Weidmann M, et al. Development of quantitative rapid isothermal amplification assay for Leishmania donovani. Diagnostics. 2021;11:1963.
Mondal D, Ghosh P, Khan MA, Hossain F, Bohlken-Fascher S, Matlashewski G, et al. Mobile suitcase laboratory for rapid detection of Leishmania donovani using recombinase polymerase amplification assay. Parasit Vectors. 2016;9:281.
Cruz I, Millet A, Carrillo E, Chenik M, Salotra P, Verma S, et al. An approach for interlaboratory comparison of conventional and real-time PCR assays for diagnosis of human leishmaniasis. Exp Parasitol. 2013;134:281–9.
Conter CC, Lonardoni MVC, Aristides SMA, Cardoso RF, Silveira TGV. New primers for the detection of Leishmania species by multiplex polymerase chain reaction. Parasitol Res. 2017;117:501–11.
Rosales-Chilama M, Diaz-Moreno N, Prieto MD, Giraldo-Parra L, Martinez-Valencia AJ, Gomez MA. Comparative assessment of DNA targets and amplification methods for Leishmania (Viannia) detection in human samples. Am J Trop Med Hyg. 2020;102:1323–7.
Antinori S, Calattini S, Piolini R, Longhi E, Bestetti G, Cascio A, et al. Is real-time polymerase chain reaction (PCR) more useful than a conventional PCR for the clinical management of leishmaniasis? Am J Trop Med Hyg. 2009;81:46–51.
Vega-Lopez F. Diagnosis of cutaneous leishmaniasis. Curr Opin Infect Dis. 2003;16:97–101.
Lachaud L, Marchergui-Hammami S, Chabbert E, Dereure J, Dedet JP, Bastien P. Comparison of six PCR methods using peripheral blood for detection of canine visceral leishmaniasis. J Clin Microbiol. 2002;40:210–5.
Hasker E, Singh SP, Malaviya P, Singh RP, Shankar R, Boelaert M, et al. Management of visceral leishmaniasis in rural primary health care services in Bihar. India Trop Med Int Health. 2010;15:55–62.
Sudarshan M, Weirather JL, Wilson ME, Sundar S. Study of parasite kinetics with antileishmanial drugs using real-time quantitative PCR in Indian visceral leishmaniasis. J Antimicrob Chemother. 2011;66:1751–5.
Srivastava P, Prajapati VK, Rai M, Sundar S. Unusual case of resistance to amphotericin B in visceral leishmaniasis in a region in India where leishmaniasis is not endemic. J Clin Microbiol. 2011;49:3088–91.
Ponte-Sucre A, Gamarro F, Dujardin JC, Barrett MP, Lopez-Velez R, Garcia-Hernandez R, et al. Drug resistance and treatment failure in leishmaniasis: a 21st century challenge. PLoS Negl Trop Dis. 2017;11:e0006052.
Khatun M, Alam SMS, Khan AH, Hossain MA, Haq JA, Alam Jilani MS, et al. Novel PCR primers to diagnose visceral leishmaniasis using peripheral blood, spleen or bone marrow aspirates. Asian Pac J Trop Med. 2017;10:753–9.
Srivastava P, Mehrotra S, Tiwary P, Chakravarty J, Sundar S. Diagnosis of Indian visceral leishmaniasis by nucleic acid detection using PCR. PLoS ONE. 2011;6:e19304.
Hu Y. Regulatory concern of polymerase chain reaction (PCR) carryover contamination polymerase chain reaction for biomedical applications. London: Intech; 2016. p. 57–68.
Nateghi Rostami M, Darzi F, Farahmand M, Aghaei M, Parvizi P. Performance of a universal PCR assay to identify different Leishmania species causative of Old World cutaneous leishmaniasis. Parasit Vectors. 2020;13:431.
Pandey N, Siripattanapipong S, Leelayoova S, Manomat J, Mungthin M, Tan-Ariya P, et al. Detection of Leishmania DNA in saliva among patients with HIV/AIDS in Trang Province, southern Thailand. Acta Trop. 2018;185:294–300.
Salam MA, Mondal D, Kabir M, Ekram AR, Haque R. PCR for diagnosis and assessment of cure in kala-azar patients in Bangladesh. Acta Trop. 2010;113:52–5.
Namazi MJ, Dehkordi AB, Haghighi F, Mohammadzadeh M, Zarean M, Hasanabad MH. Molecular detection of Leishmania species in northeast of Iran. Comp Clin Path. 2018;27:729–33.
De Silva NL, De Silva VNH, Deerasinghe ATH, Rathnapala UL, Itoh M, Takagi H, et al. Development of a highly sensitive nested PCR and its application for the diagnosis of cutaneous leishmaniasis in Sri Lanka. Microorganisms. 2022;10:990.
Deepachandi B, Weerasinghe S, Soysa P, Karunaweera N, Siriwardana Y. A highly sensitive modified nested PCR to enhance case detection in leishmaniasis. BMC Infect Dis. 2019;19:623.
Castelli G, Bruno F, Reale S, Catanzaro S, Valenza V, Vitale F. Molecular diagnosis of leishmaniasis: quantification of parasite load by a real-time PCR assay with high sensitivity. Pathogens. 2021;10:865.
Moreira OC, Yadon ZE, Cupolillo E. The applicability of real-time PCR in the diagnostic of cutaneous leishmaniasis and parasite quantification for clinical management: current status and perspectives. Acta Trop. 2018;184:29–37.
Nath-Chowdhury M, Sangaralingam M, Bastien P, Ravel C, Pratlong F, Mendez J, et al. Real-time PCR using FRET technology for Old World cutaneous leishmaniasis species differentiation. Parasit Vectors. 2016;9:255.
Trajano-Silva LAM, Pessoa ESR, Goncalves-de-Albuquerque SDC, Morais RCS, Costa-Oliveira CND, Goes TC, et al. Standardization and evaluation of a duplex real-time quantitative PCR for the detection of Leishmania infantum DNA: a sample quality control approach. Rev Soc Bras Med Trop. 2017;50:350–7.
Mohammadiha A, Mohebali M, Haghighi A, Mahdian R, Abadi AR, Zarei Z, et al. Comparison of real-time PCR and conventional PCR with two DNA targets for detection of Leishmania (Leishmania) infantum infection in human and dog blood samples. Exp Parasitol. 2013;133:89–94.
Leon CM, Munoz M, Hernandez C, Ayala MS, Florez C, Teheran A, et al. Analytical performance of four polymerase chain reaction (PCR) and real time PCR (qPCR) assays for the detection of six Leishmania species DNA in Colombia. Front Microbiol. 2017;8:1907.
Pourmohammadi B, Motazedian M, Hatam G, Kalantari M, Habibi P, Sarkari B. Comparison of three methods for diagnosis of cutaneous leishmaniasis. Iran J Parasitol. 2010;5:1–8.
Eroglu F, Uzun S, Koltas IS. Comparison of clinical samples and methods in chronic cutaneous leishmaniasis. Am J Trop Med Hyg. 2014;91:895–900.
Filgueira CPB, Moreira OC, Cantanhede LM, de Farias HMT, Porrozzi R, Britto C, et al. Comparison and clinical validation of qPCR assays targeting Leishmania 18S rDNA and HSP70 genes in patients with American tegumentary leishmaniasis. PLoS Negl Trop Dis. 2020;14:e0008750.
Diotallevi A, Buffi G, Ceccarelli M, Neitzke-Abreu HC, Gnutzmann LV, da Costa Lima MS, et al. Real-time PCR to differentiate among Leishmania (Viannia) subgenus, Leishmania (Leishmania) infantum and Leishmania (Leishmania) amazonensis: application on Brazilian clinical samples. Acta Trop. 2020;201:105178.
Dantas-Torres F, da SilvaSales KG, GomesdaSilva L, Otranto D, Figueredo LA. Leishmania-FAST15: a rapid, sensitive and low-cost real-time PCR assay for the detection of Leishmania infantum and Leishmania braziliensis kinetoplast DNA in canine blood samples. Mol Cell Probes. 2017;31:65–9.
Sevilha-Santos L, Dos Santos Junior ACM, Medeiros-Silva V, Bergmann JO, da Silva EF, Segato LF, et al. Accuracy of qPCR for quantifying Leishmania kDNA in different skin layers of patients with American tegumentary leishmaniasis. Clin Microbiol Infect. 2019;25:242–7.
Frommer M, McDonald L, Millar D, Collis C, Watt F, Grigg G, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–31.
Siah SP, Merif J, Kaur K, Nair J, Huntington PG, Karagiannis T, et al. Improved detection of gastrointestinal pathogens using generalised sample processing and amplification panels. Pathology. 2014;46:53–9.
Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–10.
Roy M, Sarkar D, Chatterjee M. Quantitative monitoring of experimental and human leishmaniasis employing amastigote-specific genes. Parasitology. 2022;149:1085–93.
Ramirez JD, Herrera G, Muskus C, Mendez C, Duque MC, Butcher R. Development of a digital droplet polymerase chain reaction (ddPCR) assay to detect Leishmania DNA in samples from cutaneous leishmaniasis patients. Int J Infect Dis. 2019;79:1–3.
Satow MM, Yamashiro-Kanashiro EH, Rocha MC, Oyafuso LK, Soler RC, Cotrim PC, et al. Applicability of kDNA-PCR for routine diagnosis of American tegumentary leishmaniasis in a tertiary reference hospital. Rev Inst Med Trop Sao Paulo. 2013;55:393–9.
Rodriguez-Cortes A, Ojeda A, Francino O, Lopez-Fuertes L, Timon M, Alberola J. Leishmania infection: laboratory diagnosing in the absence of a “gold standard.” Am J Trop Med Hyg. 2010;82:251–6.
Singh OP, Hasker E, Sacks D, Boelaert M, Sundar S. Asymptomatic Leishmania infection: a new challenge for Leishmania control. Clin Infect Dis. 2014;58:1424–9.
Izadi S, Mirhendi H, Jalalizand N, Khodadadi H, Mohebali M, Nekoeian S, et al. Molecular epidemiological survey of cutaneous leishmaniasis in two highly endemic metropolises of Iran, application of FTA Cards for DNA extraction from Giemsa-stained slides. Jundishapur J Microbiol. 2016;9:17–21.
Gomes CM, Mazin SC, Raphael E, Cesetti MV, Albergaria G, Bächtold B, et al. Accuracy of mucocutaneous leishmaniasis diagnosis using polymerase chain reaction: systematic literature review and meta-analysis. Mem Inst Oswaldo Cruz. 2015;110:157–65.
Van der Auwera G, Ravel C, Verweij J, Bart A, Schönian G, Felger I. Evaluation of four single-locus markers for Leishmania species discrimination by sequencing. J Clin Microbiol. 2014;52:1098–104.
Brodskyn CI, Kato H, Gomez EA, Seki C, Furumoto H, Martini-Robles L, et al. PCR-RFLP analyses of Leishmania species causing cutaneous and mucocutaneous leishmaniasis revealed distribution of genetically complex strains with hybrid and mito-nuclear discordance in Ecuador. PLoS Negl Trop Dis. 2019;13:e0007403.
Shaw J, Pratlong F, Floeter-Winter L, Ishikawa E, El Baidouri F, Ravel C, et al. Characterization of Leishmania (Leishmania) waltoni n.sp. (Kinetoplastida: Trypanosomatidae), the parasite responsible for diffuse cutaneous leishmaniasis in the Dominican Republic. Am J Trop Med Hyg. 2015;93:552–8.
Van der Auwera G, Bart A, Chicharro C, Cortes S, Davidsson L, Di Muccio T, et al. Comparison of Leishmania typing results obtained from 16 European clinical laboratories in 2014. Euro Surveill. 2016;21:30418.
Rioux JA, Lanotte G, Serres E, Pratlong F, Bastien P, Perieres J. Taxonomy of Leishmania. Use of isoenzymes. Suggestions for a new classification. Ann Parasitol Hum Comp. 1990;65:111–25.
Hide M, Banuls A, Tibayrenc M. Genetic heterogeneity and phylogenetic status of Leishmania (Leishmania) infantum zymodeme MON-1: epidemiological implications. Parasitology. 2001;123:425–32.
Ovalle-Bracho C, Camargo C, Diaz-Toro Y, Parra-Munoz M. Molecular typing of Leishmania (Leishmania) amazonensis and species of the subgenus Viannia associated with cutaneous and mucosal leishmaniasis in Colombia: a concordance study. Biomedica. 2018;38:86–95.
Cupolillo E, Grimaldi G Jr, Momen H. A general classification of New World Leishmania using numerical zymotaxonomy. Am J Trop Med Hyg. 1994;50:296–311.
Herrera G, Hernandez C, Ayala MS, Florez C, Teheran AA, Ramirez JD. Evaluation of a multilocus sequence typing (MLST) scheme for Leishmania (Viannia) braziliensis and Leishmania (Viannia) panamensis in Colombia. Parasit Vectors. 2017;10:236.
Hosseini M, Nateghi Rostami M, Hosseini Doust R, Khamesipour A. Multilocus sequence typing analysis of Leishmania clinical isolates from cutaneous leishmaniasis patients of Iran. Infect Genet Evol. 2020;85:104533.
Lauthier JJ, Ruybal P, Barroso PA, Hashiguchi Y, Marco JD, Korenaga M. Development of a multilocus sequence typing (MLST) scheme for pan-Leishmania. Acta Trop. 2020;201:105189.
Schonian G, Kuhls K, Mauricio IL. Molecular approaches for a better understanding of the epidemiology and population genetics of Leishmania. Parasitology. 2011;138:405–25.
Botilde Y, Laurent T, Quispe Tintaya W, Chicharro C, Cañavate C, Cruz I, et al. Comparison of molecular markers for strain typing of Leishmania infantum. Infect Genet Evol. 2006;6:440–6.
Kuhls K, Keilonat L, Ochsenreither S, Schaar M, Schweynoch C, Presber W, et al. Multilocus microsatellite typing (MLMT) reveals genetically isolated populations between and within the main endemic regions of visceral leishmaniasis. Microb Infect. 2007;9:334–43.
Alam MZ, Haralambous C, Kuhls K, Gouzelou E, Sgouras D, Soteriadou K, et al. The paraphyletic composition of Leishmania donovani zymodeme MON-37 revealed by multilocus microsatellite typing. Microbes Infect. 2009;11:707–15.
Kocher A, Valiére S, Banuls A-L, Murienne J. High-throughput sequencing of kDNA amplicons for the analysis of Leishmania minicircles and identification of Neotropical species. Parasitology. 2017;145:585–94.
Kato H, Caceres AG, Seki C, Silupu Garcia CR, Holguin Mauricci C, Castro Martinez SC, et al. Further insight into the geographic distribution of Leishmania species in Peru by cytochrome b and mannose phosphate isomerase gene analyses. PLoS Negl Trop Dis. 2019;13:e0007496.
Bamorovat M, Sharifi I, Mohammadi MA, Eybpoosh S, Nasibi S, Aflatoonian MR, et al. Leishmania tropica isolates from non-healed and healed patients in Iran: a molecular typing and phylogenetic analysis. Microb Pathog. 2018;116:124–9.
Imai K, Tarumoto N, Amo K, Takahashi M, Sakamoto N, Kosaka A, et al. Non-invasive diagnosis of cutaneous leishmaniasis by the direct boil loop-mediated isothermal amplification method and MinION nanopore sequencing. Parasitol Int. 2018;67:34–7.
Calarco L, Barratt J, Ellis J. Detecting sequence variants in clinically important protozoan parasites. Int J Parasitol. 2020;50:1–18.
Downing T, Imamura H, Decuypere S, Clark TG, Coombs GH, Cotton JA, et al. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011;21:2143–56.
Morrison DA. Next generation systematics. Cambridge: Cambridge University Press; 2016.
Anuntasomboon P, Siripattanapipong S, Unajak S, Choowongkomon K, Burchmore R, Leelayoova S, et al. Comparative draft genomes of Leishmania orientalis isolate PCM2 (formerly named Leishmania siamensis) and Leishmania martiniquensis isolate PCM3 from the southern province of Thailand. Biology. 2022;11:515.
Carvalho KSS, da Silva Junior WJ, da Silveira Regueira Neto M, Silva VC, de Sa Leitao Paiva Junior S, Balbino VQ, et al. Application of next generation sequencing (NGS) for descriptive analysis of 30 genomes of Leishmania infantum isolates in middle-north Brazil. Sci Rep. 2020;10:12321.
Banu SS, Meyer W, Ferreira-Paim K, Wang Q, Kuhls K, Cupolillo E, et al. A novel multilocus sequence typing scheme identifying genetic diversity amongst Leishmania donovani isolates from a genetically homogeneous population in the Indian subcontinent. Int J Parasitol. 2019;49:555–67.
Imamura H, Dujardin JC. A guide to next generation sequence analysis of leishmania genomes. Methods Mol Biol. 2019;1971:69–94.
Maljkovic Berry I, Melendrez MC, Bishop-Lilly KA, Rutvisuttinunt W, Pollett S, Talundzic E, et al. Next generation sequencing and bioinformatics methodologies for infectious disease research and public health: approaches, applications, and considerations for development of laboratory capacity. J Infect Dis. 2019;221:1–16.
Imamura H, Jara M, Monsieurs P, Sanders M, Maes I, Vanaerschot M, et al. Evaluation of whole genome amplification and bioinformatic methods for the characterization of Leishmania genomes at a single cell level. Sci Rep. 2020;10:15043.
Montalvo AM, Fraga J, Monzote L, Montano I, De Doncker S, Dujardin JC, et al. Heat-shock protein 70 PCR-RFLP: a universal simple tool for Leishmania species discrimination in the New and Old World. Parasitology. 2010;137:1159–68.
Espada CR, Ortiz PA, Shaw JJ, Barral AMP, Costa JML, Uliana SRB, et al. Identification of Leishmania (Viannia) species and clinical isolates of Leishmania (Leishmania) amazonensis from Brazil using PCR-RFLP of the heat-shock protein 70 gene reveals some unexpected observations. Diagn Microbiol Infect Dis. 2018;91:312–8.
Victoir K, Dujardin J. How to succeed in parasitic life without sex? Asking Leishmania. Trends Parasitol. 2002;18:81–5.
Ghatee MA, Mirhendi H, Marashifard M, Kanannejad Z, Taylor WR, Sharifi I. Population structure of Leishmania tropica causing anthroponotic cutaneous leishmaniasis in southern Iran by PCR-RFLP of kinetoplastid DNA. Biomed Res Int. 2018;2018:6049198.
Quispe-Tintaya K, Laurent T, Decuypere S, Hide M, Bañuls A, De Doncker S, et al. Fluorogenic assay for molecular typing of the Leishmania donovani complex: taxonomic and clinical applications. J Infect Dis. 2005;192:685–92.
Kaufer A, Ellis J, Stark D. Identification of clinical infections of Leishmania imported into Australia: revising speciation with polymerase chain reaction-RFLP of the kinetoplast maxicircle. Am J Trop Med Hyg. 2019;101:590–601.
Schönian G, Nasereddin A, Dinse N, Schweynoch C, Schallig H, Presber W, et al. PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn Microbiol Infect Dis. 2003;47:349–58.
Roberts T, Barratt J, Sandaradura I, Lee R, Harkness J, Marriott D, et al. Molecular epidemiology of imported cases of leishmaniasis in Australia from 2008 to 2014. PLoS ONE. 2015;10:1–11.
de Morais RC, da Costa Oliveira CN, de Albuquerque C, Mendonca Trajano Silva LA, Pessoa ESR, Alvesda Cruz HL, et al. Real-time PCR for Leishmania species identification: evaluation and comparison with classical techniques. Exp Parasitol. 2016;165:43–50.
Kuang Z, Zhang C, Pang H, Ma Y. A rapid high-resolution melting method for differentiation of Leishmania species targeting lack gene. Acta Trop. 2018;178:103–6.
Schulz A, Mellenthin K, Schonian G, Fleischer B, Drosten C. Detection, differentiation, and quantitation of pathogenic Leishmania organisms by a fluorescence resonance energy transfer-based real-time PCR assay. J Clin Microbiol. 2003;41:1529–35.
Sirekbasan S, Polat E. Real-time PCR using high-resolution melting analysis technology for diagnosis of Leishmania and determination of types of clinical samples. Turk J Med Sci. 2018;48:1358–63.
Ceccarelli M, Diotallevi A, Buffi G, De Santi M, Fernandez-Figueroa EA, Rangel-Escareno C, et al. Differentiation of Leishmania (L.) infantum, Leishmania (L.) amazonensis and Leishmania (L.) mexicana using sequential qPCR assays and high-resolution melt analysis. Microorganisms. 2020;8:818.
Wittwer CT. High-resolution DNA melting analysis: advancements and limitations. Hum Mutat. 2009;30:857–9.
Perinoto Â, Maki R, Colhone M, Santos F, Migliaccio V, Daghastanli K, et al. Biosensors for efficient diagnosis of leishmaniasis: innovations in bioanalytics for a neglected disease. Anal Chem. 2010;82:9763–8.
Jain S, Santana W, Dolabella SS, Santos ALS, Souto EB, Severino P. Are nanobiosensors an improved solution for diagnosis of Leishmania? Pharmaceutics. 2021;13:491.
Gedda MR, Madhukar P, Shukla A, Mudavath SL, Srivastava ON, Singh OP, et al. Nanodiagnostics in leishmaniasis: a new frontiers for early elimination. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2021;13:e1675.
Frezza V, Pinto-Diez C, Fernandez G, Soto M, Martin ME, Garcia-Sacristan A, et al. DNA aptamers targeting Leishmania infantum H3 protein as potential diagnostic tools. Anal Chim Acta. 2020;1107:155–63.
Guerra-Perez N, Ramos E, Garcia-Hernandez M, Pinto C, Soto M, Martin ME, et al. Molecular and functional characterization of ssDNA aptamers that specifically bind Leishmania infantum PABP. PLoS ONE. 2015;10:e0140048.
Sattarahmady N, Movahedpour A, Heli H, Hatam G. Gold nanoparticles-based biosensing of Leishmania major kDNA genome: visual and spectrophotometric detections. Sensors Actuators B: Chem. 2016;235:723–31.
Ferreira E, Lima J, Alves-Balvedi RP, Bonan P, Medeiros E, Goulart L, et al. Leishmania spp. detection using a surface plasmon resonance biosensor. Proceedings. 2017;1(4):536.
Pedro GC, Gorza FDS, da Silva RJ, do Nascimento KTO, Medina-Llamas JC, Chavez-Guajardo AE, et al. A novel nucleic acid fluorescent sensing platform based on nanostructured films of intrinsically conducting polymers. Anal Chim Acta. 2019;1047:214–24.
Nazari-Vanani R, Heli H, Sattarahmady N. An impedimetric genosensor for Leishmania infantum based on electrodeposited cadmium sulfide nanosheets. Talanta. 2020;217:121080.
de Freitas Borges P, Fiel W, Vasconcellos V, Dorledo de Faria R. Current progresses in the development of biosensors for the diagnosis of neglected tropical diseases. Syst Biosci Eng. 2020;1:41–52.
Jarockyte G, Karabanovas V, Rotomskis R, Mobasheri A. Multiplexed nanobiosensors: current trends in early diagnostics. Sensors. 2020;20:6893.
Quesada-Gonzalez D, Merkoci A. Nanomaterial-based devices for point-of-care diagnostic applications. Chem Soc Rev. 2018;47:4697–709.
Martins BR, Barbosa YO, Andrade CMR, Pereira LQ, Simao GF, de Oliveira CJ, et al. Development of an electrochemical immunosensor for specific detection of visceral leishmaniasis using gold-modified screen-printed carbon electrodes. Biosensors. 2020;10:81.
World Health Organization. Second WHO model list of essential in vitro Diagnostics. World Health Organization. 2019
Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, Cano J, et al. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE. 2012;7:e35671.
Reithinger R, Dujardin J-C, Louzir H, Pirmez C, Alexander B, Brooker S. Cutaneous leishmaniasis. Lancet Infect Dis. 2007;7:581–96.
Elmahallawy E, Sampedro Martinez A, Rodriguez-Granger J, Hoyos-Mallecot Y, Agil A, Navarro Mari J, et al. Diagnosis of leishmaniasis. J Infect Dev Ctries. 2014;8:961–72.
Adaui Sicheri V. Molecular epidemiological approach to the understanding of the emergence and spreading of drug resistance in Neotropical Leishmania. Dissertation. Antwerp: Universiteit Antwerpen; 2011.
Mock DJ, Hollenbaugh JA, Daddacha W, Overstreet MG, Lazarski CA, Fowell DJ, et al. Leishmania induces survival, proliferation and elevated cellular dNTP levels in human monocytes promoting acceleration of HIV co-infection. PLoS Pathog. 2012;8:e1002635.
Burza S, Croft SL, Boelaert M. Leishmaniasis. Lancet. 2018;392:951–70.
Escobar MA, Martinez F, Scott Smith D, Palma GI. American cutaneous and mucocutaneous leishmaniasis (tegumentary): a diagnostic challenge. Trop Doct. 1992;22:69–78.
Pavli A, Maltezou HC. Leishmaniasis, an emerging infection in travelers. Int J Infect Dis. 2010;14:e1032–9.
de Paulo LF, Rocha GF, Luisi CM Jr, Rosa RR, Durighetto AF Jr. Mucocutaneous leishmaniasis: mucosal manifestations in an endemic country. Int J Infect Dis. 2013;17:e1088–9.
Yanik M, Gurel M, Simsek Z, Kati M. The psychological impact of cutaneous leishmaniasis. Clin Exp Dermatol. 2004;29:464–7.
Morrison B, Mendoza I, Delgado D, Reyes Jaimes O, Aranzazu N, Paniz Mondolfi AE. Diffuse (anergic) cutaneous leishmaniasis responding to amphotericin B. Clin Exp Dermatol. 2010;35:e116–9.
Alcover MM, Rocamora V, Guillen MC, Berenguer D, Cuadrado M, Riera C, et al. Case report: diffuse cutaneous leishmaniasis by Leishmania infantum in a patient undergoing immunosuppressive therapy: risk status in an endemic Mediterranean area. Am J Trop Med Hyg. 2018;98:1313–6.
Turetz M, Machado P, Ko A, Alves F, Bittencourt A, Almeida R, et al. Disseminated leishmaniasis: a new and emerging form of leishmaniasis observed in northeastern Brazil. J Infect Dis. 2002;186:1829–34.
Dassoni F, Daba F, Naafs B, Morrone A. Leishmaniasis recidivans in Ethiopia: cutaneous and mucocutaneous features. J Infect Dev Ctries. 2017;11:106–10.
Asfaram S, Fakhar M, Mohebali M, Mardani A, Banimostafavi ES, Ziaei Hezarjaribi H, et al. Asymptomatic human blood donors carriers of Leishmania infantum: potential reservoirs for visceral leishmaniasis in northwestern Iran. Transfus Apher Sci. 2017;56:474–9.
Stark D, Pett S, Marriott D, Harkness J. Post-kala-azar dermal leishmaniasis due to Leishmania infantum in a human immunodeficiency virus type 1-infected patient. J Clin Microbiol. 2006;44:1178–80.
Steverding D. The history of leishmaniasis. Parasit Vectors. 2017;10:82.
Garrido-Jareno M, Sahuquillo-Torralba A, Chouman-Arcas R, Castro-Hernandez I, Molina-Moreno JM, Llavador-Ros M, et al. Cutaneous and mucocutaneous leishmaniasis: experience of a Mediterranean hospital. Parasit Vectors. 2020;13:24.
Aliaga L, Cobo F, Mediavilla JD, Bravo J, Osuna A, Amador JM, et al. Localized mucosal leishmaniasis due to Leishmania (Leishmania) infantum: clinical and microbiologic findings in 31 patients. Medicine. 2003;82:147–58.
Alborzi A, Pouladfar GR, Ghadimi Moghadam A, Attar A, Drakhshan N, Khosravi Maharlooei M, et al. First molecular-based detection of mucocutaneous leishmaniasis caused by Leishmania major in Iran. J Infect Dev Ctries. 2013;7:413–6.
Dixit K, Singh R, Salotra P. Advancement in molecular diagnosis of post-kala-azar dermal leishmaniasis. Indian J Dermatol. 2020;65:465–72.
Mukhopadhyay D, Dalton JE, Kaye PM, Chatterjee M. Post-kala-azar dermal leishmaniasis: an unresolved mystery. Trends Parasitol. 2014;30:65–74.
Castilho TM, Shaw JJ, Lucile M, Floeter-Winter LM. New PCR assay using glucose-6-phosphate dehydrogenase for identification of Leishmania species. J Clin Microbiol. 2003;41:540–6.
Zelazny AM, Fedorko DP, Li L, Neva FA, Fischer SH. Evaluation of 7SL RNA gene sequences for the identification of Leishmania spp. Am J Trop Med Hyg. 2005;72:415–20.
McAvin JS, Swanson KI, Chan AST, Quintana M, Coleman RE. Leishmania detection in sand flies using a field-deployable real-time analytic system. Mil Med. 2012;177:460–6.
Fotouhi-Ardakani R, Ghafari SM, Ready PD, Parvizi P. Developing, modifying, and validating a TaqMan real-time PCR Technique for accurate identification of Leishmania parasites causing most leishmaniasis in Iran. Front Cell Infect Microbiol. 2021;11:731595.
Miranda A, Samudio F, Saldaña A, Castillo J, Brandão A, Calzada JE. The calmodulin intergenic spacer as molecular target for characterization of Leishmania species. Parasit Vectors. 2014;7:35.
Bhatia A, Sanyal R, Paramchuk W, Gedamu L. Isolation, characterization and disruption of the casein kinase II alpha subunit gene of Leishmania chagasi. Mol Biochem Parasitol. 1998;92:195–206.
Weirather JL, Jeronimo SMB, Gautam S, Sundar S, Kang M, Kurtz MA, et al. Serial quantitative PCR assay for detection, species discrimination, and quantification of Leishmania spp. in human samples. J Clin Microbiol. 2011;49:3892–904.
Jamjoom MB, Ashford RW, Bates PA, Chance ML, Kemp SJ, Watts PC, et al. Leishmania donovani is the only cause of visceral leishmaniasis in East Africa; previous descriptions of L. infantum and “L. archibaldi” from this region are a consequence of convergent evolution in the isoenzyme data. Parasitology. 2004;129:399–409.
da Silva RE, Sampaio BM, Tonhosolo R, da Costa APR, da Silva Costa LE, Nieri-Bastos FA, et al. Exploring Leishmania infantum cathepsin as a new molecular marker for phylogenetic relationships and visceral leishmaniasis diagnosis. BMC Infect Dis. 2019;19:895.
Chaouch M, Fathallah-Mili A, Driss M, Lahmadi R, Ayari C, Guizani I, et al. Identification of Tunisian Leishmania spp. by PCR amplification of cysteine proteinase B (cpb) genes and phylogenetic analysis. Acta Trop. 2013;125:357–65.
Rogers MB, Hilley JD, Dickens NJ, Wilkes J, Bates PA, Depledge DP, et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011;21:2129–42.
Wortmann G, Hochberg L, Houng HH, Sweeney C, Zapor M, Aronson N, et al. Rapid identification of Leishmania complexes by a real-time PCR assay. Am J Trop Med Hyg. 2005;73:999–1004.
Marcili A, Speranca MA, da Costa AP, Madeira Mde F, Soares HS, de Sanches O, et al. Phylogenetic relationships of Leishmania species based on trypanosomatid barcode (SSU rDNA) and gGAPDH genes: taxonomic revision of Leishmania (L.) infantum chagasi in South America. Infect Genet Evol. 2014;25:44–51.
Garcia L, Kindt A, Bermudez H, Llanos-Cuentas A, De Doncker S, Arevalo J, et al. Culture-independent species typing of neotropical Leishmania for clinical validation of a PCR-based assay targeting heat shock protein 70 genes. J Clin Microbiol. 2004;42:2294–7.
Baharia RK, Tandon R, Sahasrabuddhe AA, Sundar S, Dube A. Nucleosomal histone proteins of L. donovani: a combination of recombinant H2A, H2B, H3 and H4 proteins were highly immunogenic and offered optimum prophylactic efficacy against Leishmania challenge in hamsters. PLoS ONE. 2014;9:e97911.
Zackay A, Nasereddin A, Takele Y, Tadesse D, Hailu W, Hurissa Z, et al. Polymorphism in the HASPB repeat region of East African Leishmania donovani strains. PLoS Negl Trop Dis. 2013;7:e2031.
de Almeida ME, Koru O, Steurer F, Herwaldt BL, da Silva AJ. Detection and differentiation of Leishmania spp. in clinical specimens by use of a SYBR green-based real-Time PCR assay. J Clin Microbiol. 2017;55:281–90.
De Almeida ME, Steurer FJ, Koru O, Herwaldt BL, Pieniazek NJ, Da Silva AJ. Identification of Leishmania spp. by molecular amplification and DNA sequencing analysis of a fragment of rRNA internal transcribed spacer 2. J Clin Microbiol. 2011;49:3143–9.
Zhang RG, Zhang J, Jing BQ. Virulence-associated gene profiling of different Leishmania spp. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi. 2009;27:307–11.
Aguiar BG, Coelho DL, Costa DL, Drumond BP, Coelho LF, Figueiredo LC, et al. Genes that encode NAGT, MIF1 and MIF2 are not virulence factors for kala-azar caused by Leishmania infantum. Rev Soc Bras Med Trop. 2014;47:593–8.
Tupperwar N, Vineeth V, Rath S, Vaidya T. Development of a real-time polymerase chain reaction assay for the quantification of Leishmania species and the monitoring of systemic distribution of the pathogen. Diagn Microbiol Infect Dis. 2008;61:23–30.
Zauli-Nascimento RC, Miguel DC, Yokoyama-Yasunaka JK, Pereira LI, Pelli de Oliveira MA, Ribeiro-Dias F, et al. In vitro sensitivity of Leishmania (Viannia) braziliensis and Leishmania (Leishmania) amazonensis Brazilian isolates to meglumine antimoniate and amphotericin B. Trop Med Int Health. 2010;15:68–76.
Marfurt J, Nasereddin A, Niederwieser I, Jaffe CL, Beck HP, Felger I. Identification and differentiation of Leishmania species in clinical samples by PCR amplification of the miniexon sequence and subsequent restriction fragment length polymorphism analysis. J Clin Microbiol. 2003;41:3147–53.
Waki K, Dutta S, Ray D, Kolli BK, Akman L, Kawazu S-I, et al. Transmembrane molecules for phylogenetic analyses of pathogenic protists: Leishmania-specific informative sites in hydrophilic loops of trans-endoplasmic reticulum N-acetylglucosamine-1-phosphate transferase. Eukaryot Cell. 2007;6:198–210.
Hadighi R, Mohebali M, Boucher P, Hajjaran H, Khamesipour A, Ouellette M. Unresponsiveness to Glucantime treatment in Iranian cutaneous leishmaniasis due to drug-resistant Leishmania tropica parasites. PLoS Med. 2006;3:e162.
Hide M, Banuls AL. Species-specific PCR assay for L. infantum/L. donovani discrimination. Acta Trop. 2006;100:241–5.
Vallur AC, Duthie MS, Reinhart C, Tutterrow Y, Hamano S, Bhaskar KR, et al. Biomarkers for intracellular pathogens: establishing tools as vaccine and therapeutic endpoints for visceral leishmaniasis. Clin Microbiol Infect. 2014;20:O374–83.
Eslami G, Salehi R. Genetic variation in RPOIILS gene encoding RNA polymerase II largest subunit from Leishmania major. Mol Biol Rep. 2014;41:2585–9.
Giraud E, Martin O, Yakob L, Rogers M. Quantifying Leishmania metacyclic promastigotes from individual sandfly bites reveals the efficiency of vector transmission. Commun Biol. 2019;2:84.
Chiurillo MA, Sachdeva M, Dole VS, Yepes Y, Miliani E, Vazquez L, et al. Detection of Leishmania causing visceral leishmaniasis in the Old and New Worlds by a polymerase chain reaction assay based on telomeric sequences. Am J Trop Med Hyg. 2001;65:573–82.
Khosravi S, Hejazi S, Hashemzadeh M, Eslami G, Darani H. Molecular diagnosis of Old World leishmaniasis: real-time PCR based on tryparedoxin peroxidase gene for the detection and identification of Leishmania spp. J Vector Borne Dis. 2012;49:15–8.
Hitakarun A, Tan-ariya P, Siripattanapipong S, Mungthin M, Piyaraj P, Naaglor T, et al. Comparison of PCR methods for detection of Leishmania siamensis infection. Parasit Vectors. 2014;7:458.
Haouas N, Garrab S, Gorcii M, Khorchani H, Chargui N, Ravel C, et al. Development of a polymerase chain reaction-restriction fragment length polymorphism assay for Leishmania major/Leishmania killicki/Leishmania infantum discrimination from clinical samples, application in a Tunisian focus. Diagn Microbiol Infect Dis. 2010;68:152–8.
Romano A, Inbar E, Debrabant A, Charmoy M, Lawyer P, Ribeiro-Gomes F, et al. Cross-species genetic exchange between visceral and cutaneous strains of Leishmania in the sand fly vector. Proc Natl Acad Sci USA. 2014;111:16808–13.
Ibrahim ME, Barker DC. The origin and evolution of the Leishmania donovani complex as inferred from a mitochondrialcytochrome oxidase II gene sequence. Infect Genet Evol. 2001;1:61–8.
Yang BB, Chen DL, Chen JP, Liao L, Hu X, Xu JN. Analysis of kinetoplast cytochrome b gene of 16 Leishmania isolates from different foci of China: different species of Leishmania in China and their phylogenetic inference. Parasit Vectors. 2013;6:32.
Flegontov PN, Strelkova MV, Kolesnikov AA. The Leishmania major maxicircle divergent region is variable in different isolates and cell types. Mol Biochem Parasitol. 2006;146:173–9.
Zarean M, Maraghi SH, Hajjaran H, Mohebali M, Feiz-Haddad MH, Assarehzadegan MA. Correlation between clinical responses with the drug-susceptibility of parasites in Iranian cutaneous leishmaniasis caused by Leishmania major. Trop Biomed. 2016;34:338–45.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
IG was responsible for the conception and design of the review, and drafted and substantively revised the review. NCS, DS and JE substantively revised the review. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gow, I., Smith, N.C., Stark, D. et al. Laboratory diagnostics for human Leishmania infections: a polymerase chain reaction-focussed review of detection and identification methods. Parasites Vectors 15, 412 (2022). https://doi.org/10.1186/s13071-022-05524-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05524-z