
Abstract
Background
Bartonella are intracellular bacteria that are transmitted via animal scratches, bites and hematophagous arthropods. Rodents and their associated fleas play a key role in the maintenance of Bartonella worldwide, with > 22 species identified in rodent hosts. No studies have addressed the occurrence and diversity of Bartonella species and vectors for small mammals in Arctic and Subarctic ecosystems, which are increasingly impacted by invasive species and climate change.
Methods
In this study, we characterized the diversity of rodent fleas using conventional PCR targeting the mitochondrial cytochrome c oxidase II gene (COII) and Bartonella species in rodents and shrews (n = 505) from northern Canada using conventional PCR targeting the ITS (intergenic transcribed spacer) region and gltA (citrate synthase) gene. Metagenomic sequencing of a portion of the gltA gene was completed on a subset of 42 rodents and four rodent flea pools.
Results
Year, total summer precipitation the year prior to sampling, average minimum spring temperature and small mammal species were significant factors in predicting Bartonella positivity. Occurrence based on the ITS region was more than double that of the gltA gene and was 34% (n = 349) in northern red-backed voles, 35% (n = 20) in meadow voles, 37% (n = 68) in deer mice and 31% (n = 59) in shrews. Six species of Bartonella were identified with the ITS region, including B. grahamii, B. elizabethae, B. washoensis, Candidatus B. rudakovii, B. doshiae, B. vinsonii subsp. berkhoffii and subsp. arupensis. In addition, 47% (n = 49/105) of ITS amplicons had < 97% identity to sequences in GenBank, possibly due to a limited reference library or previously unreported species. An additional Bartonella species (B. heixiaziensis) was detected during metagenomic sequencing of the gltA gene in 6/11 rodents that had ITS sequences with < 97% identity in GenBank, highlighting that a limited reference library for the ITS marker likely accounted for low sequence similarity in our specimens. In addition, one flea pool from a northern red-backed vole contained multiple species (B. grahamii and B. heixiaziensis).
Conclusion
Our study calls attention to the usefulness of a combined approach to determine the occurrence and diversity of Bartonella communities in hosts and vectors.
Graphical Abstract

Similar content being viewed by others


Background
The genus Bartonella consists of a diverse group of emerging zoonotic bacteria that infect a range of hosts, including mammals, reptiles and birds [1,2,3]. These intracellular pathogens occupy endothelial cells and erythrocytes, leading to immune evasion and chronic asymptomatic bacteremia [4]. Bartonella infections frequently relapse following antibiotic treatment, in part due to the niches occupied outside of the blood. Several species of Bartonella are known to cause disease in humans and other mammals, resulting in endocarditis [5, 6], myocarditis [7, 8], splenomegaly [9] and neurological disease [10]. However, most experience mild symptoms including fever and headache [11]. The development of molecular diagnostic methods has resulted in a steady increase in the number of documented cases of bartonellosis and has drastically expanded the group of known reservoirs, vectors and Bartonella species [12].
Rodents are arguably the most important reservoirs for Bartonella, with > 98 species documented with infections [13]. There are at least 22 species of rodent-associated Bartonella [14], and several have been implicated in human infections, including B. elizabethae, B. tribocorum, B. grahamii, B. rochalimae, B. vinsonii and B. washoensis [13, 14]. Rodents can be co-infected with multiple species, suggesting that recombination events account for some of the diversity of rodent-associated Bartonella [15, 16]. Transmission occurs via blood-feeding arthropods or through the inoculation of bacteria during animal bites and scratches [17, 18]. Among the ectoparasites found on rodents, fleas are crucial vectors for Bartonella transmission. Under experimental conditions, rodent fleas (Xenopsylla ramesis) have been shown to acquire and transmit Bartonella spp., which suggests that rodent fleas may be competent vectors in the wild [19]. The microbiome of Bartonella-positive rodent fleas is also dominated by Bartonella lineages, suggesting that they may out-compete other community members [20]. Bartonella DNA has been detected in other rodent ectoparasites, including lice, ticks and mites [21, 22]; however, their vector competency is poorly understood.
Many complications can arise when trying to characterize the diversity of Bartonella within hosts and vectors. Co-infections are not uncommon, concentration of DNA for each Bartonella species can vary among tissues, recombination events can be observed in a single gene, and annealing affinity can cause primer sets to exhibit amplification bias towards species [12, 14, 23]. The citrate synthase gene (gltA) is the most frequently used marker for Bartonella spp. identification and has the largest reference library and best discriminatory power for Bartonella identification [12, 24]. However, some primers for the gltA gene can exhibit cross reactivity to host DNA (such as Rattus and Mus) and other bacterial species, reducing its analytical specificity [12, 25]. Alternatively, the intergenic transcribed spacer (ITS) region has the highest detection frequency for Bartonella DNA in blood and tissues [12]. There are limitations for this target, since primers may lack specificity and the locus is prone to hypervariability, containing insertions and deletions that complicate alignment and phylogenetic analysis [12, 26, 27]. In this study, we compare both targets to investigate the detection and discriminatory power for Bartonella spp. identification. As both conventional PCR approaches typically detect the most abundant Bartonella species present in samples, we also used a metagenomic approach with the gltA gene to identify all Bartonella sequence diversity within rodent hosts and vectors [16].
Despite the expansive published literature on rodent-associated Bartonella, no studies to our knowledge have investigated the occurrence and diversity of these pathogens in small mammals from Arctic and Subarctic ecosystems. Recently, B. vinsonii and B. henselae were reported in fleas from goose nests and Arctic foxes on the mainland of Nunavut, revealing new hosts, potential vectors and a complex web of transmission involving migratory geese and associated ectoparasites in a tundra ecosystem [28]. Thus, we determined the occurrence and diversity of endemic Bartonella spp. in small mammals collected from the Northwest Territories (NT) and Nunavut (NU) (Canada) and molecularly and morphologically identified the range of potential rodent flea vectors in northern ecosystems.
Methods
Sample collection
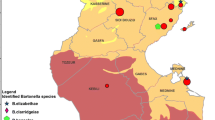
Snap-trapped rodents were collected during the summers of 2017, 2018 and 2019 using line transects placed near the communities of Yellowknife, Fort Liard, Fort Simpson, Fort Smith, Fort Resolution and Inuvik (NT, Canada) (Fig. 1). Similarly, line transects were placed near Cambridge Bay (NU, Canada) and Salluit (QC, Canada). All rodents were frozen at − 20 °C and submitted to the Zoonotic Parasite Research Unit at the Western College of Veterinary Medicine (Saskatoon, Saskatchewan, Canada). During necropsy, each rodent was morphologically identified as a northern collared lemming (Dicrostonyx groenlandicus), Ungava collared lemming (Dicrostonyx hudsonius), northern red-backed vole (Myodes rutilus), meadow vole (Microtus pennsylvanicus), deer mouse (Peromyscus maniculatus) or shrew. Shrews were not identified to species. Thawed carcasses were sexed and weighed and biometric measurements were collected, including length of the right hind foot, body length without the tail and body length with the tail. Any fleas found on the carcasses were pooled for each animal in 1.5-ml Eppendorf tubes containing 1 ml of 100% ethanol. The liver, lung, spleen and kidney of each animal were collected and placed in a single 1.5-ml Eppendorf tube. Organs were stored at − 20 °C until DNA extraction. No fleas were found on northern collared lemmings during our snap-trapping efforts (sample size was small); however, fleas were collected from four Ungava collared lemmings snap-trapped in Salluit. Organs from Ungava collared lemmings were not included in the study, as specimens were used for museum preparations. A flowchart outlining the methods used on small mammals is provided in Fig. 2.

A Location of traplines in the Northwest Territories, Canada. B Composition of small mammal communities collected from each location. C Occurrence of Bartonella infections in small mammals from each location based on positive ITS PCR results. D Species of Bartonella detected in each location based on ITS sequences

Flowchart of study methods. A Small mammals were necropsied, and liver, lung, spleen and kidney were collected. B Approximately 10 mg of each tissue was pooled, and DNA was extracted. C Samples were tested via a conventional PCR targeting ITS and gltA markers. D Bartonella species were identified via ITS amplicons and Sanger sequencing. E A subset of samples [including those that failed during Sanger sequencing, those that were unidentified (< 97% identity) and those that were identified (≥ 97% identity)] were selected for the modified gltA PCR. F Samples that successfully amplified underwent deep sequencing via MiSeq, and amplicon sequence variants were identified. G Fleas were removed from small mammals and morphologically and molecularly identified. Fleas included in the modified gltA PCR originated from northern red-backed voles collected from Nunavut. Created with BioRender.com
Flea identification
All fleas were identified using morphological features as per Holland [29]. For molecular identification, fleas of the same species from the same rodent were pooled together. Genomic DNA was extracted from these pools using the DNeasy Blood & Tissue Kit (Qiagen Inc; Hilden, Germany). Conventional PCR targeting ~ 615 bp of the mitochondrial cytochrome c oxidase II gene (COII) was conducted using the primers COII-2a (5’ATA GAK CWT CYC CHT TAA TAG AAC A 3’) and COII-9b (5’ GTA CTT GCT TTC AGT CAT CTW ATG 3’) [30]. A 25-µl reaction mixture was used containing 10× PCR buffer (200 mM Tris–HCl pH 8.4, 500 mM KCl; Invitrogen), 2.5 µl 50 mM MgCl2, 0.5 µl dNTPs (Invitrogen), 0.25 µl each primer, 0.1 µl Taq DNA Polymerase (Invitrogen) and 5 µl of genomic DNA. PCRs were conducted using the following conditions: 96 °C for 5 min followed by 35 cycles of 94° for 30 s, 55 °C for 30 s, 72 °C for 30 s and a final extension of 72 °C for 5 min. Results were visualized by agarose gel electrophoresis, and amplicons produced were then purified using the QIAquick PCR Purification Kit (Qiagen Inc.) and sequenced (Sanger sequencing; Macrogen; Seoul, South Korea).
Identifying Bartonella DNA in small mammal tissues
DNA was extracted from pooled organs (liver, lung, spleen and kidney) from each individual animals using the DNeasy Blood & Tissue Kit (Qiagen Inc.). Each pooled sample was tested via conventional PCRs targeting the 16S-23S rRNA ITS region and the gltA gene. Positive amplification controls were gBlocks™ gene fragments (Integrated DNA Technologies; Coralville, IA, USA) designed from the complete genome of Bartonella quintana (BX897700.1). The primers used to amplify ~ 767 bp of the gltA gene were CS443f (5’ GCT ATG TCT GCA TTC TAT CA 3’) and CS1210r (5’ GAT CYT CAA TCA TTT CTT TCC A 3’) [31]. The gltA PCR was conducted using the following conditions: 94 °C for 2 min followed by 45 cycles of 94 °C for 30 s, 48 °C for 1 min, 72 °C for 1 min and a final extension of 72 °C for 7 min. The primers used to amplify 450–720 bp of the ITS region (size is species dependent) were 325 s (5′ CTT CAG ATG ATG ATC CCA AGC CTT CTG GCG 3′) and 1100as (5′ GAA CCG ACG ACC CCC TGC TTG CAA AGC A 3′) [32]. The ITS PCR was conducted using the following conditions: 94 °C for 5 min followed by 35 cycles of 94 °C for 1 min, 66 °C for 1 min, 72 °C for 1 min and a final extension of 72 °C for 10 min. For both PCRs, a 25-µl reaction mixture was used containing 2.5 µl 10× PCR buffer (200 mM Tris–HCl, pH 8.4, 500 mM KCl; Invitrogen), 1.25 µl 50 mM MgCl2, 1 µl 2 mM dNTPs (Invitrogen), 1 µl each primer (10 µM), 0.1 µl Taq DNA Polymerase (Invitrogen) and 2.5 µl genomic DNA. ITS amplicons obtained from PCR-positive samples were purified using the QIAquick PCR Purification Kit (Qiagen Inc.) and sequenced (Macrogen). No sequencing of gltA amplicons was performed. For ITS sequences to be identified as a species of Bartonella, there had to be ≥ 97% identity to sequences reported in GenBank [33].
Metagenomic sequencing of gltA amplicons
A subset of 60 rodents and shrews was selected for deep sequencing of gltA amplicons (Fig. 2) based on whether they successfully amplified via conventional gltA PCR. Animals were included that were ITS PCR positive, but where Sanger sequencing of the PCR product failed, those that generated ITS sequences with < 97% identity to sequences available in GenBank and those that yielded ITS sequences that were ≥ 97% to Bartonella species (providing a comparison of those that had been clearly identified with the ITS region and those that were not). Four flea pools (Amalaraeus dissimilis) collected from northern red-backed voles in Nunavut were also selected for deep sequencing as they had previously been tested with the ITS and gltA PCRs. Amplification of a 487-bp fragment from the gltA gene was performed with primers described in Norman et al. [34] modified with the addition of Illumina adaptors: gltA-MiseqF (5’ TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG GGG ACC AGC TCA TGG TGG 3’) and gltA-MiseqR (5’ GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GAA TGC AAA AAG AAC AGT AAA CA 3’). A 487-bp gltA amplicon was generated with 2 μl genomic DNA in 50-µl reactions containing 2.5 U Platinum Taq DNA polymerase (Invitrogen,), 2.5 mM MgCl2, 50 mM KCl, 10 mM Tris/HCl pH 8.3, 250 μM each of dNTPs and 10 pmol each of primers gltA F and gltA R. Reactions were incubated at 95 °C for 3 min followed by 40 cycles of (10 s at 94 °C, 10 s at 57 °C and 30 s at 72 °C) and a final extension of 5 min at 72 °C. Amplicons were purified using 40 μl NucleoMag beads (TaKara Bio USA), and index PCR was performed according to Illumina 16S metagenomic protocol Part # 15,044,223 Rev. B with a slight modification in index PCR clean-up step by using a magnetic bead volume to 40 μl for each sample. Libraries were quantified using the Qubit ds BR kit (Invitrogen), normalized to 4 nM, pooled and loaded as an 8-pmol library with 25% phiX (Illumina) using a Miseq V2 500 cycle kit (Illumina) in a 2 × 250 sequencing run.
Amplification primer sequences were removed from demultiplexed FASTQ data using cutadapt [35], and reads were subsequently trimmed for quality with Trimmomatic [36] using a quality score of 30 and a minimum length of 100. Paired reads were merged, denoised and dereplicated using DADA2 [37] within QIIME2 (version qiime2-2019.10) using a truncation length of 200 to allow assembly of complete gltA amplicon sequences (~ 338 bp without primer sequences). The resulting amplicon sequence variants (ASV) and .biom files from DADA2 were exported for further analysis. The .biom files were converted to feature tables containing read counts for each unique variant detected in each sample. Within a sample, the number of reads attributed to each sequence variant was converted into a proportion of the total reads, and a specific sequence variant was only considered to be present if it accounted for ≥ 1% of the reads within a sample. Species affiliation of the ASV sequences was initially determined based on alignment of ASV to the NCBI GenBank non-redundant nucleotide database and further resolved by phylogenetic analysis of ASV sequences and gltA sequences selected from GenBank. Multiple sequence alignments were conducted with Muscle, and bootstrapped trees were generated using the neighbor-joining method in Mega (version 10.1.8).
Statistical analyses
Occurrence and 95% confidence intervals (CI) were calculated using EpiTools epidemiological calculators [38]. Possible associations between predictor variables (species, sex, biometric measurements, presence/absence of fleas, weight, sampling region, year) and the outcome variable (Bartonella positivity) were evaluated using multiple linear regression. Similarly, associations between Bartonella positivity and climatic factors for both the concurrent year and the year prior to sampling were assessed via multiple linear regression (year itself was not included as a variable in these models). Climate factors included annual and seasonal values [spring (January–May); summer (June–August)] for average maximum and minimum temperature (°C) and total precipitation (mm). Data were collected from Yellowknife A Weather Station (62°27′47.000" N, 114°26′25.000" W) and obtained from the Government of Canada (climate.weather.gc.ca), as climate data from all locations in the study were not available. Models for three outcome variables were tested, including Bartonella positivity with (i) ITS region, (ii) gltA gene and (iii) positive result with both target sequences. Analyses were conducted in SPSS (version 28; IMB Corp. 2021).
Results
Small mammal collection
A total of 446 rodents and 59 shrews was collected, including 349 northern red-backed voles, 20 meadow voles, 68 deer mice and 9 northern collared lemmings. Most of the small mammals originated from the NT, including 82 from Yellowknife, 113 from Fort Smith, 99 from Fort Liard, 115 from Fort Resolution, 13 from Fort Simpson and 72 from Inuvik (Fig. 1). Small mammals were collected over 3 years in the NT, including the summers of 2017 (n = 45), 2018 (n = 273) and 2019 (n = 187). Northern collared lemmings were collected from Cambridge Bay, NU (n = 9). Fleas were collected from the carcasses of 69 rodents from the NT (16%; CI95 12–19), including 59 northern red-backed voles (17%), 4 meadow voles (20%) and 6 deer mice (9%). Fleas from four Ungava collared lemmings were collected in Salluit (Quebec).
Flea Identification Targeting the COII Gene
Fleas from three families were identified, including Leptopsyllidae, Ceratophyllidae and Hystrichopsyllidae. The fleas collected from small mammals are listed in Table 1. A subset of fleas was mounted on slides and kept as references [J.B. Wallis/R.E. Roughley Museum of Entomology (WRME), Department of Entomology, University of Manitoba]. Molecular identification with the COII gene verified the family and genus of most fleas collected from rodents (Table 1); however, reference data for COII sequences were limited in GenBank and species were most closely matched to species within the same genus that have ranges located further south. Following molecular identification, prepared slides were re-examined and the morphological identities of flea species verified.
Bartonella Detection by PCR Targeting gltA and ITS
The occurrence of Bartonella (ITS PCR positives) in small mammals from the NT was 34% (n = 170; CI95 30–38). Occurrence based on gltA PCR (positives) was 16% (n = 79; CI95 13–19), and only 12% of small mammals were positive on both PCRs (n = 60; 95% CI = 9–15). Results for Bartonella genetic markers for each rodent species are compared in Table 2. Unlike the ITS PCR, conventional PCR targeting the gltA gene did not produce any positives for deer mice or meadow voles. Occurrence in animals (ITS positives) was 22% in 2017 (n = 10/45; 95% CI = 13–36); 26% in 2018 (n = 72/273; 95% CI = 22–32) and 47% in 2019 (n = 88/187; 95% CI = 40–54).
Of the 105 ITS PCR products that yielded high-quality sequence data (ranging from 230 to 606 bp), only 56 (53%) provided a species identification (≥ 97% identity with Bartonella sequences in GenBank, Fig. 2). The most common species of Bartonella identified in both northern red-backed and meadow voles was B. grahamii (Table 3). In deer mice, the most common species detected were B. vinsonii subsp. arupensis and B. grahamii. In shrews, the most common species detected was B. vinsonii subsp. berkhoffii. The occurrence of Bartonella (ITS positives) in small mammals for each location sampled in the NT was 48% for Fort Liard (n = 47/99; 95% CI = 38–57), 21% for Fort Resolution (n = 24/115; 14–29), 62% for Fort Simpson (n = 8/13; 36–82), 31% for Fort Smith (n = 35/113; 23–40), 40% for Inuvik (n = 29/72; 30–52) and 32% for Yellowknife (n = 26/82; 23–42) (Fig. 1).
Factors associated with occurrence of Bartonella
Year (β = 0.2, 95% CI = 0.1–0.3) was significantly associated with Bartonella positivity in small mammals across all three models, with the highest exposure in 2019. No additional biological factors were associated with small mammal exposure in the model that defined Bartonella positivity as a PCR positive result via the ITS region (R2 = 0.06, df = 1, P < 0.001). Rodent species (β = 0.1, 95% CI = 0.03–0.1) became statistically significant alongside year (β = 0.2, 95% CI = 0.1–0.3) in the model that defined Bartonella positivity with the gltA gene (R2 = 0.1, df = 2, P = 0.001). Similarly, when Bartonella positivity was defined as a positive result on PCR assays for both the ITS and gltA markers, rodent species (β = 0.1, 95% CI = 0.02–0.09) and year (β = 0.1, 95% CI = 0.09–0.2) were statistically significant (R2 = 0.09, df = 2, P = 0.005). Across all three models, total summer precipitation for the year prior to sampling and average minimum spring temperature during the year of sampling were statistically significant [ITS (R2 = 0.05, df = 2, P < 0.001), gltA (R2 = 0.1, df = 2, P < 0.001), PCR positive for both (R2 = 0.08, df = 2, P < 0.001)], with colder minimum spring temperatures and higher precipitation during the year prior to sampling linked to more PCR-positive small mammals.
Metagenomic sequencing of the gltA region
Of the 60 small mammal samples and 4 flea pools selected for metagenomic gltA amplicon sequencing, 44 of the small mammals (73%) and all flea samples successfully amplified on the modified gltA PCR. Our two no-template controls yielded 0 and 67 reads. An average of 12,272 raw reads per sample were obtained (median 11,696; range 707 to 27,116). Following quality filtering, two northern red-backed voles were removed from further analysis because of extremely low sequence read counts (< 50 reads) leaving 46 samples (42 rodents, 4 flea pools). Twenty-two unique amplicon sequence variant (ASV) sequences were identified. Four ASV sequences were removed from further analysis since they were determined not to be gltA sequences based on alignment to reference data. The average total ASV count per sample was 5662 (median 5642, range 337 to 13,974).
Seventeen unique ASVs were detected that accounted for at least 1% of the counts in at least one sample. A phylogenetic analysis was performed based on a 309-bp alignment of the gltA ASV sequences and selected reference sequences. Four of the ASVs clustered with B. grahamii, nine with B. heixiaziensis, one with Candidatus B. rudakovii and three with B. vinsonii, all with good bootstrap support (Fig. 3). Four ASVs (NT-heix-7, NT-heix-8, NT-heix-9 and NT-grah-4) were detected only in fleas. ASV sequences resembling B. grahamii were most common, detected in 28 of the 42 rodents and accounting for 100% of sequence reads in each sample. There was 89% agreement for sequences that were identified as B. grahamii on both ITS and modified gltA PCRs (18 rodents via ITS PCR and 16 of the same rodents via modified gltA PCR) (Table 4). ASVs resembling B. heixiaziensis were the second most common variants, detected in 11 of the rodent samples (100% of counts in each sample). There was no agreement between sequences obtained via ITS and modified gltA PCRs for these rodents, likely because of the absence of ITS sequences for B. heixiaziensis in GenBank. There was no agreement between ASVs that resembled B. vinsonii and Candidatus B. rudakovii, despite the availability of ITS sequences in GenBank. Multiple species were not detected in rodents, and only one flea pool contained more than one species (B. grahamii and B. heixiaziensis). Of the 46 samples that underwent deep sequencing, 24 (52%) contained multiple ASVs.

Phylogenetic relationships of gltA sequence variants with previously published reference sequences. Study sequences amplified from small mammals and flea pools are shown in red. Accession numbers for reference sequences are indicated. The tree was constructed from a 309-bp alignment using the neighbor-joining method. The tree is drawn to scale, with branch lengths indicating differences per sequence according to the scale bar. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown next to the branches
Sequence data have been deposited to the NCBI Sequence Read Archive and are associated with BioProject PRJNA813524.
Discussion
This study supports the diversity of fleas on northern small mammals and identifies a complex community of rodent-associated Bartonella present in northern Canada, demonstrating the utility of conventional and metagenomic approaches. Ten distinct flea species from rodents were identified morphologically. Molecular identification with the COII gene verified the genus of most flea species (n = 9/10) and highlights the need for a larger reference library for northern vectors (Table 1). Our morphological identifications are consistent with the known geographic range and the time of year that samples were collected [29]. All flea species that were identified infesting small mammals from the NT and NU have been previously recorded on small mammals from Alaska and Yukon [39]. The list of flea species likely contains competent vectors for Bartonella, as rodent fleas can transmit the bacteria under experimental conditions [19, 31, 40]. All rodent flea pools (Amalaraeus dissimilis) that underwent deep sequencing contained Bartonella DNA, though this does not provide definitive proof of vector competence. As seen with B. henselae and cat scratch disease, a source of flea-borne transmission involves the indirect inoculation of infected flea feces, which can remain viable for up to 72 h, into cuts or abrasions on the skin [41,42,43,44]. Given the diversity of Bartonella spp. identified in small mammals and fleas during this study, similar passive mechanisms of transmission may be involved. Bartonella bacteria are dominant members of bacterial communities in several rodent fleas and mixed infections occur in the rat flea, Nosopsyllus fasciatus [16, 20]. This supports the ‘spillover phenomenon,’ where Bartonella transmission is driven by interspecies interactions and exchange of fleas with low host specificity [13]. Though our study did not test individual fleas for mixed infections, our results (deep sequencing) indicate that one flea pool contained two species of Bartonella, revealing that conventional molecular methods underestimate Bartonella diversity in pooled vector samples.
Conventional PCR targeting the ITS region identified six species of Bartonella in rodents and shrews from the NT (Table 3). Rodents are known reservoirs for many of these species and often develop asymptomatic and persistent infections [19]. All Bartonella spp. detected via ITS PCR are zoonotic and have been associated with human cases of disease, except for Candidatus Bartonella rudakovii [11]. In addition, several have been shown to infect domestic dogs, including B. vinsonii subsp. berkhoffii, B. elizabethae and B. washoensis [45, 46]. Cases of bartonellosis (B. vinsonii subsp. berkhoffii, B. henselae and B. rochalimae) have also been identified in Arctic foxes from the neighboring territory of Nunavut, Canada [28, 47]. To our knowledge, there have been no published reports of human bartonellosis in the NT, as it is not a reportable disease.
The occurrence of Bartonella (ITS PCR positives) in small mammals from northern Canada was 34% (Table 2), which is lower than numbers previously reported in wild rodents in the southern Canadian province of Saskatchewan (57%) [48]. Bartonella grahamii was the most common species detected in red-backed voles by our study and Jardine et al. [48]. In our study, small mammal species appeared to influence Bartonella spp. diversity, as B. grahamii was more prevalent in voles while B. vinsonii was more prevalent in deer mice and shrews. This may reflect differences in habitat use, interspecies interactions and variability in host specificity of ectoparasites. Our location data support this hypothesis, as there were significant differences in distribution of small mammals and Bartonella spp. among sampling sites in the NT (Fig. 1). Additionally, multiple ASVs for the gltA gene were only detected in one geographic location (for example NT-heix-3 and NT-heix-6 in Fort Resolution or NT-heix-2 and NT-heix-5 in Inuvik; Table 4), indicating that shared environments may influence Bartonella diversity. As the climate continues to warm in northern Canada, the species that make up these small mammal communities, along with their associated Bartonella spp., may change with shifts in habitat use and the movement of southern competitors further north [49, 50].
Average minimum temperature during spring (°C) and total summer precipitation during the year prior to rodent sampling (mm) were associated with Bartonella positivity in small mammals across all three models. Occurrence was higher during years with lower spring temperatures and years that followed summers with higher precipitation. These results must be interpreted with caution, as climate data were collected from one weather station (Yellowknife), which may not accurately represent other sampling sites. Regardless, climate change is likely to impact the survival of both ectothermic insects and small mammals [51, 52]. Rodent-associated Bartonella has previously been found to be density dependent [53], and climate factors known to influence rodent survival were associated with Bartonella positivity in our study [54]. There was greater precipitation (total mm) in Yellowknife during the summer of 2018 compared to other sampling years (> 100 mm more than 2017 and 2019), which was associated with increased Bartonella positivity in small mammals during 2019. Rainfall at critical times could cause flooding of rodent habitats and burrows, increasing interactions due to limited habitat as rodents search for higher ground [55, 56]. Northern red-backed voles made up the largest percentage of our specimens (69%), and previous studies have proposed that high summer precipitation leads to a greater abundance of late summer and fall berry crops (a key food source for voles) [57]. In turn, this may lead to good overwinter survival and benefit summer reproduction the following year [58, 59]. Consequently, high precipitation during the summer of 2018 may have promoted overwintering survival of northern red-backed voles (and potentially other small mammals) and caused higher population density during 2019, thus increasing the probability of transmission of ectoparasites and Bartonella. Lower spring temperatures may also lead to high rodent density as delayed snowmelt may provide longer protection in subnivean spaces and promote rodent survival [54, 59].
Fleas are ectothermic and sensitive to changes in temperature and precipitation [60]; however, there are few studies in which the impacts of climate on flea populations in northern ecosystems have been examined. Eggs, larvae and pupae of fleas occur off the host within rodent nests, which can reduce the effects of temperature fluctuations [61]. Presumably, this buffer effect may be even more pronounced in northern ecosystems because of the insulating layer of snow. Soil moisture within rodent burrows created by outside precipitation influences the survival of pre-adult stages of flea [60]. Increased rainfall during the summer of 2018 may have increased survival of immature stages, leading to an increase in the number of Bartonella positive small mammals during the summer of 2019. The presence of fleas on rodent carcasses was not significantly associated with PCR positive animals across all three models (gltA, ITS and both). However, snap-trapped rodents are often collected up to 24 h after death, providing a window of time for ectoparasites to abandon the host, which likely caused the prevalence and abundance of fleas in our study to be underestimated.
Rodent species were not statistically significant in the first model that defined Bartonella positivity as a positive ITS result. However, it was significant in the two models that defined Bartonella positivity as a positive gltA result or a positive result on both conventional PCRs. Interpreting associations between rodent species and Bartonella positivity is complicated in this case as primers targeting some genetic markers may amplify host DNA and sensitivity of the conventional gltA PCR may be lower than that of the ITS PCR (Table 2) [12].
Only 62% (n = 105) of the 170 positive small mammal samples identified on the ITS PCR were successfully sequenced and 47% (n = 49/105) of these sequences had < 97% identity with previously reported sequences in GenBank. While it is possible that the low sequence identities were due to known challenges with ITS sequence alignment or the identification of uncharacterized species, we hypothesized that these were likely due to the limited reference library available for ITS sequences [12, 27]. Ideally, multiple loci are used to identify new Bartonella species [12]. Hence, a subsample of rodents was tested with a modified gltA PCR and metagenomic sequencing. There was high agreement between the ITS and gltA targets for most rodents that were infected with B. grahamii (Table 4). One additional species (B. heixiaziensis) was detected with the metagenomic approach (Fig. 3, Table 4). This species was originally isolated from the blood of a red-backed vole (Myodes rutilus) on Heixiazi Island (on the border of China and Russia) [62]. To our knowledge, this is the first documentation of B. heixiaziensis in North America. Bartonella heixiaziensis was the second most common species detected with deep sequencing, and there were no ITS sequences available in GenBank. Thus, we suggest that the absence of reference sequence data for the ITS region accounts for many of the specimens with low sequence similarity following conventional PCR (Table 4). During this study, two unique sequences for the ITS region were isolated from rodents that contained B. heixiaziensis DNA via deep sequencing of the gltA gene. These sequences are available in GenBank (accession numbers ON226744 and ON226745).
Conclusion
We found a diversity of previously undescribed rodent-associated Bartonella in northern Canada and a complex community of potential flea vectors present on rodents that may play a role in transmission. The majority of Bartonella spp. detected in this study are zoonotic and may contribute to human morbidity in the North (associated with exposure to vectors or reservoir hosts) [11]. In addition, we identified climatic factors associated with Bartonella positivity and addressed previously reported strengths and weaknesses of conventional PCR with genetic loci (gltA and ITS) commonly used for detection and identification of Bartonella spp. A metagenomic approach was used to detect one additional species of Bartonella (B. heixiaziensis) in northern red-backed voles that was previously unreported in North America and demonstrated that northern flea pools can contain multiple bacterial species. Future studies that aim to describe the composition of Bartonella communities in hosts and vectors accurately should use multiple loci to identify species and implement a metagenomic approach to detect the diversity of Bartonella within samples.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the Zenodo repository doi: 10.5281/zenodo.6332993. Sequences for B. heixiaziensis (accession numbers ON226744 and ON226745) and rodent fleas (ON221314—ON221322) are available in GenBank.
References
Breitschwerdt EB, Kordick DL. Bartonella infection in animals: carriership, reservoir potential, pathogenicity, and zoonotic potential for human infection. Clinical Microbiol Rev. 2000;13:428–38.
Valentine KH, Harms CA, Cadenas MB, Birkenheuer AJ, Marr HS, Braun-McNeill J, et al. Bartonella DNA in loggerhead sea turtles. Emerg Infect Dis. 2007;13:949–50.
Mascarelli PE, McQuillan M, Harms CA, Harms RV, Breitschwerdt EB. Bartonella henselae and B. koehlerae DNA in birds. Emerg Infect Dis. 2014;20:490–2.
Harms A, Dehio C. Intruders below the radar: molecular pathogenesis of Bartonella spp. Clin Microbiol Rev. 2012;25:42–78.
Roux V, Eykyn SJ, Wyllie S, Raoult D. Bartonella vinsonii subsp. berkhoffii as an agent of afebrile blood culture-negative endocarditis in a human. J Clin Microbiol. 2000;38:1698–700.
Chomel BB, Mac Donald KA, Kasten RW, Chang C, Wey AC, Foley JE, et al. Aortic valve endocarditis in a dog due to Bartonella clarridgeiae. J Clin Microbiol. 2001;39:3548–54.
Montcriol A, Benard F, Fenollar F, Ribeiri A, Bonnet M, Collart F, et al. Fatal myocarditis-associated Bartonella quintana endocarditis: a case report. J Med Case Reports. 2009;3:7325.
Shelnutt LM, Balakrishnan N, DeVanna J, Batey KL, Breitschwerdt EB. Death of military working dogs due to Bartonella vinsonii subspecies berkhoffii genotype III endocarditis and myocarditis. Mil Med. 2017;182:1864–9.
Henn JB, Liu C, Kasten RW, VanHorn BA, Beckett LA, Kass PH, et al. Seroprevalence of antibodies against Bartonella species and evaluation of risk factors and clinical signs associated with seropositivity in dogs. Amer J Vet Res. 2005;66:688–94.
Breitschwerdt EB, Maggi RG, Lantos PM, Woods CW, Hegarty BC, Bradley JM. Bartonella vinsonii subsp. berkhoffii and Bartonella henselae bacteremia in a father and daughter with neurological disease. Parasit Vectors. 2010;3:29.
Cheslock MA, Embers ME. Human bartonellosis: an underappreciated public health problem? Trop Med Infect Dis. 2019;4:69.
Kosoy M, McKee C, Albayrak L, Fofanov Y. Genotyping of Bartonella bacteria and their animal hosts: current status and perspectives. Parasitology. 2018;145:543–62.
Gutiérrez R, Krasnov B, Morick D, Gottlieb Y, Khokhlova IS, Harrus S. Bartonella infection in rodents and their flea ectoparasites: an overview. Vector-Borne Zoon Dis. 2015;15:27–39.
Buffet JP, Kosoy M, Vayssier-Taussat M. Natural history of Bartonella-infecting rodents in light of new knowledge on genomics, diversity and evolution. Future Microbiol. 2013;8:1117–28.
Berglund EC, Frank AC, Calteau A, VinnerePettersson O, Granberg F, Eriksson AS, et al. Run-off replication of host-adaptability genes is associated with gene transfer agents in the genome of mouse-infecting Bartonella grahamii. PLoS Genet. 2009;5:e1000546.
Himsworth CG, Byers KA, Fernando C, Speerin L, Lee MJ, Hill JE. When the sum of the parts tells you more than the whole: the advantage of using metagenomics to characterize Bartonella spp. infections in Norway rats (Rattus norvegicus) and their fleas. Front Vet Sci. 2020;7:584724.
Angelakis E, Rolain JM, Raoult D, Brouqui P. Bartonella quintana in head louse nits. FEMS Immunol Med Microb. 2011;62:244–6.
Ellis BA, Rotz LD, Leake JAD, Samalvides F, Bernable J, Ventura G, et al. An outbreak of acute bartonellosis (Oroya fever) in the Urubamba region of Peru, 1998. Am J Trop Med Hyg. 1999;61:344–9.
Morick D, Krasnov BR, Khokhlova IS, Gottlieb Y, Harrus S. Investigation of Bartonella acquisition and transmission in Xenopsylla ramesis fleas (Siphonaptera: Pulicidae). Mol Ecol. 2011;20:2864–70.
Jones RT, McCormick KF, Martin AP. Bacterial communities of Bartonella-positive fleas: diversity and community assembly patterns. Appl Environ Microbiol. 2008;74:1667–70.
Reeves WK, Szumlas DE, Moriarity JR, Loftis AD, Abbassy MM, Helmy IM, et al. Louse-borne bacterial pathogens in lice (Phthiraptera) of rodents and cattle from Egypt. J Parasitol. 2006;92:313–8.
Reis C, Cote M, Le Rhun D, Lecuelle B, Levin ML, Vayssier-Taussat M, et al. Vector competence of the tick Ixodes ricinus for transmission of Bartonella birtlesii. PLoS Negl Trop Dis. 2011;5:e1186.
Paziewska A, Siński E, Harris PD. Recombination, diversity and allele sharing of infectivity proteins between Bartonella species from rodents. Microbial Ecol. 2012;64:525–36.
La Scola B, Zeaiter Z, Khamis A, Raoult D. Gene-sequence-based criteria for species definition in bacteriology: the Bartonella paradigm. Trends Microbiol. 2003;11:318–21.
Colborn JM, Kosoy MY, Motin VL, Telepnev MV, Valbuena G, Myint KS, et al. Improved detection of Bartonella DNA in mammalian hosts and arthropod vectors by real-time PCR using the NADH dehydrogenase gamma subunit (nuoG). J Clin Microbiol. 2010;48:4630–3.
Maggi RG, Breitschwerdt EB. Potential limitations of the 16S–23S rRNA intergenic region for molecular detection of Bartonella species. J Clin Microbiol. 2005;43:1171–6.
Knap N, Duh D, Birtles R, Trilar T, Petrovec M, Avšič-Županc T. Molecular detection of Bartonella species infecting rodents in Slovenia. FEMS Immunol Med Microbiol. 2007;50:45–50.
Buhler KJ, Maggi RG, Gailius J, Galloway TD, Chilton NB, Alisauskas RT, et al. Hopping species and borders: detection of Bartonella spp. in avian nest fleas and arctic foxes from Nunavut. Canada Parasit Vectors. 2020;13:469.
Holland GP. The fleas of Canada, Alaska and Greenland (Siphonaptera). Mem Entomol Soc Can. 1985;130:3–632.
Whiting MF. Mecoptera is paraphyletic: multiple genes and phylogeny of Mecoptera and Siphonaptera. Zool Scr. 2002;31:93–104.
Billeter SA, Gundi VA, Rood MP, Kosoy MY. Molecular detection and identification of Bartonella species in Xenopsylla cheopis fleas (Siphonaptera: Pulicidae) collected from Rattus norvegicus rats in Los Angeles, California. Appl Environ Microbiol. 2011;77:7850–2.
Maggi RG, Raverty SA, Lester SJ, Huff DG, Haulena M, Ford SL, et al. Bartonella henselae in captive and hunter-harvested beluga (Delphinapterus leucas). J Wildl Dis. 2008;44:871–7.
Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol. 2005;71:1501.
Norman AF, Regnery R, Jameson P, Greene C, Krause D. Differentiation of Bartonella-like isolates at the species level by PCR-restriction fragment length polymorphism in the citrate synthase gene. J Clin Microbiol. 1995;1995:1797–803.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–2.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinform. 2014;30:2114–20.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Sergeant ESG. Epitools epidemiological calculators. Ausvet pty ltd. 2019. http://epitools.ausvet.com.au. Accessed 27 Sept 2021.
Haas GE, Wilson N, Osborne TO, Zarnke RL, Johnson L, Wolff JO. Mammal fleas (Siphonaptera) of Alaska and Yukon Territory. Can J Zool. 1989;67:394–405.
Bown KJ, Bennet M, Begon M. Flea-borne Bartonella grahamii and Bartonella taylorii in bank voles. Emerg Infect Dis. 2004;10:684–7.
Chomel BB, Kasten RW, Floyd-Hawkins K, Chi B, Yamamoto K, Roberts-Wilson J, et al. Experimental transmission of Bartonella henselae by the cat flea. J Clin Microbiol. 1996;34:1952–6.
Finkelstein JL, Brown TP, O’Reilly KL, Wedincamp J Jr, Foil LD. Studies on the growth of Bartonella henselae in the cat flea (Siphonaptera: Pulicidae). J Med Entomol. 2002;39:915–9.
Chomel BB, Boulouis HJ, Breitschwerdt EB, Kasten RW, Vayssier-Taussat M, Birtles RJ, et al. Ecological fitness and strategies of adaptation of Bartonella species to their hosts and vectors. Vet Res. 2009;40:29.
Mosbacher ME, Klotz S, Klotz J, Pinnas JL. Bartonella henselae and the potential for arthropod vector-borne transmission. Vector-Borne Zoon Dis. 2011;11:471–7.
Perez C, Maggi RG, Diniz PP, Breitschwerdt EB. Molecular and serological diagnosis of Bartonella infection in 61 dogs from the United States. J Vet Intern Med. 2011;25:805–10.
Alvarez-Fernandez A, Breitschwerdt EB, Solano-Gallego L. Bartonella infections in cats and dogs including zoonotic aspects. Parasit Vectors. 2018;11:624.
Mascarelli PE, Elmore SA, Jenkins EJ, Alisauskas RT, Walsh M, Breitschwerdt EB, et al. Vector-borne pathogens in arctic foxes, Vulpes lagopus, from Canada. Res Vet Sci. 2015;99:58–9.
Jardine C, Appleyard G, Kosoy MY, McColl D, Chirino-Trejo M, Wobeser G, et al. Rodent-associated Bartonella in Saskatchewan. Canada Vector-Borne Zoon Di. 2006;5:402–9.
Morris DW, Dupuch A. Habitat change and the scale of habitat selection: shifting gradients used by coexisting Arctic rodents. Oikos. 2012;121:975–84.
Zhang X, Flato G, Kirchmeier-Young M, Vincent L, Wan H, Wang X, et al. Changes in temperature and precipitation across Canada, Chapter 4. In: Bush E, Lemmen DS, editors., et al., Canada’s changing climate report. Ottawa: Government of Canada; 2019. p. 112–93.
Parkinson AJ, Evengard B, Semenza JC, Ogden N, Børresen ML, Berner J, et al. Climate change and infectious diseases in the Arctic: establishment of a circumpolar working group. Int J Circumpolar Health. 2014;73:1.
Koltz AM, Culler LE. Biting insects in a rapidly changing Arctic. Curr Opin Insect Sci. 2021;47:75–81.
Telfer S, Clough HE, Birtles RJ, Bennett M, Carslake D, Helyar S, et al. Ecological differences and coexistence in a guild of microparasites: Bartonella in wild rodents. Ecology. 2007;88:1841–9.
Reid DG, Krebs CJ, Kenney A. Limitation of collared lemming population-growth at low-densities by predation mortality. Oikos. 1995;73:387–98.
Shelford VE. The abundance of the collared lemming (Dicrostonyx Groenlandicus (TR). VAR. Richardsoni Mer.) in the Churchill Area, 1929 to 1940. Ecology. 1943;24:472–84.
Fauteux D, Gauthier G, Berteaux D. Seasonal demography of a cyclic lemming population in the Canadian Arctic. J Anim Ecol. 2015;84:1412–22.
Krebs CJ, Boonstra R, Cowcill K, Kenney AJ. Climatic determinants of berry crops in the boreal forest of the southwestern Yukon. Botany. 2009;87:401–8.
West SD. Dynamics of colonization and abundance in central Alaskan populations of the northern red-backed vole Clethrionomys rutilus. J Mammal. 1982;63:128–43.
Boonstra R, Krebs CJ. Population dynamics of red-backed voles (Myodes) in North America. Oecologia. 2012;168:601–20.
Eisen RJ, Gage KL. Adaptive strategies of Yersinia pestis to persist during inter-epizootic and epizootic periods. Vet Res. 2009;40:01.
Ben Ari T, Neerinckx S, Gage KL, Kreppel K, Laudisoit A, Leirs H, et al. Plague and climate: scales matter. PLOS Pathog. 2011;7:e1002160.
Li D-M, Hou Y, Song X-P, Fu Y-Q, Li G-C, Li M, et al. High prevalence and genetic heterogeneity of rodent-borne Bartonella species on Heixiazi Island. China Appl Environ Microbiol. 2015;81:7981–92.
Acknowledgements
Authors thank the Canadian Museum of Nature for the donation of lemming fleas from Salluit. We also thank Dr. Ricardo Maggi from the Intracellular Pathogens Research Laboratory at North Carolina State University for cross-examining sequences with low identity in their Bartonella reference database. In addition, we thank Dr. Sarah Parker (Western College of Veterinary Medicine, University of Saskatchewan) for advice regarding statistical analyses.
Funding
NSERC Discovery Grant and Northern Research Supplement (NRS-2018-517969 and RGPIN-2018-04900), Weston Family Foundation, Northern Scientific Training Program, WCVM’s Widlife Health Research Fund, ArcticNet and Polar Knowledge Canada (NST-1718-0012).
Author information
Authors and Affiliations
Contributions
KJB collected fleas and samples from rodents, completed testing and led the writing of the manuscript. CF assisted with the modified gltA PCR and completed Illumina sequencing. JEH prepared the metagenomic sequencing data and created the phylogenetic tree. TG morphologically identified all rodent fleas. SC and HF facilitated permits, organized and collected rodents during snap-trapping efforts in the Northwest Territories. HF performed necropsies and collected samples from select NT rodents. DF contributed fleas from Salluit, Nunavik, and rodents from snap-trapping efforts in Nunavut. EJJ contributed to study design, guided writing and acquired funding. All authors contributed to writing and revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Rodent trapping in Nunavut was approved under Government of Nunavut wildlife permits (2018-013 and 2019-016) and University of Saskatchewan animal research ethics (2011–0030 and 2019–0021). Rodent and shrew trapping in the NT was approved under permit NWTWCC 2021-001 (live-trapping) and NWTWCC 2021-002 (snap-trapping). Rodent trapping in Nunavik was approved by the Ministères des Forêts, de la Faune et des Parcs du Québec (2019-03-04-188-10-S-F), Canadian Museum of Nature animal ethics committee (2018.002.01), and written supports were obtained from the Village of Salluit, Qaqqalik landholding corp., and the LNUK (contact: Putulik Papigatuk).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Buhler, K.J., Fernando, C., Hill, J.E. et al. Combining deep sequencing and conventional molecular approaches reveals broad diversity and distribution of fleas and Bartonella in rodents and shrews from Arctic and Subarctic ecosystems. Parasites Vectors 15, 366 (2022). https://doi.org/10.1186/s13071-022-05446-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05446-w