
Abstract
Background
Infections with Strongyloides stercoralis belong to the most neglected helminth diseases, and research and development (R&D) efforts on novel drugs are inadequate.
Methods
A commercially available library containing 1600 FDA-approved drugs was tested in vitro against Strongyloides ratti larvae (L3) at 100 µM. Hits (activity > 70%) were then evaluated against S. ratti adult worms at 10 µM. Morantel, prasterone, and levamisole were tested in the S. ratti rat model using dosages of 1–100 mg/kg.
Results
Seventy-one of the 1600 compounds tested against S. ratti L3 revealed activity above 70%. Of 64 compounds which progressed into the adult screen, seven compounds achieved death of all worms (benzethonium chloride, cetylpyridinium chloride, Gentian violet, methylbenzethonium chloride, morantel citrate, ivermectin, coumaphos), and another eight compounds had activity > 70%. Excluding topical and toxic compounds, three drugs progressed into in vivo studies. Prasterone lacked activity in vivo, while treatment with 100 mg/kg morantel and levamisole cured all rats. The highest in vivo activity was observed with levamisole, yielding a median effective dose (ED50) of 1.1 mg/kg.
Conclusions
Using a drug repurposing approach, our study identified levamisole as a potential backup drug for strongyloidiasis. Levamisole should be evaluated in exploratory clinical trials.
Graphical Abstract

Similar content being viewed by others

Background
Strongyloidiasis, caused by infections with the soil-transmitted helminth Strongyloides stercoralis and occasionally Strongyloides fuelleborni, is a highly neglected tropical disease (NTD) in tropical and subtropical settings. In 2017, 614 million people were estimated to be infected, with the majority of infections occurring in South-East Asian, African, and Western Pacific regions [1]. Clinical manifestations are manifold, ranging from nonspecific gastrointestinal symptoms to severe health consequences such as hyperinfection syndromes and disseminated strongyloidiasis [2].
Ivermectin is currently the best treatment option for S. stercoralis infections, characterized by a high cure rate and excellent safety profile [3]. Moxidectin, also a macrocyclic lactone belonging to the milbemycin family and recently approved for onchocerciasis, shows a similar promising efficacy and safety profile [3] and would therefore qualify as novel treatment for S. stercoralis infection. Thiabendazole and albendazole are used as backup drugs, but have considerable limitations including lower efficacy and less favorable tolerability compared to ivermectin [4]. Additional alternative drugs are not available and not on the horizon, as there is little research and development (R&D) on drugs or diagnostics in NTDs [5, 6]. This is an alarming situation, as the widespread use of ivermectin bears a risk of the emergence of drug resistance. In the laboratory, it was shown that ivermectin resistance in Strongyloides ratti could be induced by the F4 generation with corresponding upregulation of some ABC isoform genes using subtherapeutic doses in rats [7]. Given the low level of funding for R&D on NTDs [5, 6], alternative low-cost strategies have been explored to increase the pool of anthelmintic drugs, including drug repurposing [8, 9].
The aim of the present study was to evaluate the activity of 1600 Food and Drug Administration (FDA)-approved drugs against S. ratti in an attempt to identify an alternative drug against Strongyloides spp. All compounds were in an initial screen evaluated against the L3 larval stage in vitro. Active compounds were followed up first against adult worms in vitro, and promising compounds were tested in vivo.
Methods
Drugs and media
The FDA Pharmakon compound library was purchased from MicroSource Discovery Systems, Inc. (USA). Compounds were delivered in microplates (10 mM, dissolved in DMSO) and kept at −80 °C until use. For in vivo studies, morantel, levamisole, and prasterone (DHEA) were purchased from Sigma-Aldrich (Buchs, Switzerland).
Medium RPMI 1640 was purchased from Gibco/Thermo Fisher (Waltham, MA USA), and penicillin (100 U/ml), streptomycin (100 µg/ml), and inactivated fetal calf serum (iFCS) were purchased from BioConcept (Allschwil, Switzerland).
Animals and parasites
Three-week-old male Wistar rats (n = 36) were purchased from Janvier (Le Genest-Saint-Isle, France). Rats were kept for 1 week in the animal facility with food and tap water ad libitum (22 °C, 50% humidity, with a 12-h light/dark cycle, 6 a.m. to 6 p.m.) before the start of the experiments.
In vitro studies
Strongyloides ratti third-stage larvae (L3) were obtained from an established in-house life-cycle, as described by Garcia et al. [10]. For the drug assay, 30–40 L3 were placed in each well of a 96-well plate for each compound. Larvae were incubated in 175 µl culture medium with the test drugs at 100 µM in the dark and at room temperature for 72 h. For the readout, first, the total number of L3 per well was determined. Then, 50–80 µl of hot water (≈ 80 °C) was added to each well, and the moving larvae were counted. The proportion of larval death was determined.
Adult worms for the in vitro studies were collected from the rats’ intestines (n = 15 control or life-cycle rats). The intestines were opened longitudinally, washed with phosphate-buffered saline (PBS), and placed in Petri dishes containing PBS with 1% pen/strep (penicillin 100 U/ml–streptomycin 0.1 mg/ml). Plates were kept for 4 h in an incubator (37 °C, 5% CO2) (Innova CO-48, New Brunswick Scientific). Afterwards, the worms were collected, washed, and transferred to Petri dishes with medium RPMI 1640 (supplemented with 1% pen/strep and 5% FCS). The in vitro assays were prepared in duplicates in 24-well plates with 5–8 worms per well. The worms were incubated for 72 h in medium and tested in concentrations of 100 µM and 10 µM, and active compounds were also tested at two lower concentrations of 1 µM and 0.1 µM. Worms incubated in medium containing 0.5% DMSO served as controls. For evaluating the assays using a bright-field inverted microscope (Carl Zeiss Oberkochen, Germany, magnification ×4 and ×10), 200 µl of 80 °C hot water was added to each well to stimulate worm motility. A scale ranging from 0 to 3 was used, where 0 corresponds to dead worms (no motion within 5″) and 3 indicates maximal motility. Half maximal inhibitory concentration (IC50) values were calculated using CalcuSyn version 2.0 (Biosoft, Cambridge, UK).
In vivo studies
Rats were infected orally with 800 S. ratti L3. Eight days after infection, rats were randomly assigned and treated with the three test drugs at 100 mg/kg. In a second experiment, morantel and levamisole were also evaluated at a single oral dose of 10 mg/kg and 1 mg/kg (levamisole only). For the oral administration, the drugs were first dissolved in a mixture of 70% Tween 80 (Sigma-Aldrich, Buchs, Switzerland) and 30% ethanol (Merck, Darmstadt, Germany), corresponding to 10% of the final volume, and then tap water was added under constant agitation using a magnetic stirrer to reach the necessary volume. Four untreated animals served as control in each of the two experiments. Seven days after administration, rats were euthanized in CO2, and the hosting worms were counted following the procedures described for the in vitro studies.
Results and discussion
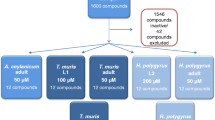
Of the 1600 compounds tested against S. ratti L3, 71 compounds were active (defined as activity > 70%) (hit rate of 4.4%) (Fig. 1, Table 1). A threshold of 70% was applied to define activity; hence, both highly active and moderately active compounds progressed further in our screen.

Screening flow, testing 1600 FDA compounds against S. ratti
The active compounds are from several therapeutic areas, including many anti-infective (and anthelmintic) agents and insecticides (Fig. 1, Table 1). Twenty-one compounds killed all S. ratti larvae after 72 h of incubation at 100 µM.
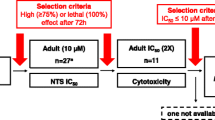
Sixty-four compounds (seven compounds were not available) were tested against adult S. ratti at 10 µM. Seven compounds achieved death of all worms (benzethonium chloride, cetylpyridinium chloride, Gentian violet, methylbenzethonium chloride, morantel citrate, ivermectin, coumaphos), and another eight compounds had activity > 70%. It is interesting to note that pyrantel pamoate was only moderately active (53%) while morantel showed high activity (100%) at 10 µM of the active ingredients. Since several of the active compounds are for topical use only (e.g., Gentian violet or cetrimonium bromide) (see Table 1) or are rather toxic (e.g., coumaphos or sodium nitroprusside), only three of the 14 compounds, namely levamisole, morantel, and prasterone, were selected for IC50 determination and in vivo studies. The activity of ivermectin in in vivo studies was presented in earlier work [11, 12].
Levamisole is an anthelmintic which has been tested in different veterinary formulations in ruminants, with moderate activity against Strongyloides papillosus in cattle [13] and Strongyloides spp. in goats [14]. An injectable formulation of levamisole was effective against Strongyloides venezuelensis in rats. A case report from a human infection documented a positive response [15]. However, a thorough evaluation of levamisole against Strongyloides spp. has not been done in either the laboratory or in human studies. The drug is misused as an adulterant and cutting agent in cocaine distribution [16]. Levamisole-adulterated cocaine use has been reported to cause severe adverse events including neutropenia and agranulocytosis, vasculitis, skin necrosis, and arthralgia, which resulted in withdrawal of levamisole as a human drug [17]. The drug was, however, found safe and well tolerated in the doses used as an anthelmintic in human clinical trials [18] and is still listed on the WHO list of essential list of medicines as anthelmintic (https://list.essentialmeds.org/). Levamisole is also receiving increasing attention as therapy for relapsing nephrotic syndrome [19].
Only a single study was found in the literature using morantel, which is a tetrahydropyrimidine like pyrantel and oxantel, against Strongyloides spp., which evaluated its activity in sheep [20]. Morantel is not used in human medicine. Lastly, prasterone is a naturally occurring androstane steroid used for menopausal symptoms [21].
The calculated IC50 values for levamisole, morantel, and prasterone against adult S. ratti at 72 h were 0.1, > 1, and 3.3 µM, respectively. The activity of levamisole against larval and adult stages was also studied at earlier time points (24 and 48 h), and the findings are presented in Table 2. Activity was clearly visible at the 24-h evaluation time point, in particular at the highest concentrations tested, and both larval and adult stages were highly affected after 48 h of incubation. The calculated IC50 values for levamisole against larval S. ratti were 2.0, 0.2, and 0.1 µM at 24, 48, and 72 h, respectively. IC50 values against adult worms were > 100, 0.4, and 0.1 µM at 24, 48, and 72 h, respectively.
The in vivo results are summarized in Table 3. At 100 mg/kg, morantel and levamisole cured all rats, while prasterone was not active (worm burden reduction of 8%). At a lower dose of 10 mg/kg, worm burden reductions of 45% were obtained for morantel, while levamisole still cured all rats. At 1 mg/kg, levamisole achieved a worm burden reduction of 44%.
Conclusions
The use of a drug repurposing approach, screening a large library of 1600 approved drugs in vitro followed by in vivo studies, enabled the identification of levamisole and morantel as alternative drug candidates for strongyloidiasis. Since morantel is not approved for human use, a long and costly preclinical and clinical development process would be required. A shorter pathway would be foreseen for levamisole. Exploratory clinical trials are necessary to evaluate whether levamisole could serve as a backup drug in case of treatment failures with ivermectin and moxidectin.
Availability of data and materials
The data supporting the conclusions of this article are included within the article. Raw data are available upon request from the corresponding author.
Abbreviations
- DMSO:
-
Dimethyl sulfoxide
References
Buonfrate D, Bisanzio D, Giorli G, Odermatt P, Fürst T, Greenaway C, et al. The global prevalence of Strongyloides stercoralis infection. Pathogens. 2020;9:468.
Vasquez-Rios G, Pineda-Reyes R, Pineda-Reyes J, Marin R, Ruiz EF, Terashima A. Strongyloides stercoralis hyperinfection syndrome: a deeper understanding of a neglected disease. J Parasit Dis. 2019;43:167–75.
Hofmann D, Sayasone S, Sengngam K, Chongvilay B, Hattendorf J, Keiser J. Efficacy and safety of ascending doses of moxidectin against Strongyloides stercoralis infections in adults: a randomised, parallel-group, single-blinded, placebo-controlled, dose-ranging, phase 2a trial. Lancet Infect Dis. 2021;21:1151–60.
Henriquez-Camacho C, Gotuzzo E, Echevarria J, White AC Jr, Terashima A, Samalvides F, et al. Ivermectin versus albendazole or thiabendazole for Strongyloides stercoralis infection. Cochrane Database Syst Rev. 2016;2016:Cd007745.
Benazzouz SM, Malkinson J, Tóth G. Current trends in and future potential of crowdfunding to finance R&D of treatments for neglected tropical diseases. Drug Discov Today. 2021;26:1563–8.
Chapman N, Doubell A, Barnsley P, Goldstein M, Oversteegen L, Chowdhary V, et al. Neglected disease research and development: uneven progress. Policy Cures Res. 2019.
Sengthong C, Yingklang M, Intuyod K, Haonon O, Pinlaor P, Jantawong C, Hongsrichan N, Laha T, Anutrakulchai S, Cha’on U, Sithithaworn P, Pinlaor S. Repeated ivermectin treatment induces ivermectin resistance in Strongyloides ratti by upregulating the expression of ATP-binding cassette transporter genes. Am J Trop Med Hyg. 2021. https://doi.org/10.4269/ajtmh.21-0377.
Panic G, Vargas M, Scandale I, Keiser J. Activity profile of an FDA-approved compound library against Schistosoma mansoni. PLoS Negl Trop Dis. 2015;9(7): e0003962.
Panic G, Duthaler U, Speich B, Keiser J. Repurposing drugs for the treatment and control of helminth infections. Int J Parasitol Drugs Drug Resist. 2014. https://doi.org/10.1016/j.ijpddr.2014.07.002.
Garcia LS, Shimizu RY, Bernard CN. Detection of Giardia lamblia, Entamoeba histolytica/Entamoeba dispar, and Cryptosporidium parvum antigens in human fecal specimens using the triage parasite panel enzyme immunoassay. J Clin Microbiol. 2000;38:3337–40.
Keiser J, Häberli C. Evaluation of commercially available anthelminthics in laboratory models of human intestinal nematode infections. ACS Infect Dis. 2021;7:1177–85.
Keiser J, Thiemann K, Endriss Y, Utzinger J. Strongyloides ratti: in vitro and in vivo activity of tribendimidine. PLoS Negl Trop Dis. 2008;2: e136.
Guerrero J, Seibert BP, Newcomb KM, Michael BF, Garcia-Naranjo F, Rogiers M. Controlled and clinical evaluations of the anthelmintic activity of a levamisole pour-on formulation against gastrointestinal nematodes in cattle. Am J Vet Res. 1984;45:1086–9.
Godara R, Sharma RL, Sodhi SS. Efficacy of fenbendazole, levamisole and ivermectin against gastrointestinal nematodes in Jamunapari goats. J Parasit Dis. 2011;35:219–21.
Onile B, Komolafe F, Oladiran B. Severe strongyloidiasis presenting as occult gastro-intestinal tract malignancy. Ann Trop Med Parasitol. 1985;79:301–4.
George TC, Freet DJ, Cross JM, Huzar TF. Levamisole-induced vasculitis. Jaapa. 2019;32:23–7.
Conrad F, Hirsiger S, Winklhofer S, Baumgartner MR, Stämpfli P, Seifritz E, et al. Use of levamisole-adulterated cocaine is associated with increased load of white matter lesions. J Psychiatry Neurosci. 2021;46:E281–91.
Albonico M, Bickle Q, Ramsan M, Montresor A, Savioli L, Taylor M. Efficacy of mebendazole and levamisole alone or in combination against intestinal nematode infections after repeated targeted mebendazole treatment in Zanzibar. Bull World Health Organ. 2003;81:343–52.
Gruppen MP, Bouts AH, Jansen-van der Weide MC, Merkus MP, Zurowska A, Maternik M, et al. A randomized clinical trial indicates that levamisole increases the time to relapse in children with steroid-sensitive idiopathic nephrotic syndrome. Kidney Int. 2018;93:510–8.
Reinecke RK, Louw JP. Overberg research projects. XVI. First-stage larval reduction test versus the controlled anthelmintic test to assess the efficacy of anthelmintics against nematode parasites of sheep. J S Afr Vet Assoc. 1994;65:108–12.
Drugs for menopausal symptoms. Med Lett Drugs Ther. 2020;62:124–8.
Acknowledgements
We kindly acknowledge the help and expertise from Fadri Christoffel, Kevin Varghese, Yvan Meier and Andreas Gasser.
Funding
We gratefully acknowledge financial support from the Swiss National Science Foundation (no. 320030_175585).
Author information
Authors and Affiliations
Contributions
JK designed the studies. CH conducted and supervised the experiments. JK wrote the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experiments were authorized by the veterinary office from Canton Basel Stadt (authorization no. 2070) based on the Swiss national and cantonal regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Keiser, J., Häberli, C. Assessment of FDA-approved drugs against Strongyloides ratti in vitro and in vivo to identify potentially active drugs against strongyloidiasis. Parasites Vectors 14, 615 (2021). https://doi.org/10.1186/s13071-021-05117-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-05117-2