
Abstract
Background
Panstrongylus megistus is the most important vector of Chagas disease in Brazil. Studies show that the principal factor hindering the control of triatomines is reinfestation of houses previously treated with insecticides. Studies at the microgeographic level are therefore necessary to better understand these events. However, an efficient molecular marker is not yet available for carrying out such analyses in this species. The aim of the present study was to identify and characterize microsatellite loci for future population genetic studies of P. megistus.
Methods
This study work consisted of five stages: (i) sequencing of genomic DNA; (ii) assembly and selection of contigs containing microsatellites; (iii) validation of amplification and evaluation of polymorphic loci; (iv) standardization of the polymorphic loci; and (v) verification of cross-amplification with other triatomine species.
Results
Sequencing of males and females generated 7,908,463 contigs with a total length of 2,043,422,613 bp. A total of 2,043,690 regions with microsatellites in 1,441,091 contigs were obtained, with mononucleotide repeats being the most abundant class. From a panel of 96 loci it was possible to visualize polymorphisms in 64.55% of the loci. Of the 20 loci genotyped, the number of alleles varied from two to nine with an average of 4.9. Cross-amplification with other species of triatomines was observed in 13 of the loci.
Conclusions
Due to the high number of alleles encountered, polymorphism and the capacity to amplify from geographically distant populations, the microsatellites described here show promise for utilization in population genetic studies of P. megistus.
Graphical Abstract

Similar content being viewed by others

Background
Triatomines are hematophagous insect vectors of Trypanosoma cruzi (Chagas, 1909), the etiological agent of Chagas disease, which is considered the principal parasitic disease of Latin America in terms of economic impact and public health [1,2,3].
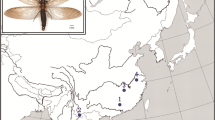
Among the autochthonous triatomines of Brazil, Panstrongylus megistus (Burmeister, 1835) is considered the vector of greatest epidemiological importance of Chagas disease due to its high capacity to invade and colonize domestic buildings, high levels of infection with T. cruzi, eclectic blood-feeding and a wide geographical distribution in Brazil, ranging from the state of Maranhão to the state of Rio Grande do Sul [4,5,6,7]. In recent years, the presence of P. megistus has also been reported in the Brazialn states of Acre and Rondônia [8].
It has become apparent that the principal factor hindering the control of triatomines is reinfestation of houses previously treated with insecticides, possibly due to insects that survive spraying (residual foci) or triatomines from sylvatic foci [9]. Analysis of this problem requires an investigation at the microgeographic level. Microsatellite molecular markers have been used with success to evaluate the gene flow of triatomine populations between natural and artificial environments (intradomestic and peridomestic) [10,11,12,13,14,15,16,17,18,19,20,21,22]. It has been reported that populations of T. infestans originating from three Bolivian Andean locations did not show any difference in preference for insects in the artificial environment and those in the natural environment at the same location, suggesting that the source of infestation in the dwellings is wild insects [15]. De Rosas et al. [12] demonstrated that the level of genetic structure of Argentine T. infestans populations is approximately 400 m, indicating the possibility of passive dispersion within that distance. To reduce the odds of reinfestation, these authors suggested that insecticide treatment should occur on a larger radius around the infested area. In northern Argentina, an evaluation of the genetic structure of T. infestans populations revealed putative sources of reinfestation and its dynamics [22]. These studies illustrate that microsatellites can be useful for understanding the factors that favor the infestation/reinfestation of domiciles.
Given the absence of tools capable of capturing at a fine-scale the process of (re)colonization of P. megistus in the artificial environment, the aim of this work was to isolate and characterize microsatellite loci from P. megistus, using Illumina HiSeq genome sequencing.
Methods
This work was perfomed in five stages: (i) genome sequencing of P. megistus; (ii) assembly and selection of contigs with microsatellites; (iii) validation of the amplification and evaluation of the polymorphisms in the loci; (iv) standardization of the polymorphic loci; and (v) verification of cross-amplification with other triatomines species.
Sequencing, identification of regions with microsatellites and development of the primers
Two pools of five P. megistus were used, one of female individuals and another of males, from a colony derived from the municipality of Santana do Riacho, Minas Gerais, Brazil (19°10′8″S, 43°42′50″W) maintained in the insectary of the Laboratório de Referência em Triatomíneos e Epidemiologia da Doença de Chagas were selected for sequencing. DNA was extracted from the reproductive organs of the insects using the Genomic DNA Extraction and Purification Kit® (Promega, Madison, WI, USA) following the protocol described by the manufacturer. The libraries were constructed using the TruSeq DNA PCR Free 350 bp kit (Macrogen Inc., Seoul, South Korea) according to the manufacturer’s instructions, and the sequencing of each pool was performed separately using the Illumina HiSeq X platform (Illumina, Inc., San Diego, CA, USA). The raw sequence reads were evaluated in terms of read quality with PRINSEQ [23]. Data filtering and trimming (adaptor removal and Phred quality score cut-off ≥ 25) were performed with Trimmomatic [24]. Contigs were assembled de novo with a kmer size of 77 using SOAPdenovo2 [25]. Microsatellite regions were identified with MISA software [26] with the following parameters: minimum of ten repeats of 1 base, six repeats of 2 bases, five repeats of 3 bases, five repeats of 4 bases, five repeats of 5 bases and five repeats with 6 bases each. Microsatellites were selected based on: (i) minimum of six repeats of perfect, di- or trinucleotides; and (ii) identified in contigs with a minimum size of 2 kb. Additionally, Primer3 [27, 28] was used in order to identify primer annealing sites flanking the repeat regions. Only intergenic regions with a predicted amplicon size of between 150 and 400 bp were selected for further analysis.
Standardization of amplification and evaluation of polymorphism in microsatellite regions
Amplification and polymorphism of the loci selected were evaluated in six specimens of P. megistus: (i) two from a colony derived from the municipality of Juquiá, São Paulo, Brazil (24°19′15″S, 47°38′6″W); (ii) two from a colony formed from insects captured in diverse localities in the state of Minas Gerais, Brazil; and (iii) two others from the same colony used for genome sequencing, which came from Santana do Riacho.
The DNA was extracted from two legs of each insect following an adapted protocol of the Wizard® Genomic DNA Purification Kit (Promega) [29]. The DNA was quantified using a NanoDrop One Microvolume UV–Vis spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and stored at − 20 ºC until used.
In order to standardize and optimize the specificity of the PCR amplification, different dilutions of enzymes and cofactors, as well different annealing temperatures (range: 54 °C to 65 °C), were tested. The PCRs were performed in a final volume of 10 µl containing the Master Mix GoTaq Green (Promega), 10 nmol of forward primer, 10 nmol of the reverse primer and approximately 10 ng of template DNA. The reactions were performed on a Veriti thermocycler (Applied Biosystems, Foster City, CA, USA) under the following conditions: an initial denaturation at 95 °C, 4 min; followed by 95 °C/30 s, 54 °C to 65 °C/30 s, 72 °C/30 s for 35 cycles; and a final extension at 72 °C/5 min. The amplicons were run on 8% polyacrylamide gels stained with silver nitrate. The approximate size of the fragments was determined using the molecular marker ΦX 174 DNA HaeIII (Promega).
Characterization of polymorphic microsatellite loci
Among the evaluated loci that were found to be polymorphic under the conditions described in the previous section, 20 were selected for standardization (Table 1) based on an amplicon size of between 100 and 300 bp and annealing temperature, for use in future multiplex PCRs.
Fifteen specimens of P. megistus captured in the municipality of Jaboticatubas, Minas Gerais, Brazil (19°30′50″S, 43°44′42″W) by Belisário et al. [30] were used. These samples were divided into two groups in order to evaluate intra-populational variability: nine insects from the locality Fazenda Santo Antônio (group I); and one insect each from the following localities: Barreiro do Papagaio, Fazenda Espada, Capão Grande II, Fazenda Borges, Guarazinho, and Fazenda Boiça (group II). The second group also included the insects from Santana do Riacho and Juquiá described in section Standardization of amplification and evaluation of polymorphism in microsatellite regions.
Four other species of triatomines were used to evaluate cross-amplification: (i) one specimen of Panstrongylus diasi Pinto & Lent 1946 from the insect collection of the Instituto René Rachou Fiocruz Minas/Belo Horizonte, Brazil; (ii) one specimen of Panstrongylus lignarius (Walker, 1873); (iii) one specimen of Triatoma tibiamaculata (Pinto, 1926); and (iv) one specimen of Triatoma sordida (Stal 1859). These last three insects were derived from the colonies of the Laboratório de Referência em Triatomíneos e Epidemiologia da Doença de Chagas of the Instituto René Rachou, Fiocruz Minas.
The DNA was extracted from two legs from each individual as described in section Standardization of amplification and evaluation of polymorphism in microsatellite regions. The PCRs were performed in a total final volume of 10 µl containing 5× Colorless GoTaq® Flexi (Promega), 3 mM MgCl2, 10 nmol of the fluorescently-labeled forward primer, 10 nmol of the reverse primer and approximately 10 ng of template DNA. The reactions were performed in a Veriti® 96-well thermocycler (Applied Biosystems) using the following cycling conditions: an initial denaturation at 95 °C, 4 min; then 95 °C/30 s, 65 °C/ 30 s, 72 °C/30 s for 35 cycles; 95 °C/30 s, 53 °C/ 30 s, 65 °C/30 s, 72 °C/30 s for 3 cycles; and a final extension of 72 °C for 5 min. The products were diluted in the ratio of 1:10 in UltraPure Distilled Water (Invitrogen, Carlsbad, CA, USA) and genotyped on an ABI 3730 Sequencer (Life Technologies, Carlsbad, CA, USA) using the size standard GeneScan Liz 500 of the sequencing platform of Sequenciamento de DNA por Eletroforese Capilar of the Instituto René Rachou. The chromatograms were analyzed using the software Geneious (ver. 10.1.3) [31]. The number of alleles with observed heterozygosity (OH) and expected heterozygosity (EH) (Arlequin ver. 3.5.2.2) [32] and the presence of null alleles (MICRO-CHEKER ver. 2.2.3) [33] were evaluated.
Results
Sequencing of the P. megistus genome yielded 463,151,518 reads (90.41% > Q30) for the female pool and 338,531,204 reads (89.54% > Q30) for the male pool. The GC-content was 34.53 and 35.06%, respectively. The de novo assembly was made using only reads with a quality > 25, generating a total of 7,908,463 contigs with a total length of 2,043,422,613 bp. The N50 (sequence length of the shortest contig at 50% of the total genome length) of the assembly was 1034 and the final GC-content was 33.14%. We identified a total of 2,043,690 microsatellite regions located in 1,441,091 contigs with mononucleotide repeats being the most abundant (1,054,968, corresponding to 51.62%) and pentanucleotide repeats present at a lower quantity (1337, corresponding to 0.06%). In accordance with the parameters described in the Methods section, 79 different microsatellite regions were selected for further analysis.
Among the 96 primer pairs selected for PCR amplification, 79 resulted in amplicons visualized on polyacrylamide gels after standardization. The annealing temperature after standardization varied from 60 °C to 67 °C (Additional file 1: Table S1). It was possible to visualize the presence of polymorphism on the gel for 64.55% (51) of the loci. Among these 51 loci, 20 markers were selected for characterization (Table 1). It was not possible to standardize the amplification of 17 primer pairs due either to low specificity or the absence of amplicons.
In the samples of P. megistus, the number of alleles per locus varied from two (Pm051, Pm071 and Pm079) to nine (Pm049), with a mean of 4.9 (Table 2). Among the 20 loci genotyped, two (Pm051 in group II and Pm058 in group I) were monomorphic and for one (Pm054) it was not possible to visualize peaks on the chromatogram (Table 3).
The OH of group I varied from 0.00000 (Pm002) to 0.55556 (Pm049) (mean 0.29630), while the EH varied from 0.20915 (Pm071) to 0.77778 (Pm083) (mean 0.57807). In group II, the OH varied from 0.00000 (Pm002) to 0.60000 (Pm049) (mean 0.24504), while the EH varied from 0.10000 (Pm071) to 0.88421 (Pm049) (mean 0.61446) (Table 3).
Of the 20 loci analyzed, group I had null alleles at seven loci (Pm002, Pm008, Pm018, Pm048, Pm051, Pm076 and Pm079) and group II had null alleles at five loci (Pm002, Pm027, Pm048, Pm079 and Pm083).
Regarding cross-amplification, 13 loci were amplified from other species. The Pm030 marker was amplified from all the samples tested. However, the number of alleles was lower than that in P. megistus, varying from one to four, with a mean of 1.5 (Table 2). OH and EH were not calculated because there was only one specimen for each of the species evaluated.
Discussion
This study is novel in two aspects: in the development of primers to microsatellites of P. megistus and in the methodology used. Unlike previously published studies on the identification of microsatellite markers in triatomines, in which the methodology most commonly used is enriched libraries and the subsequent use of probes for the selection of microsatellite regions [34,35,36,37,38,39,40], the present study is the first to use next-generation sequencing. This methodology was chosen due to the limited information available on the genome of P. megistus, the principal species transmitting T. cruzi in Brazil.
For the design of the initial primers, we chose to synthesize only primers to regions flanking dinucleotide and trinucleotide repeats because these latter arrangements have higher mutation rates compared to other microsatellite classes [41]. Therefore, they will be more informative in future population genetic studies.
To make the PCR reactions more specific, we tested different annealing temperatures. The optimal temperatures utilized were those observed prior to the absence of bands in the polyacrylamide gel, as well as prior to the dilution of enzymes and cofactors. Modification of such determinants can alter the success of PCRs [42]. Even after these attempts, it was not possible to standardize the PCR for 17.7% of the 96 loci selected due to the absence of fragments or the presence of nonspecific fragments. In these cases, the primer may have been designed to a non-conserved region or have more than one binding site.
Two loci (Pm051 in group II and Pm058 in group I) exhibited monomorphic patterns and in another locus (Pm054) it was not possible to visualize peaks in the chromatogram. However, working with a larger sample number can increase the chances of observing heterozygous and polymorphic individuals [43]. Accordingly, these three loci will be evaluated in a larger sample set in a future study to verify the absence of polymorphism, since it was possible to visualize amplicons from these loci in polyacrylamide gels.
The quantity of alleles encountered in the 20 loci tested differed from that that encountered in other studies on triatomines: in T. dimidiata, the mean allele number (AN) was reported to be 16, varying from six to 27 alleles at eight loci [35]; in T. infestans, the mean AN was 9.7, varying from five to 17 alleles at 13 loci [36]; in Rhodnius pallescens, the mean observed AN was nine alleles, varying from two to 20 at ten loci analyzed [34]; in T. sordida, the mean AN was 7.4, varying from one to 12 at ten loci [44]; in T. pseudomaculata, the mean AN was 6.6, varying from two to 15 alleles at seven loci [39]; and for T. brasiliensis, the mean observed AN was five at seven standardized loci, ranging from one to 13 alleles per locus [40]. This difference can be explained by the sample size in each study, ranging from 34 [35] to 171 [44] samples, while we used 19 samples of P. megistus.
The presence of four loci in P. diasi and eight in P. lignarius was expected due to the conservation of some genomic regions in phylogenetically related species. This has also been reported in other studies [35, 36, 38,39,40, 44]. However, the present work is the first to report amplification in species from different genera (T. sordida and T. tibiamaculata).
The values observed for OH and EH may be indicative of the presence of excessive homozygotes in the groups tested, population structure or the presence of null alleles [45]. The analysis of null alleles demonstrated low occurrence in both groups (35 and 25% in groups I and I, respectively). The presence of null alleles can occur due to mutations in the flanking sequences of the microsatellite regions, thus preventing binding of the primers. The regions not amplified result in apparently homozygous samples when present in a heterozygous state [46, 47].
In the last 40 years, microsatellites have been the most used molecular marker to access polymorphisms of a wide variety of organisms. One of the difficulties in applying this methodology is the need for prior knowledge of the genome. Currently, this problem can be overcome by using the genotyping-by-sequencing (GBS) as a marker. However, GBS requires a greater amount of DNA, throughput and the cost is higher [48].
Conclusions
Due to the high polymorphism and number of alleles encountered in each locus, as well as the capacity to amplify from geographically distant populations, we conclude that the markers developed in this study show promise for population genetic studies of P. megistus. Thus, we hope to help to elucidate the reinfestation processes in the artificial environment by this vector.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author.
Abbreviations
- AN:
-
Allele number
- EH:
-
Expected heterozygosity
- OH:
-
Observed heterozygosity
References
Rassi A, Dias JC, Marin-Neto JA. Challenges and opportunities for primary, secondary, and tertiary prevention of Chagas’ disease. Heart. 2009;957:524–34.
Dias JC. Facing chagas disease. Rev Soc Bras Med Trop. 2017;50:285–6.
World Health Organization. London conference on neglected tropical diseases. Geneva: World Health Organization. 2017. https://www.unitingtocombatntds.org/. Accessed 8 Jan 2019
Aragão MB. Aspectos climáticos da doença de Chagas. II Área de ocorrência do Panstrongylus megistus (Burmeister, 1835). Rev Bras Malar. 1961;13:171–93.
Carcavallo RU, Galvão C, Lent H. Triatoma jurbergi sp. n. do norte do estado do Mato Grosso, Brasil (Hemiptera, Reduviidae, Triatominae) com uma atualização das sinonímias e outros táxons. Mem Inst Oswaldo Cruz. 1998;93:459–64.
Villela MM, Rodrigues VL, Casanova C, Dias JC. Análise da fonte alimentar de Panstrongylus megistus (Hemiptera, Reduviidae, Triatominae) e sua atual importância como vetor do Trypanosoma cruzi, no Estado de Minas Gerais. Rev Soc Bras Med Trop. 2010;43:125–8.
Gurgel-Gonçalves R, Galvao C, Costa J, Peterson AT. Geographic distribution of Chagas disease vectors in Brazil based on ecological niche modeling. J Trop Med. 2012;2012:1–15.
Castro MA, de Souza Castro GV, de Souza JL, de Souza CR, Ramos LJ, de Oliveira J, et al. First report of Panstrongylus megistus (Hemiptera, Reduviidae, Triatominae) in the State of Acre and Rondônia, Amazon. Brazil Acta Trop. 2018;182:158–60.
Gaspe MS, Gurevitz JM, Gürtler RE, Dujardin JP. Origins of house reinfestation with Triatoma infestans after insecticide spraying in the Argentine Chaco using wing geometric morphometry. Infect Genet Evol. 2013;17:93–100.
Dias FB, de Paula AS, Belisário CJ, Lorenzo MG, Bezerra CM, Harry M, et al. Influence of the palm tree species on the variability of Rhodnius nasutus Stål, 1859 (Hemiptera, Reduviidae, Triatominae). Infec Genet Evol. 2011;11:869–77.
Gómez-Palacio A, Triana O, Jaramillo-O N, Dotson EM, Marcet PL. Eco-geographical differentiation among Colombian populations of the Chagas disease vector Triatoma dimidiata (Hemiptera: Reduviidae). Infect Genet Evol. 2013;20:352–61.
De Rosas AR, Segura EL, Fusco O, Guiñazú AL, García BA. Fine-scale genetic structure in populations of the Chagas’ disease vector Triatoma infestans (Hemiptera, Reduvidae). Genetica. 2013;141:107–17.
Garcia B, de Rosas AR, Blariza M, Grosso C, Fernandez C, Stroppa M. Molecular Population genetics and evolution of the Chagas’ disease vector Triatoma infestans (Hemiptera: Reduviidae). Curr Genomics. 2013;14:316–23.
Foley EA, Khatchikian CE, Hwang J, Ancca Juárez J, Borrini Mayori K, Quıspe Machaca VR, et al. Population structure of the Chagas disease vector, Triatoma infestans, at the urban–rural interface. Mol Ecol. 2013;22:5162–71.
Brenière SF, Salas R, Buitrago R, Brémond P, Sosa V, Bosseno MF, et al. Wild populations of Triatoma infestans are highly connected to intra-peridomestic conspecific populations in the Bolivian Andes. PLoS ONE. 2013;8:e80786.
Piccinali RV, Gürtler RE. Fine-scale genetic structure of Triatoma infestans in the Argentine Chaco. Infect Genet Evol. 2015;34:143–52.
Stevens L, Monroy MC, Rodas AG, Hicks RM, Lucero DE, Lyons LA, et al. Migration and gene flow among domestic populations of the Chagas insect vector Triatoma dimidiata (Hemiptera: Reduviidae) detected by microsatellite loci. J Med Entomol. 2015;52:419–28.
Khatchikian CE, Foley EA, Barbu CM, Hwang J, Ancca-Juárez J, Borrini-Mayori K, et al. Population structure of the Chagas disease vector Triatoma infestans in an urban environment. PLoS Negl Trop Dis. 2015;9:e0003425.
Almeida CE, Faucher L, Lavina M, Costa J, Harry M. Molecular individual-based approach on Triatoma brasiliensis: inferences on triatomine foci, Trypanosoma cruzi natural infection prevalence, parasite diversity and feeding sources. PLoS Negl Trop Dis. 2016;10:e0004447.
Belisário CJ, Pessoa GC, Silva EM, Rosa AC, Ferreira RE, Bedin C, et al. Genetic characterization of residual Triatoma infestans populations from Brazil by microsatellite. Genetica. 2017;145:105–14.
Bezerra CM, Belisário CJ, Pessoa GC, Rosa AC, Barezani CP, Ferreira FC, et al. Microsatellite variation revealed panmictic pattern for Triatoma brasiliensis (Triatominae: Reduviidae) in rural northeastern Brazil: the control measures implications. BMC Genet. 2020;21:1–2.
Marcet PL, Mora MS, Cutrera AP, Jones L, Gürtler RE, Kitron U, et al. Genetic structure of Triatoma infestans populations in rural communities of Santiago del Estero, northern Argentina. Infect Genet Evol. 2008;8:835–46.
Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:2047–117.
Thiel T, Michalek W, Varshney R, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 2003;106:411–22.
Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–91.
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115.
Borges EC, Dujardin JP, Schofield CJ, Romanha AJ, Diotaiuti L. Genetic variability of Triatoma brasiliensis (Hemiptera: Reduviidae) populations. J Med Entomol. 2000;37:872–7.
Belisário CJ, Dias JV, Diotaiuti L. Profile of the Trypanosoma cruzi vector infestation in Jaboticatubas, State of Minas Gerais. Brazil Rev Soc Bras Med Trop. 2013;46:779–82.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–7.
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004;4:535–8.
Harry M, Poyet G, Roman C, Solignac M. Isolation and characterization of microsatellite markers in the bloodsucking bug Rhodnius pallescens (Heteroptera, Reduviidae). Mol Ecol. 1998;7:1784–6.
Anderson JM, Lai JE, Dotson EM, Cordon-Rosales C, Ponce C, Norris DE, et al. Identification and characterization of microsatellite markers in the Chagas disease vector Triatoma dimidiata. Infect Genet Evol. 2002;1:243–8.
Marcet PL, Lehmann T, Groner G, Gürtler RE, Kitron U, Dotson EM. Identification and characterization of microsatellite markers in the Chagas disease vector Triatoma infestans (Heteroptera: Reduviidae). Infect Genet Evol. 2006;6:32–7.
Garcia BA, Zheng L, Rosas AR, Segura EL. Isolation and characterization of polymorphic microsatellite loci in the Chagas’ disease vector Triatoma infestans (Hemiptera: Reduviidae). Mol Ecol Notes. 2004;4:568–71.
Harry M, Roose CL, Vautrin D, Noireau F, Romaña CA, Solignac M. Microsatellite markers from the Chagas disease vector, Rhodnius prolixus (Hemiptera, Reduviidae), and their applicability to Rhodnius species. Infect Genet Evol. 2008;8:381–5.
Harry M, Dupont L, Romana C, Demanche C, Mercier A, Livet A, et al. Microsatellite markers in Triatoma pseudomaculata (Hemiptera, Reduviidae, Triatominae), Chagas’ disease vector in Brazil. Infect Genet Evol. 2008;8:672–5.
Harry M, Dupont L, Quartier M, Diotaiuti L, Walter A, Romana C. New perspectives for population genetics of Chagas’ disease vectors in the Northeastern Brazil: Isolation of polymorphic microsatellite markers in Triatoma brasiliensis. Infect Genet Evol. 2009;9:633–7.
Chakraborty R, Kimmel M, Stivers DN, Davison LJ, Deka R. Relative mutation rates at di-, tri-, and tetranucleotide microsatellite loci. Proc Natl Acad Sci USA. 1997;94:1041–6.
Markoulatos P, Siafakas N, Moncany M. Multiplex polymerase chain reaction: a practical approach. J Clin Lab Anal. 2002;16:47–51.
Machado EL, Silva SA. Desenho e validação de iniciadores microssatélites SSR para mamoneira. Pesqui Agropecu Bras. 2013;48:1457–63.
Belisário CJ, Pessoa GC, dos Santos PF, Dias LS, Rosa AC, Diotaiuti L. Markers for the population genetics studies of Triatoma sordida (Hemiptera: Reduviidae). Parasites Vectors. 2015;8:1–3.
Hartl DL, Clark AG, Clark AG. Principles of population genetics. Sunderland: Sinauer Associates; 1997.
Varshney RK, Graner A, Sorrells ME. Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 2005;23:48–55.
Guichoux E, Lagache L, Wagner S, Chaumeil P, Léger P, Lepais O, et al. Current trends in microsatellite genotyping. Mol Ecol Resour. 2011;11:591–611.
Grover A, Sharma PC. Development and use of molecular markers: past and present. Crit Rev Biotechnol. 2016;36(2):290–302.
Acknowledgements
The authors express their thanks to the technicians of the DNA Sequencing Platform at IRR/Fiocruz Minas and Bioinformatics Platform RPT04B–IRR/Fiocruz Minas.
Funding
This study was financed in by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior–Brasil (CAPES)–Finance Code 001, CNPq (460585/2014-2) and Instituto René Rachou/Fiocruz Minas.
Author information
Authors and Affiliations
Contributions
CJB, LGD and JCR designed the study; CJB and LGD coordinated the research; FCF performed the experiments; CJB, LGD and FCF analyzed the data; LBK sequenced the genome; FSM, LOG and JCR performed the bioinformatics analysis; FCF and CJB were major contributors to the writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Forward (F) and reverse (R) primer sequences, repeat motif, annealing temperature (AT) and presence (+) or absence (−) of polymorphism of the Panstrongylus megistus microsatellite loci.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ferreira, F.C., Gonçalves, L.O., Ruiz, J.C. et al. Identification and characterization of microsatellite markers for population genetic studies of Panstrongylus megistus (Burmeister, 1835) (Triatominae: Reduviidae). Parasites Vectors 14, 273 (2021). https://doi.org/10.1186/s13071-021-04771-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-04771-w