
Abstract
Background
Visceral leishmaniasis (VL) caused by dimorphic Leishmania species is a parasitic disease with high socioeconomic burden in endemic areas worldwide. Sustaining control of VL in terms of proper and prevailing immunity development is a global necessity amid unavailability of a prophylactic vaccine. Screening of experimental proteome of the human disease propagating form of Leishmania donovani (amastigote) can be more pragmatic for in silico mining of novel vaccine candidates.
Methods
By using an immunoinformatic approach, CD4+ and CD8+ T cell-specific epitopes from experimentally reported L. donovani proteins having secretory potential and increased abundance in amastigotes were screened. A chimera linked with a Toll-like receptor 4 (TLR4) peptide adjuvant was constructed and evaluated for physicochemical characteristics, binding interaction with TLR4 in simulated physiological condition and the trend of immune response following hypothetical immunization.
Results
Selected epitopes from physiologically important L. donovani proteins were found mostly conserved in L. infantum, covering theoretically more than 98% of the global population. The multi-epitope chimeric vaccine was predicted as stable, antigenic and non-allergenic. Structural analysis of vaccine-TLR4 receptor docked complex and its molecular dynamics simulation suggest sufficiently stable binding interface along with prospect of non-canonical receptor activation. Simulation dynamics of immune response following hypothetical immunization indicate active and memory B as well as CD4+ T cell generation potential, and likely chance of a more Th1 polarized response.
Conclusions
The methodological approach and results from this study could facilitate more informed screening and selection of candidate antigenic proteins for entry into vaccine production pipeline in future to control human VL.

Similar content being viewed by others


Background
Leishmania spp. are obligate intracellular pathogens of phagocytic host cells. Two species, Leishmania donovani and L. infantum cause visceral leishmaniasis (VL), a neglected tropical disease and second only to malaria in parasitic cause of death. With a chance of case fatality of 100% in an inadequate treatment scenario, over 90% of VL cases occur in relatively poor communities of Bangladesh, India, Sudan, South Sudan, Ethiopia and Brazil [1]. The VL elimination program known as Kala-azar Elimination Programme (KEP) has contributed to a remarkable decline in the incidence of VL over recent years in the Indian subcontinent and now it is approaching the maintenance phase of VL elimination [2]. However, sustained elimination cannot be possible without proper and prevailing immunity development in the endemic population against Leishmania parasites in the post-elimination era due to the chance of reservoir mediated re-emergence of the disease [3]. A vaccination strategy can induce long-term protection with proper immunity in order to prevent development of disease in the most economical way, regardless of its mode of implementation.
In recent years, enormous progress has been made in the design of vaccines against leishmaniasis using live-attenuated or killed parasites, cellular extracts, and individual and/or recombinant antigens of parasites. The first-generation vaccine, which includes live-attenuated, killed and fractionated parasites, is the only class of human prophylactic VL vaccine that entered phase III clinical trials so far. However, this vaccine failed to achieve satisfactory results [4]. The second-generation vaccines are produced from recombinant Leishmania antigens (single peptides/polypeptides). Among several approaches, LEISH-F3, a multicomponent vaccine formulated with GLA-SE adjuvant showed promising results in phase I as a robust immune response inducer in healthy people [5]. Earlier, LEISH-F1 in combination with MPL-SE adjuvant also showed strong antigen-specific immune response in healthy people living in a L. donovani endemic area [6]. More recently, a third-generation DNA vaccine approach that employed simian adenovirus expressing a novel synthetic gene encoding Leishmania antigens, hence termed as ChAd63-KH, has shown potentiality to be a safe and immunogenic therapeutic vaccine for human VL and post kala-azar dermal leishmaniasis (PKDL) in a phase I trial [7]. Despite the ongoing progresses in vaccine development, the priority objective has not yet been achieved, i.e. the development of safe, effective, durable and low-cost prophylactic vaccine for human visceral leishmaniasis [8].
Besides producing memory lymphocytes towards a long-term immunity pathway, an ideal vaccine against Leishmania will stimulate parasite-specific cellular immunity that include a strong Th1 response to eliminate infections. In this regard, the use of epitopes or epitope-containing peptides is advantageous since epitopes can be evaluated for immuno-recognition and epitope-specific response. Since epitopes/peptides themselves remain poorly immunogenic, the approaches that have been gaining interest are based on the development of peptide-based formulations in combination with potent adjuvant components (peptide, lipids, virus particles, nanoparticles etc.) [9]. However, mapping of epitopes in immunogenic proteins remains crucial in peptide vaccine development. In addition to in vitro methods of epitope mapping such as phage display library, immunodominance and peptide competition assays, immunoinformatic mapping can be a powerful approach to facilitate screening of desired epitopes in immunogenic proteins [9]. Recent findings of leishmaniasis vaccine research also suggest that in silico predicted MHC class I and class II restricted epitope-containing peptides derived from Leishmania antigens alone, as a cocktail, as a chimeric peptide or in combination with adjuvant can be substantially immunogenic in vitro and/or in vivo [10,11,12,13]. Thus, the application of immunoinformatics-based pipeline can facilitate large-scale screening of peptide epitopes from Leishmania proteome for rational design of potent vaccines.
While derivation of potentially immunogenic peptides can be performed by analyzing (in vitro and/or in silico) either the whole parasite proteome, proteins known to elicit immunological outcome, or the known peptide libraries [9], two essential criteria have been suggested for consideration to initially select potential vaccine antigens for leishmaniasis: (i) known antigen that is expressed in the disease-causing mammalian stage of the parasite; and (ii) selected adjuvants that elicit a cellular, Th1-biased immune response for the immunizations in humans [14]. The human stage-associated Leishmania proteins that facilitate intracellular survival and infective process of the parasite thus constitute attractive targets for anti-Leishmania vaccine design. In order to adapt in mammalian host, the promastigote stage of Leishmania undergoes morphological and metabolic changes when transformed into amastigote stage upon entry and invasion. This is accompanied by a cascade of programmed changes in mRNA abundance, translation rate, and/or protein processing. However, interpretation of Leishmania transcriptome is likely controversial on whether relative changes in mRNA abundance is substantial [15, 16], constitutive or negligible [17,18,19]. Moreover, stage-specific upregulation for some transcripts [20] does not necessarily reflect in altered functional protein profile because of post-transcriptional [21] and post-translational regulation [22] evident for Leishmania species. While mRNA abundance may not be a perfect indicator of protein levels in eukaryotes [19, 23, 24], the relationship between mRNA and protein abundance is suggested to be dynamically changing as L. donovani adapts to amastigote condition, with correlation in changes for only a small proportion [22]. Moreover, the changes in protein level can also vary between clinical and cultured amastigotes [25]. In this scenario, experimentally evaluated proteome analysis can better highlight the key changes, which have important implications for diagnostics, drug target identification and vaccine design.
Reverse vaccinology [26] has been becoming increasingly popular in supported vaccine design by the combined use of genomics, transcriptomics, proteomics and immunoinformatics. Here, we propose an approach to design a subunit vaccine based exclusively on mass spectrometry (MS)-driven comparative proteomic information associated with amastigotes, since genome/transcriptome information can be deviant as Leishmania adapts to amastigote condition. Invasion and survival of L. donovani in the mammalian host largely involves the export of virulence factors and immune-modulatory components into the host cytosol. However, constant exposure of these secretory proteins to host immune system can lead to immunological tolerance and strong parasite-specific humoral response, which can be problematic for vaccine design. Hence, we limited our focus on rational screening of immunogenic T cell-specific epitopes in such secretory proteins, which have substantial coverage of endemic population as well. We then combined the epitopes into a single recombinant protein molecule. We explored into the physicochemical properties and receptor binding interaction of the multi-epitope vaccine, followed by molecular dynamics simulation of the vaccine-receptor complex and simulation of immune response. Overall, we showed that the experimental proteome data-driven immunoinformatic approach can facilitate informed screening of potential subunit vaccine candidates from truly produced human stage-associated parasitic proteins of pathological/physiological importance. The immunogenic potential evaluated in silico can also provide rationality for experimental validation of the modeled subunit vaccine.
Methods
All the computational tasks using online and offline tools in this study were carried out with the concurrent version of the tools between September 2018 and December 2018. Graphpad Prism v.7 software was used for descriptive data calculation, comparison of means and to reproduce graphs using software generated numeric values when applicable. A two-tailed P-value of < 0.05 was considered as significant. The methodological flowchart of the study is given in Fig. 1. The details of the methodological steps are given in Additional file 1: Text S1.

Methodological flowchart in multi-epitope subunit vaccine design
Antigen selection
Literature reports on the proteome profile of L. donovani were screened in the PubMed (NCBI) database to index articles published between year 2000 and 2018, and reports on MS-driven comparative abundance of proteins in promastigotes and amastigotes were selected to generate a local database of proteins. Sequences of identical or closely similar L. donovani proteins, which had more abundance in amastigotes, were retrieved. An MS-derived secretome profile of L. donovani was also retrieved from the published literature [27]. Secretory proteins in the local database were screened by both cross-matching the secretome and proteome data using ViroBLAST [28], as well as by evaluating combined prediction of SignalP [29], SecretomeP [30] and TMHMM [31]. Antigenicity of the potential secretory proteins was estimated using ANTIGENpro [32] and VaxiJen [33] programs.
Epitope screening
For screening of cytotoxic T-lymphocyte (CTL, 9-mer) epitopes and helper T-lymphocyte (HTL, 15-mer) epitopes, both affinity and allele coverage were considered. Initially, CTL and HTL epitopes were screened for above cut-off prediction scores in NetCTL [34] and lower percentile rank in IEDB (Immune Epitope Database) recommended MHC-II consensus module (http://tools.iedb.org/tcell/), respectively. Epitopes that are superior in human leukocyte antigen (HLA) cross-allele coverage were preliminarily selected. Then, both CTL and HTL epitope sets were filtered through specific (MHC-I and MHC-II modules of IEDB) and common (VaxiJen) immunogenicity prediction tools. CTL epitopes were further filtered in TAPpred [35] for more accurate prediction of TAP transporter binder. As per requirement of a proper anti-Leishmania immune response, T cell epitopes containing B cell recognition region (predicted by Bepipred [36]) were excluded, and all IL-10 inducing epitopes (predicted by IL-10Pred [37]) were removed. BLAST search against the non-redundant human protein database was carried out to rule out self-epitopes, while search against the RefSeq [38] protein database of Leishmania was performed to find out the conservancy of the epitopes in other Leishmania species.
In order to calculate the theoretical population coverage (TPC) (http://tools.iedb.org/population/) of each epitope, binding affinity to HLA allele-specific MHC molecules was set at percentile rank cut-off of 1.0 for CTL epitopes (IEDB recommended), and IC50 cut-off of 100 nM for HTL epitopes (10-times lower than the IEDB recommended value). Our target was to reach more than 90% population coverage by both CTL and HTL epitope sets in each of three most endemic areas of VL (India, Sudan and Brazil) with maximum number of alleles per epitope, while keeping the subunit length at minimum. In accordance, CTL epitopes with minimum TPC of 40% and at least eight HLA alleles were selected for vaccine construction. On the other hand, most of the screened HTL epitopes had more than 80% coverage in our observation, and therefore HTL epitopes having greater than 90% coverage were selected.
Chimeric vaccine construction and evaluation
The vaccine construct was arranged by joining the CTL and HTL epitopes with linkers [39, 40], and preceded by a synthetic peptide adjuvant [41]. The selection of chimeric arrangement was based on antigenicity (ANTIGENpro and VaxiJen) and allergenicity (AlgPred [42] and AllerTOP [43]) scores, retaining of cleavage, TAP transporter- and MHC-binding propensity of target epitopes, and generation of none/least number of non-specific and/or IL-10 inducing epitopes due to recombination. The capability to induce IFN-γ and IL-10 by the chimera was predicted by scanning in IFNepitope [44] and IL-10Pred module, respectively. Simulation of immune response based exclusively on the chimeric construct was performed in C-ImmSim [45] server, whereas two previously reported candidate Leishmania vaccine peptides [46, 47] were used to evaluate whether C-ImmSim prediction corroborates to the dynamicity of antigenic constructs. For structural analysis, the tertiary structure of the construct was produced in I-TASSER [48] modeling server followed by refinement using YASARA [49] force-field and GalaxyRefine [50] web tools. Stability of vaccine construct, a prerequisite for antigen processing, was assessed using physicochemical features predicted by both sequence-based (ProtParam [51]) and structural (SCooP [52] and CamSol [53]) analysis tools. Furthermore, chimeric vaccine-specific linear (Bepipred and BCPREDS [54]) and conformational (Ellipro [55]) B cell epitopes were predicted. The structural model was used to dock to Toll-like receptor 4 by using ClusPro [38] docking server and the binding interactions were analyzed. Molecular dynamics (MD) simulation was performed by using Desmond v5.3 (Schrödinger, LLC, New York, USA) software to check the conformational stability of vaccine-receptor docked complex.
In silico cloning
To validate the immunoinformatic findings, evaluation of immuno-reactivity through serological analysis is the preliminary step and this requires expression of the candidate vaccine. For this purpose, codon optimization was carried out by using JCAT [56]. A cloning model was then developed for this construct by using SnapGene (GSL Biotech, California, USA) tool and by inserting the optimized coding sequence into a plasmid vector.
Results
Screening of secretory amastigote proteins from experimental studies
Following literature screening, three out of 28 PubMed indexed experimental studies on L. donovani proteome were considered for the study. Two of the studies compared proteomic abundance of promastigotes with that of amastigotes as Leishmania adapts to the changes in conditions resembling the host [57, 58], while the other one compared splenic amastigotes to axenic amastigotes [25]. A total of 118 out of 134 proteins, which had a relative increase of at least 1.5-fold or were reported exclusively in the protein profile of amastigotes and/or splenic amastigotes, were found to have an identity of 90% or above for absolute query coverage with L. donovani proteins of similar functional annotations. After cross-matching of this group of proteins to 151 L. donovani secretory proteins revealed experimentally [27], 16 proteins were found to have an identity percentage and query coverage of 96.75 ± 1.1% and 99.44 ± 1.13%, respectively. Based on the presence of classical or non-classical secretion signal sequences along with minimum (no more than one) transmembrane helices, one common and an additional 17 secretory proteins were included to the pool. Among 33 amastigote-associated potential secretory proteins, 26 were selected based on their antigenicity probability scores of ≥ 0.5 as predicted by both ANTIGENpro and VaxiJen (Table 1, Additional file 2: Data S1).
Multi-epitope subunit L. donovani vaccine: construction and properties
A total of 79 CTL 9-mer epitopes were initially screened in NetCTL. Among them, only nine epitopes from six proteins were predicted to be non-self, highly immunogenic and high-to-moderate TAP-transporter binder non-B cell epitopes. These epitopes covered theoretically, an average of 66.46 ± 7.88% and a cumulative of 98.57% of the world population. Similarly, HTL 15-mer epitopes were screened to ensure both affinity and coverage. Fourteen selected HTL epitopes from eight proteins were finally predicted to be non-self, highly immunogenic non-B cell epitopes, with a mean theoretical coverage of 96.62 ± 1.35% and a cumulative of 99.52% of the world population. All the CTL and HTL epitopes except for H2-10 and H2-13 were conserved (100% identical) in L. infantum, whereas, less conservancy was found in representative proteins of L. major (13/23) and L. mexicana (10/23). Properties of individual CTL and HTL epitopes are given in Tables 2 and 3, respectively.
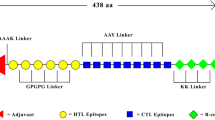
The construct of 397 amino acid residues comprised of the 9 CTL and 14 HTL epitopes, with AAY and GPGPG linkers added in the intra-epitopic positions of CTL and HTL epitopes, respectively. It preceded in N-terminal by TLR4 peptide adjuvant, APPHALS, linked by EAAK linker to the vaccine. The selected rearranged model had the antigenicity score of 0.8 calculated by ANTIGENpro, and 0.74 (bacteria model) and 0.65 (parasite model) by VaxiJen. Furthermore, the construct was found to be non-allergenic for human use. When re-analyzed by the screening tools, all the original CTL and HTL epitopes were found consistent with the pre-screening immunogenicity, cleavage and TAP binding properties in the rearranged model. On the other hand, the arrangement of the construct resulted in generation of only three regions (15-mer overlapping) of IL-10 inducing epitopes and three non-specific CTL epitopes (9-mer) comparable to the potency of target epitopes (Additional file 3: Figure S1).
IFN-γ epitopes
Prediction on IFN-γ induction capacity revealed a total of 117 epitopes (15-mer) with positive scores. This prediction was consistent with the C-ImmSim simulated immune response in terms of high IFN-γ production after hypothetical immunization (three doses) in a population characterized by a combination of frequent and VL susceptible HLA alleles [59,60,61]. Since the hypothetical cytokine levels in simulated immune response represent only the outcome of algorithmically set dynamic cellular interactions for a defined time period after antigen priming [37], it was important to evaluate whether the simulation module can respond dynamically to different constructs [45]. Therefore, we simulated immune responses of two additional peptide vaccine candidates: peptide 1 (L. infantum derived fusion peptide [46]) and peptide 2 (L. donovani GP63 derived peptide [47]), which were experimentally found to exhibit varying cytokine response in comparison to soluble Leishmania antigen (SLA). Substantial difference was observed in terms of immunosuppressive IL-10 and TGF-β induction capacity between peptide 1 and peptide 2; however, determination of statistical significance was not possible in the simulation module. Nevertheless, the outcome can be considered consistent with the general trend of in vitro immune response (compared to SLA), with peptide 2 being more prominent IL-10 inducer compared to SLA as reported in [47]. In terms of cytokine induction potential, simulation outcome of our designed construct conformed more closely to that of peptide 1, which did not induce IL-10 level higher than that by SLA in vitro [46] (Fig. 2).

Simulation of cytokine response. Illustration of cytokine induction potential by control peptide 1 (L. infantum derived fusion peptide) (a), vaccine construct designed in this study (b) and control peptide 2 (L. donovani GP63 derived peptide) (c) by independent simulation of immune response. Hypothetical administration of the peptides was performed in three doses four weeks apart with 1000 units/dose
Tertiary structure of the chimeric protein and cloning model
Since there was no significant template hit, the choice of 3D model among outputs generated by I-TASSER was based on: (i) cluster size of model replicas; (ii) frequency of model in simulation trajectory; and (iii) C-score. The selected model has the highest C-score of − 1.56 which is close to the I-TASSER recommended score (− 1.5) for accuracy, and has the highest frequency in the top cluster by size. After further refinement of the protein topology, the PROCHECK [62] server returned a G-score of − 0.04, which indicates that the backbone and side chain of the model correspond to high-probability stereochemical conformations. The model scored 1.73 in X-ray resolution scale by MolProbity [63], with no poor rotamers and bad bonds, negligible all atom steric overlaps (0.5%) and an increase in Ramachandran-favored residue number from 79.2% (unrefined) to 92.4% (refined) with a subsequent decrease in outliers (Additional file 4: Figure S2). The vaccine construct has a molecular weight of 42.1 kDa, with a basic nature (isoelectric point: 9.16). The score obtained for instability index was 27.26, which implied the stable nature of the vaccine in vitro. The estimated value of aliphatic index was 75.39 which indicated its thermo-stability. The folded structure has a melting temperature of 73.9 °C and folding free energy of − 17.7 kcal/mol at neutral pH in humans. Additionally, this model was found to have substantial solubility with a score of 0.38 in folded state in contrast to the unfolded intrinsic score of − 3.06, which suggests that hydrophobic residues in this model tend to ideally form the stable core leaving hydrophilic residues much on the solvent accessible surface. The half-life of the construct in mammalian reticulocytes was estimated as 4.4 h in vitro, compared with 20 h and 10 h in yeasts and Escherichia coli in vivo, respectively.
In terms of chimera-specific B cell response, Bepipred predicted six B cell epitopes of 8–12 residues in length above the threshold score, while BCPREDS predicted 11 non-overlapping and linear 20-mer B cell epitopes with specificity scores > 0.99. Residues in those linear epitopes accounted for 41% residues of the 08 non-overlapping conformational epitopes (Fig. 3, Additional file 5: Table S1).

Refined tertiary structure of the chimeric protein. The secondary structure elements consist of helix (34%), sheet (10%), turns (39%) and coil (17%). Residue positions in B cell conformational epitope that overlap linear epitopes are depicted as CPK shapes
This sequence was used to generate in silico cloning model for E. coli (K12) expression. After optimization of the codon, the codon adaptation index (CAI) value of the chimera was 0.98, while the GC content was 56.09%. For insertion into the E. coli pET28a(+) expression vector, two restriction sites for XhoI and NdeI enzymes were added in the 3ʹ- and 5ʹ-end, respectively, of the vaccine coding strand enclosed by 6-histidine residues at both ends (Additional file 6: Figure S3).
Molecular docking of vaccine in TLR4
Molecular docking of the vaccine construct with TLR4 in ClusPro 2.0 docking server generated 30 models ranked by cluster size of the representative pose. The selected docked complex had the largest cluster size (ClusPro recommended) with second-lowest binding energy score (− 1282.3) among the top ten models. The chimeric construct seemed to occupy partially into the lateral concave surface, but not the convex surface, with strong hydrophobic interactions mostly with the beta-sheet adjacent residues at the C-terminal domain of TLR4 ectodomain (ECD) and also with its adapter protein, MD2, with support of several hydrogen bonds, thus establishing ligand mediated cross-link between TLR4 and MD2 (Fig. 4).

Docked complex of TLR4 with vaccine construct. Accompanying structural monomers include second TLR4 ECD (TLR4*), MD-2 adapter and second adapter (MD-2*). Residual participants of receptor monomers interacting with vaccine (green) are represented by yellow (hydrophobic) and red (hydrogen bond) CPK shapes
Molecular dynamics (MD) simulation of vaccine-TLR4 complex
Molecular dynamics simulation of the docked complex was performed by using OPLS_2005 force field. Using the Simulation Quality Analysis tool of the Desmond software, the mean potential energy for the complex was obtained as − 6.4e5 kilocal/mol (Additional file 7: Figure S4). The radius of gyration (Rg) obtained for the docked complex showed that the mean distance in rotating complex from the center of mass is 4.31 nanometers (SD: 0.2 nanometers) about which the model becomes consistent after 4 ns (Fig. 5a). The number of intermolecular hydrogen bond (H-bond) between the side chains of vaccine protein and TLR4 initially fluctuated probably due to solvent effect before matching the trend of Rg in reaching steadiness after 4 ns. This suggests the role of H-bonds in the overall compactness of the complex (Fig. 5b). The trends of Rg and H-bond plots indicate that 6–8 strong H-bonds were persistent over simulation period between vaccine and TLR4, and this might be crucial for stable binding.

Molecular dynamics simulation of docked complex. For a time duration of 10 ns, plots of the radius of gyration (Rg) (a), hydrogen bond (b), RMSD of the backbone atoms fitted to complex (green) and ligand-free receptor (black), with respect to initial structure (c), and RMSF for side-chain atoms fitted to complex (green) and ligand-free receptor (black) with respect to initial structure of TLR4 ECD (627 aa; divided into three domains of leucine rich repeats or LRR) (d). Receptor positions interacting with vaccine (final frame) are represented with red circles
The root mean square deviation (RMSD) of the vaccine-TLR4 complex for backbone atoms over the simulation period was 4.0 Å (SD: 0.49 Å), while it was 3.2 Å (SD: 0.35 Å) for ligand-free TLR4 atoms (Fig. 5c), suggesting comparably higher (paired t-test: P < 0.0001) RMSD of the complex backbone. The root mean square fluctuation (RMSF) for side-chain atoms of vaccine-bound TLR4 (1.9 Å, SD: 0.7 Å, range: 0.8–7.0 Å) was higher (Wilcoxon matched-pairs test, P < 0.0001) than unbound TLR4 (1.6 Å, SD: 0.5 Å, range: 0.7–4.2 Å). The RMSF indicates overall less fluctuations for atoms interacting with vaccine residues, while atoms at vaccine unbound regions of the N-terminal and central domain underwent high fluctuations (Fig. 5d). Although the trends toward reaching convergence were very similar, higher RMSD value of the complex than the vaccine-unbound TLR4 indicates structural mobility in the complex due to vaccine interaction and this is likely attributable to the higher RMSD of vaccine protein along the MD simulation time. In congruence, rearrangement of several bonds between the vaccine and TLR4 was observed between pre-simulation and post-simulation models, while the total number of non-covalent bonds increased from 41 in pre-simulation model to 64 in post-simulation model (not shown). Visualization of the interacting residues also indicates that, in comparison to unbound (and also pre-simulation) structure, the post-simulation bonding rearrangement is coupled with increased number of H-bond at the C-terminal domains between TLR4 and TLR4* (second TLR4 ECD) (Additional file 8: Figure S5). This implies likely chance of positive interactions between the TLR4 monomers in physiological condition following vaccine interaction. Overall, the conformation of vaccine-bound receptor supports structural flexibility, which might be in favor of biological response of the receptor.
Immune simulation to predict secondary response
Hypothetical administration of three doses of vaccine construct four weeks apart with 1000 unit/dose was performed to simulate the immune response generated by immunization. In silico immune simulation plots hinted at antigenic recognition and subsequent response in terms of antibody production, and active as well as memory B cell and T cell generation in the population with a VL susceptible HLA profile after hypothetical immunization. The primary response to the proposed chimera can be characterized by a marked increase in chimera-specific IgM and IgG production. After subsequent doses, a corresponding decrease in antigen concentration indicates gradual increase in memory B cell production with persistence. Furthermore, expansion of CD4+ T lymphocytes with memory development following the initial dose was observed. CD8+ T lymphocytes response was also high for the susceptible population reaching at its peak after the second dose. Repeated exposure of 12 doses, on the other hand, did not seem to cause clonal expansion of any epitope-specific T cells- as indicated by the Simpson’s index D, which is inversely related to diversity (Additional file 9: Figure S6).
Discussion
Proteomics-driven identification of potential vaccine candidates can be a sound approach for selecting promising antigens, which are elicited against environmental stimuli analogous to host response upon pathogen invasion and are physiologically relevant for pathogens within the host [64]. Availability of pathogen proteome information upon infection of the host can provide opportunities for in silico mining of novel vaccine candidates, and this approach has been utilized for in silico design of an epitope-based vaccine against Theileria parasites of ruminants [65]. For a dimorphic human parasite like Leishmania, it is important to target human stage-associated antigenic proteins that are physiologically important for parasites to infect and establish in a new host. In recent years, several studies utilized immunoinformatic approaches of epitope screening in designing epitope-based vaccines. Khatoon et al. [66], Singh et al. [67] and Vakili et al. [68] have previously reported the theoretical potential of in silico designed vaccines for visceral leishmaniasis. Notably, in a recent study by Vakili et al. [69], the group further evaluated successfully the immunogenic potential of the multi-epitope vaccine, derived in part from known antigens, by administering the chimeric construct in experimental mice. This suggests that the in silico designed vaccines with epitopes derived from appropriate protein targets have the potential to progress toward advanced phases of vaccine development for visceral leishmaniasis. While the in silico studies by Khatoon et al. [66] and Singh et al. [67] largely utilized available genomic databases of L. donovani to select vaccine targets, Dikhit et al. [11, 70] performed thorough investigations involving in silico, in vitro and in vivo analysis to screen and validate immunogenic epitopes obtained from proteins that are increasingly expressed at the infective parasite stage. Such highly expressed proteins are likely important for physiological and/or infective process of the parasite and thus can be more effective vaccine targets. In this study, we took an approach to select such amastigote proteins in terms of contrasting abundance or specificity (abundant up to the level of detection) from comparative proteome profiles of L. donovani promastigotes and amastigotes. Based on the propensity of those proteins for secretion in vitro and/or having secretory signal sequence, we further combined immunoinformatic tools to identify candidate antigens that have secretory potential. A comparison of the methodological and outcome features among several studies that have employed in silico design and evaluation of epitope-based candidate vaccines against visceral leishmaniasis to date is summarized in Table 4. Overall, our reported vaccine construct was found to be comparable to the earlier exclusively-in silico reports in terms of antigenicity, population coverage and receptor interaction. However, experimental studies remain crucial to validate the immunogenic potential of the designed vaccine.
Analyzing amastigote secretome through intra-macrophagic studies is considered difficult, while significant difference in secretome between amastigotes and promastigotes is unlikely due to relatively low stage-specific differences in gene expression [27]. However, due to the dynamicity in the relationship between mRNA and protein abundance as L. donovani adapts to the amastigote condition, comparative levels of abundance of these secretory proteins can be a more reliable indicator. Hence, our screening approach is relevant within the context. Perhaps, the most studied amastigote-specific vaccine candidate in L. donovani happens to be a cellular stress countering abundant surface antigen, A2, which has shown to confer whole or epitope-specific efficacy in multiple immunization models [8, 71]. The vaccine construct reported in the present study comprised of immunogenic T cell-specific epitopes (as predicted immunoinformatically) from 13 amastigote-associated proteins. Five of them are known to associate with virulence in the mammalian host (fructose-1,6-bisphosphatase, putative protein disulfide isomerase, putative lipophosphoglycan biosynthetic protein, leishmanolysin and cysteine protease), while others have putative roles in countering the host-induced stress response (thioredoxin-like protein, glutathione peroxidase, stress-inducible protein STI1 homolog), host-microbicidal activity regulation (proteasome endopeptidase) and protein synthesis (elongation factor 2). Three proteins were uncharacterized according to the proteomic studies. Protein domain and homology (to proteins of other Leishmania species) suggest that two of these proteins may potentially play a role in drug resistance phenotype (E9BUW4) and protection from intracellular stress (E9BDB8), while the specific function of alpha/beta hydrolase domain-containing protein (E9BQ40) in amastigotes has not yet been deciphered. On the other hand, lack of reports on experimental evaluation of immunogenicity of several Leishmania proteins, which have been included in our set of antigenic proteins is apparent. Among the 13 proteins of current interest, only six (elongation factor 2, proteasome endopeptidase complex, putative protein disulfide isomerase, leishmanolysin, cysteine protease and putative lipophosphoglycan biosynthetic protein) or their species homologs are known to have proven immunoreactive properties (Table 1). Nevertheless, the increased abundance of the unexplored proteins suggests their likely role of pathological/physiological significance in host invasion and/or survival. The antigenicity scores further corroborate to the potentiality of these proteins as antigenic. Immunological evaluation of these amastigote stage-associated proteins may unravel novel Leishmania antigens in future.
In the context of functional roles of selected proteins, our designed vaccine has the potential to benefit the host by generating appropriate immune response both in the early and progressive phase of systemic infection. Furthermore, almost all of the epitopes were found in corresponding proteins of L. infantum, indicating potential cross-protection against this visceralizing species. Most of the VL cases are reported from the endemic zones of the Indian subcontinent, East Africa and South America. Thus, in designing an epitope-based subunit vaccine, it is important to estimate the fractions of population in the target endemic zones based on HLA genotypic frequencies. The immunogenic non-self CTL epitopes in the vaccine modeled here is estimated to cover 96.8%, 91.7% and 93.9% of the allelic populations of Brazil, India and Sudan, respectively, with experimentally evaluated truly binding affinity [72], while for HTL epitopes, it is almost 100% for each of these populations. The vaccine construct has antigenic properties while it was not found to be an allergen. The structure was found thermodynamically stable and surface-soluble, while the core is hydrophobic, a favorable feature for antigen processing. Vaccine-specific, but not parasite protein-specific humoral response was predicted, and this can be used as a biomarker of vaccine efficacy [46, 73] without eliciting a parasite-specific B cell response. Moreover, the construct structure showed a good binding affinity in previously reported binding cavity of TLR4 [74,75,76,77].
The structural interface between TLR4 and the peptide adjuvant (APPHALS) used here has been extensively studied before. The position occupied by the adjuvant peptide in the TLR4-MD2 complex has been suggested to be varying depending on its position in the vaccine model and the canonical activation of the receptor is thought to be mechanized by insertion of peptide adjuvant in MD2 [78]. Since we used already activating but hypo-responsive TLR4-MD2 crystal structure removed of LPS for docking [79], it was not possible to speculate about the agonistic behavior of the bound vaccine. Nevertheless, our docking model is suggestive of non-MD2 (non-canonical) binding of adjuvant linked peptide, in which the vaccine intrinsic segment may have more affinity than the peptide adjuvant for binding to TLR4. The binding interface along with the molecular dynamics (MD) simulation of the docked complex in the solvent system hint at a sufficiently stable cross-link of TLR4 and MD2 with no major bond rearrangement between TLR4 and MD2, and between TLR4* and MD2 heterodimer formations. Although, the simulation time was short, this is reasonable as none of the vaccine residues interacted at crucial MD2-binding sites [74]. On the other hand, H-bond was found to increase between TLR4 ECDs (where vaccine is bound to one TLR4 ECD) in the vaccine-bound form compared to the unbound TLR4, which suggests potential event of positive interactions and movement between the ECDs. Additionally, reduction in electrostatic surface potential at the vaccine-bound TLR4 interface was observed after docking, which was consistent in post-simulation structural interface. Simultaneously, it was observed that a homo-dimer destabilizing His458-His458* repulsion [76] at pre-dock TLR4 was nullified and superseded post-dock by a solvent stable pi-hydrophobic interaction. It is thus possible that a change in the interpolated charge difference between pre-dock and post-dock TLR4 interface could have contributed to the bonding rearrangement between TLR4 ECDs. Notably, this rearrangement also involved participation of other critical histidine (His431, His555) residues at the TLR4-TLR4* interface [80] unlike the unbound structure (Additional file 8: Figure S5). Overall, these events are congruent with non-canonical TLR4 activation model mediated by microbial peptides, metals and cationic lipid nano-carriers, which are suggested to not confer canonical interaction with other monomers but to induce bond rearrangement among receptor monomers upon interaction [74,75,76,77]. Although the exact mechanism remains to be elucidated, our observations suggest that the vaccine construct may possess a characteristic peptide feature of a non-canonical TLR4 ligand [81, 82], which may facilitate TLR4-TLR4* dimerization for downstream activation of immune cells. The trends of backbone RMSD, Rg and H-bond of the vaccine-bound complex over the simulation period complied with structural flexibility rather than rigidity of the complex. The RMSF values of the complex side-chain indicate that the higher fluctuations in TLR4 were of those residues, which are vaccine-unbound and located in the solvent exposed loop mostly at or around glycines [83]. Increased residual fluctuation at LRR10-12 and around Gly397 may also be attributed to the mutations introduced at the position 299 and 399 in TLR4 structure (4G8A), as reported in [79], which was used to dock the vaccine protein. Nevertheless, it is unlikely that vaccine interaction would induce dissociation in structural interface of natural TLR4-MD2 since none of the highly fluctuating TLR4 residues had any direct interaction with the vaccine or MD2.
Simulation outcome of hypothetical immunization in VL susceptible HLA alleles (hypothetical heterozygous combination) was consistent with the predicted immunogenicity of the vaccine. Furthermore, we showed that the simulation outcome can be dynamic for different constructs when we used the same criteria in the simulation program and the same HLA profile to test two known vaccine candidates for VL. Importantly, for these peptides, IL-10 production was reported previously as either prominent (peptide-2) or lessened (peptide-1) in comparison to SLA in vitro. It is not expected that simulation results will reflect experimental outcomes; however, we observed a general trend of difference in immunosuppressive cytokine (e.g. IL-10) induction potential between the two peptides from the simulation outcome, with peptide-2 having more potent IL-10 induction capacity. Although statistical significance could not be inferred from the simulation plots, the difference seems consistent with the experimental result. Understandably, the predicted epitopes (not shown) in the simulation program did not comply mostly with our target set of epitopes due to the difference in the epitope prediction algorithm [45]. However, when compared to the simulation outcome of the known peptides, the general trend was comparable to both peptides for IFN-γ induction, while TGF-β and IL-10 were predicted to be considerably less pronounced than that by peptide-2. Besides IL-10, TGF-β has potent immunosuppressive properties, enhances disease progression and may prevent cure and protective immunity development against leishmaniasis [84, 85]. Thus, the simulation prediction of higher propensity of the construct to induce a more Th1-polarized response rather than Th2 is consistent with our desired immunogenicity.
Despite the difference in the epitope set, simulation dynamics over time can be extrapolated for the estimated set of epitopes of our construct since it is also comprised of diverse T cell epitopes and vaccine-specific B cell immunogenic regions as predicted by several immunoinformatic tools. It has been proposed previously that the simulation dynamics can be consistent with a realistic immunization process in terms of primary and secondary immune responses [45]. Likewise, clearance of antigen, production of antibody, development and persistence of memory B cells as well as CD4+ T cells over several months were assumed in the simulation outcome. For primary activation and maintenance of CD8+ T cells, CD4+ T cells (both Th1 and Th2 type) [86] are believed to be required [87, 88], where cytokines such as IFN-γ, IL-2 and IL-4 could be involved [89,90,91,92]. The simulation outcome suggests chance for expression of high levels of IFN-γ and IL-2, which may potentiate CD8+ T cell expansion. On the other hand, it is unlikely that the vaccine would trigger clonal expansion of epitope-specific T cells since we combined potent epitopes from several amastigote-associated proteins of comparable affinity, and it was consistent with the simulation dynamics for repeated exposure of 12 doses, as indicated by Simpsonʼs index (D). Rather, high level of IL-2 production can be expected for diverse epitope-mediated immune response functional over long time in vaccine-mediated immunity.
Experimental validation is utmost to prove this computational work. Next phases of the reverse vaccinology approach would ideally involve assessing the recombinant immunogenic protein expressed in the E. coli (strain K12) system as proposed here, in vitro stimulation of peripheral blood mononuclear cells from active VL patients as well as healthy endemic people for cytokine production, and evaluation in challenge models. While a multi-epitope vaccine molecule generated by using a reverse vaccinology approach can induce specific responses in in vivo and in vitro assays, a single recombinant molecule can also reduce the cost of production [93, 94]. The in silico designed vaccine reported here confers substantial immunogenic potential to be considered for in vitro experimental evaluation in the next phase of the study.
Conclusions
Screening and design of large-scale subunit/peptide vaccine candidates can be facilitated by a reverse vaccinology approach prior to experimental validation. This modelling study took a systematic approach to apply a series of immunoinformatic tools to extract T cell-specific epitopes from MS-driven human stage-associated L. donovani proteins with secretory potential, and design a subunit vaccine with a broad population coverage. Development of such a prophylactic vaccine for VL may complement therapeutic strategies against active infections as well. Overall, collective approaches of in silico, in vitro and in vivo investigations are utmost to develop a universal subunit vaccine against human VL.
Availability of data and materials
Data supporting the conclusions of this article are included within the article and its additional files.
Abbreviations
- VL:
-
visceral leishmaniasis
- KEP:
-
Kala-azar Elimination Programme
- MS:
-
mass spectrometry
- CTL:
-
cytotoxic T-lymphocyte
- HTL:
-
helper T-lymphocyte
- HLA:
-
human leukocyte antigen
- TPC:
-
theoretical population coverage
- CAI:
-
codon adaptation index
- ECD:
-
ectodomain
- MD:
-
molecular dynamics
- Rg:
-
radius of gyration
- RMSD:
-
root mean square deviation
- RMSF:
-
root mean square fluctuation
References
WHO. Leishmaniasis in high-burden countries: an epidemiological update based on data reported in 2014. Wkly Epidemiol Rec. 2016;91:285–96.
Hirve S, Kroeger A, Matlashewski G, Mondal D, Banjara MR, Das P, et al. Towards elimination of visceral leishmaniasis in the Indian subcontinent—translating research to practice to public health. PLoS Negl Trop Dis. 2017;11:e0005889.
Engwerda CR, Matlashewski G. Development of Leishmania vaccines in the era of visceral leishmaniasis elimination. Trans R Soc Trop Med Hyg. 2015;109:423–4.
Teixeira MC, Oliveira GG, Santos PO, Bahiense TC, da Silva VM, Rodrigues MS, et al. An experimental protocol for the establishment of dogs with long-term cellular immune reactions to Leishmania antigens. Mem Inst Oswaldo Cruz. 2011;106:182–9.
Coler RN, Duthie MS, Hofmeyer KA, Guderian J, Jayashankar L, Vergara J, et al. From mouse to man: safety, immunogenicity and efficacy of a candidate leishmaniasis vaccine LEISH-F3+GLA-SE. Clin Transl Immunol. 2015;4:e35.
Chakravarty J, Kumar S, Trivedi S, Rai VK, Singh A, Ashman JA, et al. A clinical trial to evaluate the safety and immunogenicity of the LEISH-F1+MPL-SE vaccine for use in the prevention of visceral leishmaniasis. Vaccine. 2011;29:3531–7.
Osman M, Mistry A, Keding A, Gabe R, Cook E, Forrester S, et al. A third generation vaccine for human visceral leishmaniasis and post kala azar dermal leishmaniasis: first-in-human trial of ChAd63-KH. PLoS Negl Trop Dis. 2017;11:e0005527.
Iborra S, Solana JC, Requena JM, Soto M. Vaccine candidates against Leishmania under current research. Expert Rev Vaccines. 2018;17:323–34.
De Brito RCF, Cardoso JMO, Reis LES, Vieira JF, Mathias FAS, Roatt BM, et al. Peptide vaccines for leishmaniasis. Front Immunol. 2018;9:1043.
Agallou M, Athanasiou E, Koutsoni O, Dotsika E, Karagouni E. Experimental validation of multi-epitope peptides including promising MHC class I- and II-restricted epitopes of four known Leishmania infantum proteins. Front Immunol. 2014;5:268.
Dikhit MR, Kumar A, Das S, Dehury B, Rout AK, Jamal F, et al. Identification of potential MHC class-II-restricted epitopes derived from Leishmania donovani antigens by reverse vaccinology and evaluation of their CD4+ T-cell responsiveness against visceral leishmaniasis. Front Immunol. 2017;8:1763.
Alves-Silva MV, Nico D, Morrot A, Palatnik M, Palatnik-de-Sousa CB. A chimera containing CD4+ and CD8+ T-cell epitopes of the Leishmania donovani nucleoside hydrolase (NH36) optimizes cross-protection against Leishmania amazonesis infection. Front Immunol. 2017;8:100.
Martins VT, Lage DP, Duarte MC, Carvalho AM, Costa LE, Mendes TA, et al. A recombinant fusion protein displaying murine and human MHC class I- and II-specific epitopes protects against Leishmania amazonensis infection. Cell Immunol. 2017;313:32–42.
Duthie MS, Favila M, Hofmeyer KA, Tutterrow YL, Reed SJ, Laurance JD, et al. Strategic evaluation of vaccine candidate antigens for the prevention of visceral leishmaniasis. Vaccine. 2016;34:2779–86.
Saxena A, Lahav T, Holland N, Aggarwal G, Anupama A, Huang Y, et al. Analysis of the Leishmania donovani transcriptome reveals an ordered progression of transient and permanent changes in gene expression during differentiation. Mol Biochem Parasitol. 2007;152(1):53–65.
Almeida R, Gilmartin BJ, McCann SH, Norrish A, Ivens AC, Lawson D, et al. Expression profiling of the Leishmania life cycle: cDNA arrays identify developmentally regulated genes present but not annotated in the genome. Mol Biochem Parasitol. 2004;136:87–100.
Leifso K, Cohen-Freue G, Dogra N, Murray A, McMaster WR. Genomic and proteomic expression analysis of Leishmania promastigote and amastigote life stages: the Leishmania genome is constitutively expressed. Mol Biochem Parasitol. 2007;152:35–46.
Akopyants NS, Matlib RS, Bukanova EN, Smeds MR, Brownstein BH, Stormo GD, et al. Expression profiling using random genomic DNA microarrays identifies differentially expressed genes associated with three major developmental stages of the protozoan parasite Leishmania major. Mol Biochem Parasitol. 2004;136:71–86.
Haile S, Papadopoulou B. Developmental regulation of gene expression in trypanosomatid parasitic protozoa. Curr Opin Microbiol. 2007;10:569–77.
Rosenzweig D, Smith D, Opperdoes F, Stern S, Olafson RW, Zilberstein D. Retooling Leishmania metabolism: from sand fly gut to human macrophage. FASEB J. 2008;22:590–602.
Clayton CE. Life without transcriptional control? From fly to man and back again. EMBO J. 2002;21:1881–8.
Lahav T, Sivam D, Volpin H, Ronen M, Tsigankov P, Green A, et al. Multiple levels of gene regulation mediate differentiation of the intracellular pathogen Leishmania. FASEB J. 2011;25:515–25.
Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol. 2007;25:117–24.
Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003;4:117.
Pescher P, Blisnick T, Bastin P, Spath GF. Quantitative proteome profiling informs on phenotypic traits that adapt Leishmania donovani for axenic and intracellular proliferation. Cell Microbiol. 2011;13:978–91.
Seib KL, Zhao X, Rappuoli R. Developing vaccines in the era of genomics: a decade of reverse vaccinology. Clin Microbiol Infect. 2012;18(Suppl. 5):109–16.
Silverman JM, Chan SK, Robinson DP, Dwyer DM, Nandan D, Foster LJ, et al. Proteomic analysis of the secretome of Leishmania donovani. Genome Biol. 2008;9:R35.
Deng W, Nickle DC, Learn GH, Maust B, Mullins JI. ViroBLAST: a stand-alone BLAST web server for flexible queries of multiple databases and user’s datasets. Bioinformatics. 2007;23:2334–6.
Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6.
Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel. 2004;17:349–56.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80.
Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, Felgner PL, et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26:2936–43.
Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8:4.
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007;8:424.
Bhasin M, Raghava GP. Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Sci. 2004;13:596–607.
Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2.
Nagpal G, Usmani SS, Dhanda SK, Kaur H, Singh S, Sharma M, et al. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci Rep. 2017;7:42851.
Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–5.
Pandey RK, Bhatt TK, Prajapati VK. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating Anopheles salivary protein. Sci Rep. 2018;8:1125.
Pentel PR, LeSage MG. New directions in nicotine vaccine design and use. Adv Pharmacol. 2014;69:553–80.
Shanmugam A, Rajoria S, George AL, Mittelman A, Suriano R, Tiwari RK. Synthetic Toll like receptor-4 (TLR-4) agonist peptides as a novel class of adjuvants. PLoS ONE. 2012;7:e30839.
Saha S, Raghava GPS. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006;34(Suppl. 2):W202–9.
Dimitrov I, Bangov I, Flower DR, Doytchinova I. AllerTOP vol 2 - a server for in silico prediction of allergens. J Mol Model. 2014;20:2278.
Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013;8:30.
Rapin N, Lund O, Bernaschi M, Castiglione F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE. 2010;5:e9862.
Dias DS, Ribeiro PAF, Martins VT, Lage DP, Costa LE, Chavez-Fumagalli MA, et al. Vaccination with a CD4(+) and CD8(+) T-cell epitopes-based recombinant chimeric protein derived from Leishmania infantum proteins confers protective immunity against visceral leishmaniasis. Transl Res. 2018;200:18–34.
Elfaki MEE, Khalil EAG, De Groot AS, Musa AM, Gutierrez A, Younis BM, et al. Immunogenicity and immune modulatory effects of in silico predicted L donovani candidate peptide vaccines. Hum Vaccin Immunother. 2012;8:1769–74.
Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008;9:40.
Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins. 2009;77(Suppl. 9):114–22.
Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41:W384–8.
Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531–52.
Pucci F, Kwasigroch JM, Rooman M. SCooP: an accurate and fast predictor of protein stability curves as a function of temperature. Bioinformatics. 2017;33:3415–22.
Sormanni P, Aprile FA, Vendruscolo M. The CamSol method of rational design of protein mutants with enhanced solubility. J Mol Biol. 2015;427:478–90.
El-Manzalawy Y, Dobbs D, Honavar V. Predicting linear B-cell epitopes using string kernels. J Mol Recognit. 2008;21:243–55.
Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A, et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008;9:514.
Grote A, Hiller K, Scheer M, Munch R, Nortemann B, Hempel DC, et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005;33:W526–31.
Bente M, Harder S, Wiesgigl M, Heukeshoven J, Gelhaus C, Krause E, et al. Developmentally induced changes of the proteome in the protozoan parasite Leishmania donovani. Proteomics. 2003;3:1811–29.
Sardar AH, Kumar S, Kumar A, Purkait B, Das S, Sen A, et al. Proteome changes associated with Leishmania donovani promastigote adaptation to oxidative and nitrosative stresses. J Proteomics. 2013;81:185–99.
Singh T, Fakiola M, Oommen J, Singh AP, Singh AK, Smith N, et al. Epitope-binding characteristics for risk versus protective DRB1 alleles for visceral leishmaniasis. J Immunol. 2018;200:2727–37.
Faghiri Z, Tabei SZ, Taheri F. Study of the association of HLA class I antigens with kala-azar. Hum Hered. 1995;45:258–61.
Kaye PM, Aebischer T. Visceral leishmaniasis: immunology and prospects for a vaccine. Clin Microbiol Infect. 2011;17:1462–70.
Laskowski RA, MacArthur MW, Thornton JM. PROCHECK: validation of protein-structure coordinates. International tables for crystallography. Dordrecht: Kluwer Academic Publishers; 2012.
Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21.
Zielke RA, Wierzbicki IH, Baarda BI, Gafken PR, Soge OO, Holmes KK, et al. Proteomics-driven antigen discovery for development of vaccines against Gonorrhea. Mol Cell Proteomics. 2016;15:2338–55.
Kar PP, Srivastava A. Immuno-informatics analysis to identify novel vaccine candidates and design of a multi-epitope based vaccine candidate against Theileria parasites. Front Immunol. 2018;9:2213.
Khatoon N, Pandey RK, Prajapati VK. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep. 2017;7:8285.
Singh G, Pritam M, Banerjee M, Singh AK, Singh SP. Genome based screening of epitope ensemble vaccine candidates against dreadful visceral leishmaniasis using immunoinformatics approach. Microb Pathog. 2019;136:103704.
Vakili B, Eslami M, Hatam GR, Zare B, Erfani N, Nezafat N, et al. Immunoinformatics-aided design of a potential multi-epitope peptide vaccine against Leishmania infantum. Int J Biol Macromol. 2018;120:1127–39.
Vakili B, Nezafat N, Zare B, Erfani N, Akbari M, Ghasemi Y, et al. A new multi-epitope peptide vaccine induces immune responses and protection against Leishmania infantum in BALB/c mice. Med Microbiol Immunol. 2020;209:69–79.
Dikhit MR, Kumar A, Amit A, Dehury B, Nathsharma YP, Ansari MY, et al. Mining the proteome of Leishmania donovani for the development of novel MHC class I restricted epitope for the control of visceral leishmaniasis. J Cell Biochem. 2018;119:378–91.
Fernandes AP, Coelho EAF, Machado-Coelho GLL, Grimaldi G, Gazzinelli RT. Making an anti-amastigote vaccine for visceral leishmaniasis: rational, update and perspectives. Curr Opin Microbiol. 2012;15:476–85.
Paul S, Weiskopf D, Angelo MA, Sidney J, Peters B, Sette A. HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. J Immunol. 2013;191:5831–9.
Martins VT, Duarte MC, Lage DP, Costa LE, Carvalho AM, Mendes TA, et al. A recombinant chimeric protein composed of human and mice-specific CD4(+) and CD8(+) T-cell epitopes protects against visceral leishmaniasis. Nat Immunol. 2017;39:1.
Oblak A, Pohar J, Jerala R. MD-2 determinants of nickel and cobalt-mediated activation of human TLR4. PLoS ONE. 2015;10:e0120583.
Schmidt M, Raghavan B, Muller V, Vogl T, Fejer G, Tchaptchet S, et al. Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat Immunol. 2010;11:814–9.
Lonez C, Irvine KL, Pizzuto M, Schmidt BI, Gay NJ, Ruysschaert JM, et al. Critical residues involved in Toll-like receptor 4 activation by cationic lipid nanocarriers are not located at the lipopolysaccharide-binding interface. Cell Mol Life Sci. 2015;72:3971–82.
Chu M, Zhou M, Jiang C, Chen X, Guo L, Zhang M, et al. Staphylococcus aureus phenol-soluble modulins alpha1-alpha3 act as novel Toll-like receptor (TLR) 4 antagonists to inhibit HMGB1/TLR4/NF-kappaB signaling pathway. Front Immunol. 2018;9:862.
Negahdaripour M, Nezafat N, Eslami M, Ghoshoon MB, Shoolian E, Najafipour S, et al. Structural vaccinology considerations for in silico designing of a multi-epitope vaccine. Infect Genet Evol. 2018;58:96–109.
Ohto U, Yamakawa N, Akashi-Takamura S, Miyake K, Shimizu T. Structural analyses of human Toll-like receptor 4 polymorphisms D299G and T399I. J Biol Chem. 2012;287:40611–7.
Berglund NA, Kargas V, Ortiz-Suarez ML, Bond PJ. The role of protein–protein interactions in Toll-like receptor function. Prog Biophys Mol Biol. 2015;119:72–83.
Yan M, Peng J, Jabbar IA, Liu X, Filgueira L, Frazer IH, et al. Activation of dendritic cells by human papillomavirus-like particles through TLR4 and NF-kappaB-mediated signalling, moderated by TGF-beta. Immunol Cell Biol. 2005;83:83–91.
Black M, Trent A, Tirrell M, Olive C. Advances in the design and delivery of peptide subunit vaccines with a focus on Toll-like receptor agonists. Expert Rev Vaccines. 2010;9:157–73.
Peters GH, Bywater RP. Computational analysis of chain flexibility and fluctuations in Rhizomucor miehei lipase. Protein Eng. 1999;12:747–54.
Bhowmick S, Mazumdar T, Ali N. Vaccination route that induces transforming growth factor beta production fails to elicit protective immunity against Leishmania donovani infection. Infect Immun. 2009;77:1514–23.
Wilson ME, Jeronimo SM, Pearson RD. Immunopathogenesis of infection with the visceralizing Leishmania species. Microb Pathog. 2005;38:147–60.
Ekkens MJ, Shedlock DJ, Jung E, Troy A, Pearce EL, Shen H, et al. Th1 and Th2 cells help CD8 T-cell responses. Infect Immun. 2007;75:2291–6.
Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–9.
Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–6.
Whitmire JK, Tan JT, Whitton JL. Interferon-gamma acts directly on CD8+ T cells to increase their abundance during virus infection. J Exp Med. 2005;201:1053–9.
Sercan O, Stoycheva D, Hammerling GJ, Arnold B, Schuler T. IFN-gamma receptor signaling regulates memory CD8+ T cell differentiation. J Immunol. 2010;184:2855–62.
Huang H, Hao S, Li F, Ye Z, Yang J, Xiang J. CD4+ Th1 cells promote CD8+ Tc1 cell survival, memory response, tumor localization and therapy by targeted delivery of interleukin 2 via acquired pMHC I complexes. Immunology. 2007;120:148–59.
Carvalho LH, Sano G, Hafalla JC, Morrot A, Curotto de Lafaille MA, Zavala F. IL-4-secreting CD4+ T cells are crucial to the development of CD8+ T-cell responses against malaria liver stages. Nat Med. 2002;8:166–70.
Bazhan SI, Antonets DV, Karpenko LI, Oreshkova SF, Kaplina ON, Starostina EV, et al. In silico designed ebola virus T-cell multi-epitope DNA vaccine constructions are immunogenic in mice. Vaccines. 2019;7:2.
Mustafa AS. In silico analysis and experimental validation of Mycobacterium tuberculosis-specific proteins and peptides of Mycobacterium tuberculosis for immunological diagnosis and vaccine development. Med Princ Pract. 2013;22(Suppl. 1):43–51.
Moreira DS, Murta SMF. Involvement of nucleoside diphosphate kinase b and elongation factor 2 in Leishmania braziliensis antimony resistance phenotype. Parasit Vectors. 2016;9:641.
Kushawaha PK, Gupta R, Sundar S, Sahasrabuddhe AA, Dube A. Elongation Factor-2, a Th1 stimulatory protein of Leishmania donovani, generates strong IFN-γ and IL-12 response in cured Leishmania-infected patients/hamsters and protects hamsters against Leishmania challenge. J Immunol. 2011;187:6417–27.
Schluter A, Wiesgigl M, Hoyer C, Fleischer S, Klaholz L, Schmetz C, et al. Expression and subcellular localization of cpn60 protein family members in Leishmania donovani. Biochim Biophys Acta. 2000;1491:65–74.
Folgueira C, Requena JM. A postgenomic view of the heat shock proteins in kinetoplastids. FEMS Microbiol Rev. 2007;31:359–77.
Agallou M, Athanasiou E, Samiotaki M, Panayotou G, Karagouni E. Identification of immunoreactive Leishmania infantum protein antigens to asymptomatic dog sera through combined immunoproteomics and bioinformatics analysis. PLoS ONE. 2016;11:e0149894.
Rey-Ladino JA, Joshi PB, Singh B, Gupta R, Reiner NE. Leishmania major: molecular cloning, sequencing, and expression of the heat shock protein 60 gene reveals unique carboxy terminal peptide sequences. Exp Parasitol. 1997;85:249–63.
Chawla B, Jhingran A, Singh S, Tyagi N, Park MH, Srinivasan N, et al. Identification and characterization of a novel deoxyhypusine synthase in Leishmania donovani. J Biol Chem. 2010;285:453–63.
Duarte MC, Lage DP, Martins VT, Costa LE, Lage LMR, Carvalho AMRS, et al. A vaccine combining two Leishmania braziliensis proteins offers heterologous protection against Leishmania infantum infection. Mol Immunol. 2016;76:70–9.
Duarte MC, Lage DP, Martins VT, Costa LE, Carvalho A, Ludolf F, et al. A vaccine composed of a hypothetical protein and the eukaryotic initiation factor 5a from Leishmania braziliensis cross-protection against Leishmania amazonensis infection. Immunobiology. 2017;222:251–60.
Lari N, Jalal R, Minuchehr Z, Rajabian Noghondar M. Identifying miltefosine-resistant key genes in protein-protein interactions network and experimental verification in Iranian Leishmania major. Mol Biol Rep. 2019;46:5371–88.
Jakkula P, Qureshi R, Iqbal A, Sagurthi SR, Qureshi IA. Leishmania donovani PP2C: kinetics, structural attributes and in vitro immune response. Mol Biochem Parasitol. 2018;223:37–49.
Guerfali FZ, Laouini D, Guizani-Tabbane L, Ottones F, Ben-Aissa K, Benkahla A, et al. Simultaneous gene expression profiling in human macrophages infected with Leishmania major parasites using SAGE. BMC Genomics. 2008;9:238.
Morales MA, Watanabe R, Dacher M, Chafey P, Osorio y Fortea J, Scott DA, et al. Phosphoproteome dynamics reveal heat-shock protein complexes specific to the Leishmania donovani infectious stage. Proc Natl Acad Sci USA. 2010;107:8381–6.
Rafati S, Gholami E, Hassani N, Ghaemimanesh F, Taslimi Y, Taheri T, et al. Leishmania major heat shock protein 70 (HSP70) is not protective in murine models of cutaneous leishmaniasis and stimulates strong humoral responses in cutaneous and visceral leishmaniasis patients. Vaccine. 2007;25:4159–69.
Carrillo E, Crusat M, Nieto J, Chicharro C, Thomas MdC, Martínez E, et al. Immunogenicity of HSP-70, KMP-11 and PFR-2 leishmanial antigens in the experimental model of canine visceral leishmaniasis. Vaccine. 2008;15:1902–11.
Forget G, Gregory DJ, Olivier M. Proteasome-mediated degradation of STAT1α following infection of macrophages with Leishmania donovani. J Biol Chem. 2005;280(34):30542–9.
Dea-Ayuela MA, Rama-Iniguez S, Bolas-Fernandez F. Proteomic analysis of antigens from Leishmania infantum promastigotes. Proteomics. 2006;6:4187–94.
Kolli BK, Kostal J, Zaborina O, Chakrabarty AM, Chang KP. Leishmania-released nucleoside diphosphate kinase prevents ATP-mediated cytolysis of macrophages. Mol Biochem Parasitol. 2008;158:163–75.
König J, Fairlamb AH. A comparative study of type I and type II tryparedoxin peroxidases in Leishmania major. FEBS J. 2007;274:5643–58.
Ramos CS, Yokoyama-Yasunaka JKU, Guerra-Giraldez C, Price HP, Mortara RA, Smith DF, et al. Leishmania amazonensis META2 protein confers protection against heat shock and oxidative stress. Exp Parasitol. 2011;127:228–37.
Achour YB, Chenik M, Louzir H, Dellagi K. Identification of a disulfide isomerase protein of Leishmania major as a putative virulence factor. Infect Immun. 2002;70:3576–85.
Kushawaha PK, Gupta R, Tripathi CDP, Sundar S, Dube A. Evaluation of Leishmania donovani protein disulfide isomerase as a potential immunogenic protein/vaccine candidate against visceral leishmaniasis. PLoS ONE. 2012;7:e35670.
Gerbaba TK, Gedamu L. Cathepsin B gene disruption induced Leishmania donovani proteome remodeling implies cathepsin B role in secretome regulation. PLoS ONE. 2013;8:e79951.
Khoshgoo N, Zahedifard F, Azizi H, Taslimi Y, Alonso MJ, Rafati S. Cysteine proteinase type III is protective against Leishmania infantum infection in BALB/c mice and highly antigenic in visceral leishmaniasis individuals. Vaccine. 2008;26:5822–9.
Rochette A, Raymond F, Ubeda J-M, Smith M, Messier N, Boisvert S, et al. Genome-wide gene expression profiling analysis of Leishmania major and Leishmania infantum developmental stages reveals substantial differences between the two species. BMC Genomics. 2008;9:255.
Patino LH, Muskus C, Ramirez JD. Transcriptional responses of Leishmania (Leishmania) amazonensis in the presence of trivalent sodium stibogluconate. Parasit Vectors. 2019;12:348.
Angerer H. The superfamily of mitochondrial Complex1_LYR motif-containing (LYRM) proteins. Biochem Soc Trans. 2013;41:1335–41.
Joshi PB, Kelly BL, Kamhawi S, Sacks DL, McMaster WR. Targeted gene deletion in Leishmania major identifies leishmanolysin (GP63) as a virulence factor. Mol Biochem Parasitol. 2002;120:33–40.
Bhowmick S, Ravindran R, Ali N. gp63 in stable cationic liposomes confers sustained vaccine immunity to susceptible BALB/c mice infected with Leishmania donovani. Infect Immun. 2008;76:1003–15.
Saini S, Ghosh AK, Das S, Singh R, Abhishek K, Verma S, et al. Reduced pathogenicity of fructose-1,6-bisphosphatase deficient Leishmania donovani and its use as an attenuated strain to induce protective immunogenicity. Vaccine. 2018;36:1190–202.
Gannavaram S, Sharma P, Duncan RC, Salotra P, Nakhasi HL. Mitochondrial associated ubiquitin fold modifier-1 mediated protein conjugation in Leishmania donovani. PLoS ONE. 2011;6:e16156.
Garg G, Ali V, Singh K, Gupta P, Ganguly A, Sahasrabuddhe AA, et al. Quantitative secretome analysis unravels new secreted proteins in amphotericin B resistant Leishmania donovani. J Proteomics. 2019;207:103464.
Das A, Ali N. Correction: combining cationic liposomal delivery with MPL-TDM for cysteine protease cocktail vaccination against Leishmania donovani: evidence for antigen synergy and protection. PLoS Negl Trop Dis. 2015;9:e0004185.
Descoteaux A, Avila HA, Zhang K, Turco SJ, Beverley SM. Leishmania LPG3 encodes a GRP94 homolog required for phosphoglycan synthesis implicated in parasite virulence but not viability. EMBO J. 2002;21:4458–69.
Martins TVF, Zeraik AE, Alves NO, de Oliveira LL, de Oliveira Mendes TA, DeMarco R, et al. Lipophosphoglycan 3 From Leishmania infantum chagasi binds heparin with micromolar affinity. Bioinform Biol Insights. 2018;12:1177932218763363.
Abdian N, Gholami E, Zahedifard F, Safaee N, Rafati S. Evaluation of DNA/DNA and prime-boost vaccination using LPG3 against Leishmania major infection in susceptible BALB/c mice and its antigenic properties in human leishmaniasis. Exp Parasitol. 2011;127:627–36.
Denise H, Poot J, Jimenez M, Ambit A, Herrmann DC, Vermeulen AN, et al. Studies on the CPA cysteine peptidase in the Leishmania infantum genome strain JPCM5. BMC Mol Biol. 2006;7:42.
Acknowledgements
The publication of this study has been sponsored by Georg-August-Universität Göttingen.
Funding
This study did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
MAAK conceptualized and designed the study, analyzed the data and drafted the manuscript. JQA, KF and RC performed the screening, curated the data and helped in visualization of the results. PG and FH validated the results and edited the manuscript. AAEW and DM supervised the study and critically reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Text S1.
Methodological details. Reasoning and description of tools used in the study.
Additional file 2: Data S1.
Database of screened proteins from proteomic studies, and of MHC I and MHC II molecules considered for epitope prediction.
Additional file 3: Figure S1.
Proposed vaccine construct. The peptide adjuvant precedes CTL (H1) and HTL (H2) epitopes. Non-specific CTL epitopes and IL-10 inducing HTL epitopes are underlined in black and blue, respectively.
Additional file 4: Figure S2.
Ramachandran plot of the refined structure of the vaccine construct.
Additional file 5: Table S1.
Account of conformational epitopes in the vaccine construct.
Additional file 6: Figure S3.
In silico restriction cloning of vaccine construct. The vaccine coding region is colored red and the green arrow underneath indicates direction of transcription of open reading frame.
Additional file 7: Figure S4.
Simulation quality analysis. a The plot of thermodynamic properties as a function of simulation time over a period of 10 ns. b Tabular summary of thermodynamic properties. Abbreviations: T.E, total energy; P.E, potential energy.
Additional file 8: Figure S5.
Bond re-arrangement at the C-terminal domain of TLR4-TLR4* junction. a Vaccine-unbound interface. b Vaccine-bound interface (post-simulation). Hydrogen bonds are represented as conventional bonds. Interacting surface of TLR4 (encircled) highlights residual charges ranging from positive (blue) to negative (magenta). Inset pictures (right) indicate a transition in the mode of interaction between His458 and His458*.
Additional file 9: Figure S6.
Simulated immune response following hypothetical immunization. a Immunoglobulin production. b B cell response and memory development. c TH (helper T) cell population per state. d TC (cytotoxic T) cell population per state. e Cytokine levels at regular dose intervals for 12 doses. The inset plot indicates the level of leukocyte growth factor (IL-2) and the potential for clonal expansion (D) after each dose.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Khan, M.A.A., Ami, J.Q., Faisal, K. et al. An immunoinformatic approach driven by experimental proteomics: in silico design of a subunit candidate vaccine targeting secretory proteins of Leishmania donovani amastigotes. Parasites Vectors 13, 196 (2020). https://doi.org/10.1186/s13071-020-04064-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04064-8