
Abstract
Background
Snails of the genus Biomphalaria are intermediate hosts of Schistosoma mansoni, the causative agent of the human intestinal schistosomiasis. Two Biomphalaria species (Biomphalaria pfeifferi and Biomphalaria camerunensis) are involved in the transmission in Cameroon, where the disease is present nationwide. However, difficulty in the identification of both vectors impedes proper assessment of the epidemiological burden caused by each species. To overcome this issue, we designed a PCR-based molecular diagnostic tool to improve the identification of these species.
Methods
We analyzed the internal transcribed spacer 2 (ITS2) region of Biomphalaria ribosomal DNA (rDNA) using polymerase chain reaction amplification (PCR) and restriction fragment length polymorphism (RFLP).
Results
The amplification of the ITS2 region of Biomphalaria snails resulted in a 490 bp fragment and produced two profiles for each species after digestion with the restriction enzyme Hpa II. The profile 1 (Bc-HpaII-1: 212-bp and 139-bp bands) for B. camerunensis, was common in all the sampling points; the profile 2 (Bc-HpaII-2: 212-bp and 189-bp bands), was only observed in the Lake Monoun Njindoun sampling site. Biomphalaria pfeifferi profile 1 (Bpf-HpaII-1: 211-bp and 128-bp bands) was common in most of B. pfeifferi sampling points; the profile 2 (Bpf-HpaII-2: 289-bp and 128-bp bands) was only observed in Mokolo (Far North Cameroon).The second restriction enzyme TaqαI, revealed three band profiles, Bc-TaqαI-1 (243-bp, 136-bp and 118-bp bands) and Bc-TaqαI-2 (244-bp, 136-bp and 99-bp) for B. camerunensis and Bpf-TaqαI-1 (242-bp, 135-bp and 107-bp bands) for B. pfeifferi. Sequencing analysis revealed the occurrence of six haplotypes for B. camerunensis and three haplotypes for B. pfeifferi. The level of gene flow was low and the Biomphalaria populations were not in demographic expansion according to neutrality tests (Tajima’s D and Fu’s Fs).
Conclusions
The PCR-RFLP technique revealed genetic diversity in Biomphalaria snails, and the combination with the morphological method could improve the identification of B. pfeifferi and B. camerunensis in Cameroon. This could help focus on the infection to evaluate the transmission risk with respect of the different species and to develop efficient and cost-effective control measures.

Similar content being viewed by others


Background
Schistosomiasis, a chronic disease caused by trematodes of the genus Schistosoma, is an important public health problem in many tropical and subtropical areas of the world [1,2,3]. Indeed, transmission of schistosomiasis is proven in 78 countries, and in 2018 the total number of people in need of preventive chemotherapy was 229.2 million, of which 124.4 million were school-aged children [4].
In Cameroon in 2009, more than 5 million people were at risk of infection with schistosomiasis, and 2 million were known to be currently infected [5]. Intestinal schistosomiasis is the most widely distributed type of schistosomiasis in the equatorial area. This type is caused by the presence in the intestinal vascular system of Schistosoma mansoni Sambon, 1907. The Cameroon National Control Programme against schistosomiasis has adopted a periodic large-scale deworming campaign using praziquantel, associated with health education, as control measures. This strategy has considerably reduced morbidity and mortality levels among affected populations. However, most of the old foci remain active, and new foci are observed, likely due to the migration of the infected individuals from endemic areas to schistosomiasis free sites where intermediate hosts are present [6,7,8,9]. To overcome this situation and accelerate the progress towards elimination, the Cameroon strategic plan was revised, including the implementation of other control measures such as: (i) access to clean water; (ii) sanitation improvement; and (iii) control of intermediate host populations [10, 11]. This latter control approach requires a better understanding of the distribution, biology and population dynamics of intermediate hosts of schistosomes.
In Cameroon, S. mansoni transmission involves two snail species, Biomphalaria camerunensis (Boettger, 1941) that has also been recorded in central Africa (from Ghana at the West to the Democratic Republic of Congo at the East) [11, 12] and Biomphalaria pfeifferi (Krauss, 1848) that is also present throughout sub-Saharan Africa and also in one country in North Africa (Algeria) [12]. Biomphalaria pfeifferi is widely distributed throughout the country and is known as the main intermediate host of S. mansoni. Conversely, B. camerunensis has until now only been reported under the 6° north latitude and appears to play a minor role in the transmission of S. mansoni [13]. However, recent compatibility studies highlighted a relatively high susceptibility and cercarial emission rate in some B. camerunensis populations as high as those observed among B. pfeifferi, suggesting that they might play a more important role in the transmission of S. mansoni [14].
Biomphalaria pfeifferi and B. camerunensis are very similar morphologically, which makes their routine identification difficult. The last study on the distribution of these snail species in Cameroon was performed nearly 30 years ago and the results are not reflective to the current situation, as there have been significant ecological and human changes in the respective foci. A clear distinction of these Biomphalaria spp. snails might help sharpen disease mapping and evaluate the transmission risk in localities where they are found. For many decades, the identification of snail intermediate hosts had been mainly based on the comparison of morphological and morphometric characters of shells [15,16,17]. Although the shell morphology remains an important taxonomic instrument [18, 19], studies employing alternative tools such as molecular identification seem to be more accurate for closely related species [20,21,22,23,24,25]. In the last two decades, molecular tools have been evaluated for both snail identification and phylogenetic studies [26,27,28,29]; the polymerase chain reaction and restriction fragment length polymorphism (PCR-RFLP) analysis of the internal transcribed spacer (ITS) region of rDNA and the analysis of cytochrome c oxidase subunit 1 region of mitochondrial DNA have proven to be cheaper and powerful for the identification of several Biomphalaria species [30,31,32,33,34,35]. More recently, the PCR-RFLP technique has been used to distinguish Biomphalaria species in Brazil and it appears to be an alternative molecular tool to their morphological identification [36].
The aim of this study was to test the efficiency of the PCR-RFLP protocol developed for identification of Biomphalaria species in South America [34, 36], for the separation of the two main Biomphalaria spp. currently found in Cameroon. Furthermore, sequencing of rDNA fragments enabled us to assess the genetic diversity and population structure of both species in Cameroon.
Methods
Snail sampling sites and collection method
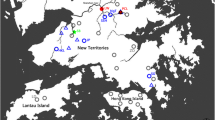
Snail sampling was conducted from July to August 2017 in eighteen sites all located in five administrative Regions of Cameroon: nine sites in the Centre, two in the South, three in the West, two in the East and one in the Far North (Table 1, Fig. 1). Sites mentioned in previous studies were visited [13, 14], while new sites were chosen in additional streams favourable to the survival of molluscs. Snails were collected using a long-handled dip net by systematically combing the aquatic vegetation and identified using morphological criteria previously described by Brown [37]. For each sample, some individuals were taken at random and kept alive for further parasites screening, while others were individually fixed in labelled tubes containing 95% ethanol and transferred to the Parasitology and Ecology Laboratory of the University of Yaoundé I, Cameroon, where they were stored at − 20 °C until processing for molecular studies.

Map showing the different collection sites. Red dots indicate collection sites
DNA extraction
Total DNA was extracted from the foot of 470 Biomphalaria snails (around thirty individuals per sampling site) using a cethyl trimethyl ammonium bromide (CTAB)-based protocol as described by Maniatis et al. [38]. Samples were thawed and air-dried; briefly, each snail foot was homogenized with a pestle in 600 µl of CTAB buffer (CTAB 2%; 0.1 M Tris-HCl, pH 8; 0.02 M EDTA, pH 8; 1.4 M NaCl). The homogenized mixture was incubated at 60 °C for 30 min. After this incubation, the DNA was extracted using 600 µl of chloroform/isoamylic alcohol (24/1; V/V) mixture, and then precipitated with isopropanol (V/V). After a centrifugation at 10,000×g for 15 min, the DNA pellet was washed with 70% ethanol, air-dried, and re-suspended in sterile water. DNA samples were stored at − 20 °C until PCR amplification.
PCR amplification of the ITS2 region of Biomphalaria spp
The ITS2 region of Biomphalaria spp. was amplified by PCR, using the primers ITS2F (5ʹ-CGT CCG TCT GAG GGT CGG TTT GC-3ʹ) [30] and ETTS1 (5ʹ-TGC TTA AGT TCA GCG GGT-3ʹ) [39] hybridizing in the conserved areas of the 5.8S and 28S ribosomal genes, respectively. The PCR amplification was undertaken in a final reaction volume of 20 μl containing 2 µl of extracted DNA, 2 µl of TBE PCR buffer (10×), 0.8 μl of each primer (10 µM), 0.4 μl of dNTPs mixture (10 mM), 0.06 μl of Taq DNA polymerase (5 U/µl) and 13.94 μl of sterile water. DNA was amplified in a Techne TC-412 (Bibby Scientific Limited, Staffordshire, UK) thermal cycler under the following cycling conditions: initial denaturation at 95 °C for 3 min 30 s, followed by 35 cycles each at 95 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s, and a final extension step at 72 °C for 10 min. The amplicons were resolved on 2% agarose gels stained with ethidium bromide and visualized under UV light.
Purification, sequencing and in silico RFLP
Purification of some amplified DNA samples was made using the enzymatic PCR clean up method with Exonuclease I (ExoI) and shrimp alkaline phosphatase (SAP) (New England Biolabs, Boston, MA, USA) which offer an easy way to remove the remaining primers and dNTPs left from the PCR reaction. The DNA concentration was then measured using a NanoDrop™ spectrophotometer (Thermo Fisher Scientific, Wilmington, USA) and samples complying with the minimal concentration (ng/µl) were directly sequenced commercially (GENEWIZ, Liverpool, UK). The sequences obtained were visualized and edited using BioEdit software, then confirmed via alignment to similar sequences using the nucleotide BLAST tool on GenBank. Once confirmed, the putative B. pfeifferi and B. camerunensis sequences were subjected to in silico RFLP using the Restriction Mapper online tool (http://www.restrictionmapper.org/) to choose restriction enzymes which could clearly distinguish the two Biomphalaria species.
Digestion and revelation of profiles
The two enzymes which exhibited good digestion profiles after in silico RFLP (HpaII and TaqαI) were used for the digestion of the PCR products. The reaction was carried out in a final volume of 25 µl, containing 2 μl of amplified DNA, 1 μl of restriction enzyme, 5 μl of the manufacturerʼs buffer and 17 μl of sterile water, at 37 °C for 2 h. After digestion, the products were resolved on 8% polyacrylamide gel stained with ethidium bromide and visualized under UV light.
Phylogenetic and population structure analyses
All the sequences were aligned in BioEdit with the ClustalW [40] algorithm for phylogenetic analyses. The evolutionary history was inferred using the Maximum Likelihood method based on the Jukes-Cantor model [41]. Initial trees for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood approach, and then selecting the topology with highest log likelihood value. Bootstrap values supporting the nodes were computed as the percentage of trees in which associated taxa clustered together. Evolutionary analyses were conducted in MEGA7 [42].
Estimation of genetic diversity, including polymorphic sites (S), haplotype diversity (Hd), nucleotide diversity (π), were performed using DnaSP5.10.01 [43]. Haplotype diversity was considered as the probability that two randomly sampled alleles are different, and nucleotide diversity was considered as the average number of nucleotide differences per site in pairwise comparisons among DNA sequences [44]. Genetic differentiation among populations (FST), Fu’s Fs statistics and Tajima’s D [45] values were also estimated using the software DnaSP 5.10.01 [43]. To better visualize the phylogenetic relationships among haplotypes, a haplotype network was constructed among the defined haplotypes using TCS v1. 21 software [46] and mutations steps were generated using DnaSP 5.10.01 [43] and MEGA7 [41].
Results
ITS2 amplification and sequencing
The PCR amplification of Biomphalaria ITS2 region resulted in a product of 490 bp. After sequencing, editing and aligning to a reference sequence, four amplicon sizes were obtained: 478 bp and 485 bp for B. camerunensis; 486 bp and 497 bp for B. pfeifferi.
Digestion and revelation of profiles
The digestion of PCR products using restriction enzymes (HpaII and TaqαI) revealed different profiles for the two Biomphalaria species.
Biomphalaria camerunensis individuals exhibited two profiles, each with two bands: profile 1 (Bc-HpaII-1: 212-bp and 139-bp bands) was common in all the sampling points; profile 2 (Bc-HpaII-2: 212-bp and 189-bp bands) was only observed in the Lake Monoun Njindoun sampling site in the West Region and was displayed by ~ 90% of the individuals sampled there. Individuals of B. pfeifferi, also presented two profiles, each with two bands: profile 1 (Bpf-HpaII-1: 211-bp and 128-bp bands), was common in all B. pfeifferi sampling points; whereas profile 2 (Bpf-HpaII-2: 289-bp and 128-bp bands) was only observed in Mokolo (Far North Cameroon) and was present in 95% of the sampled individuals (Fig. 2).

Polyacrylamide gel showing RFLP profiles obtained after the digestion with HpaII of Biomphalaria ITS2. Lane 1: B. pfeifferi profile 1; Lanes 2–5: B. pfeifferi profile 2; Lane 6: B. camerunensis profile 1; Lane M: molecular weight marker (100 bp); Lanes 7–10: B. camerunensis profile 1; Lane 11: no sample; Lanes 12–14: B. camerunensis profile 1; Lanes 15, 16: B. camerunensis profile 2; Lanes 17–19: B. pfeifferi profile 1
Figure 3 shows the different profiles obtained for the two Biomphalaria species after the digestion of ITS2 amplicons with TaqαI enzyme. Individuals of the species B. camerunensis showed two profiles, each represented on the gel by three bands: profile 1 (Bc-TaqαI-1: 243-bp, 136-bp and 118-bp bands) was common in all the sampling sites; while profile 2 (Bc-TaqαI-2: 244-bp, 136-bp and 99-bp bands) was only observed in samples from Lake Monoun Njindoun (these were the same individuals that displayed profile 2 with the HpaII enzyme). All the individuals of B. pfeifferi presented a three-band profile (242 bp, 135 bp and 107 bp).

Polyacrylamide gel showing RFLP profiles obtained after the digestion with TaqαI of Biomphalaria ITS2. Lanes 1, 2: B. camerunensis profile1; Lane 3: B. camerunensis profile 2; Lane 4: B. camerunensis profile1; Lanes 5, 6: B. camerunensis profile 2; Lanes 7–10: B. camerunensis profile1; Lane 11: B. Pfeifferi; Lane 12: molecular weight marker (25 bp); Lanes 13–16: B. pfeifferi
Phylogenetic analyses
The analysis involved 44 nucleotide sequences of 424 bp plus two reference nucleotide sequences, one for B. pfeifferi from the South-East of Nigeria [47] and one for B. camerunensis from Sangmelima in Cameroon [48]. A clear segregation of the two Biomphalaria species (bootstrap 100) was observed. One cluster included B. pfeifferi from Mokolo in the Far North Region, Nkolbisson and Ngoa Ekelle in the Centre Region, and another cluster included B. camerunensis from Mounassi in the Centre Region, Sangmelima in the South Region, Monou II in the East Region, Monoun Njindoun and Memom in the West Region (Fig. 4). A total of nine haplotypes were detected in the different studied populations including six for B. camerunensis and three for B. pfeifferi. In general, haplotypes of each species were strongly related with a maximum of 5 and 8 mutational steps in B. pfeifferi and B. camerunensis populations, respectively (Fig. 5). The haplotype H4 found only in Monoun Njindoun in the West Region was the only haplotype isolated from the other haplotypes of B. camerunensis and was different from the reference sequence from Cameroon with 22 mutational steps (Fig. 6). When comparing the haplotypes of the two species, 30 mutational steps were recorded, confirming the segregation of these species (Fig. 6).

Maximum likelihood phylogenetic tree constructed with ITS2 sequences. AY030367: reference ITS accession number of B. camerunensis; KX644870: reference ITS accession number of B. pfeifferi

Haplotype networks for B. camerunensis (a) and B. pfeifferi (b). H1-H9: different haplotypes; the sizes of the circles are proportional to the haplotype frequencies

Different mutational steps of haplotypes obtained and accession numbers of their sequences on GenBank. AY030367, reference ITS accession number of B. camerunensis; KX644870, reference ITS accession number of B. pfeifferi
Genetic diversity of Biomphalaria populations
Of the eight populations analysed, six did not display any polymorphism (all the individual having the same haplotype). Haplotype and nucleotide diversities of B. camerunensis samples were 0.798 and 0.008, respectively, while in B. pfeifferi samples, they were 0.473 and 0.004, respectively. Assessment of demographic expansion of populations, based on neutrality tests gave a negative value for Tajima’s D in Mokolo (− 0.796) and a positive value (1.718) in Monou II and these values were not significant; Fuʼs Fs test values were positive in samples of the two species (Table 2), showing that populations under study were not in expansion.
Population structure
Genetic structure of Biomphalaria spp. populations was analyzed by pairwise FST among five populations of B. camerunensis and three populations of B. pfeifferi. FST values were high and significant (P < 0.01) between all B. camerunensis pairs from Sangmelima, Monou II, Mounoun Njindoun, and Memom but low and non-significant between the two populations from Sangmelima in the South Region and Monou II in the East Region (P = 0.25) (Table 3). In B. pfeifferi populations, FST values were only significant between the samples from the Center Region (Nkolbisson, Ngoa-Ekelle) and the one from the Far North Region (Mokolo) (P < 10−3) (Table 4).
Discussion
Morphological identification of S. mansoni vector species of the genus Biomphalaria is difficult because shells are very similar in shape and color. Moreover, morphological and anatomical characters were found to vary among populations as shown in Sangmelima and Nkoteng by Peka Sangou (2010, personal communication) and Mvogo Ndongo (2012, personal communication). To scale up the fight against schistosomiasis and achieve elimination, an anti-vector campaign has recently been advocated in addition to MDA which for a very long time had been the only prevention approach [49]. The identification of the two sibling Biomphalaria spp. is tricky, but useful for the establishment of baseline data, prior to a vector control campaign. To provide more tools to achieve this goal, we designed a diagnostic assay that will facilitate the morphological identification of B. pfeifferi and B. camerunensis in Cameroon and help improve the control of these species.
Taxonomic study
The PCR-RFLP-based protocol used has clearly distinguished B. camerunensis species (profiles Bc-HpaII-1 and Bc-HpaII-2) from B. pfeifferi species (profiles Bpf-HpaII-1 and Bpf-HpaII-2) as was the case for Brazilian Biomphalaria species [35]. The same technique had already proven useful in the identification of other species such as species of the Anopheles gambiae complex [50]. This technique is a promising simple tool which can help to overcome the misidentification of sibling vectors and thus help to understand epidemiological issues. Moreover, the PCR-RFLP technique revealed an intraspecific variation, characterized by the occurrence of two ribotypes in each Biomphalaria species. As no intermediate profile was found between ribotypes, further investigation is needed to check their taxonomical status (subspecies or simple variant). The existence of intraspecific variation is in line with the observation of a high polymorphism in the susceptibility of B. camerunensis populations to S. mansoni [14]. This level of polymorphism is expected in an outbred species such as B. camerunensis [51].
The phylogenetic tree constructed with ITS2 sequences clearly segregated the individuals of the two species, thus confirming their divergence; however, within each species, the two main related branches of the tree correspond to the two different profiles obtained and are indicative of an intraspecific variation. Molecular phylogenies already helped in the classification of several mollusc taxa like bivalves [52], it is then an added value for taxonomical studies.
Molecular diversity and population dynamics
Phylogenetic studies showed that almost all the haplotypes obtained within each species, regardless of their geographical origin, were closely related. Among the five populations of B. camerunensis, Monou II showed the highest haplotype diversity (Hd: 0.600) and a low nucleotide diversity (π: 0.008), which are indicative of a rapid population growth from a small-sized ancestral population in which the time has not been enough to find a strong diversity between haplotypes [53]. The low values of Hd (0.473) and π (0.004) obtained in B. pfeifferi populations are indicative of a severe and prolonged bottleneck; however, more investigations are necessary to confirm these suggestions. In addition, pairwise FST between the samples from the different administrative regions studied were high and significant, showing no signal of gene flow between these populations. Nevertheless, the lack of polymorphism in the genetic marker used could lead to a less accurate estimation of this parameter. The low genetic diversity observed in the studied populations can result from their mating system or from their ecology, since low genetic diversity is severe in subdivided populations with seasonal variation in abundance [1]. Effective population sizes and density variation appear to be important factors in the loss of variability and Biomphalaria are known to have subdivided habitats [54]. In addition, the positive values of the Tajima’s D and Fu’s Fs neutrality tests show that all the populations studied are not currently expanding. This information shows that vector control performed now will lead to a rapid population density decrease in Biomphalaria spp., but other studies using other nuclear or mitochondrial polymorphic markers are needed to provide more accurate demographic parameters.
Conclusions
The aim of this study was to provide a simple tool that can help to easily and accurately identify B. pfeifferi and B. camerunensis from Cameroon and provide some initial data on their genetic diversity. The digestion of ITS2 DNA fragments with HpaII and TaqαI allowed to clearly distinguish the two Biomphalaria species present in Cameroon, showing that the protocol used constitutes a useful alternative that can help to accurately identify the two species and thus help in future vector control campaigns. The genetic variability observed within B. camerunensis is in line with the ability of some of its populations to transmit schistosomes at the same level as the former known principal host, B. pfeifferi. Further investigations are required to formally confirm the link between the vector competence of some B. camerunensis populations and their genetic background.
Availability of data and materials
Data supporting the conclusions of the article are included within the article. The newly generated sequences were submitted to the GenBank database under the accession numbers MN064840-MN064845 (Biomphalaria camerunensis) and MN064846-MN064848 (Biomphalaria pfeifferi). The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- MDA:
-
Mass drug administration
- DNA:
-
Deoxyribonucleic acid
- ITS:
-
Internal transcribed spacer
- PCR:
-
Polymerase chain reaction
- RFLP:
-
Restriction fragment length polymorphism
References
Brown DS. Freshwater snails of Africa and their medical importance. London: Taylor & Francis Ltd; 1980. p. 487.
Gryseels B. The relevance of schistosomiasis for public health. Trop Med Parasitol. 1989;40:134–42.
Jordan P, Webbe G, Sturrock RF. Human schistosomiasis. 3rd ed. Walligford: CAB International; 1993.
WHO. Global Health Observatory (GHO) data: Schistosomiasis. Geneva: World Health Organisation; 2020. http://www.who.int/neglected_diseases/LOP_WHO_meeting_investmentimpactcontrolNTD.pdf. Accessed 16 Mar 2020.
Tchuem Tchuente LA, NʼGoran EK. Schistosomiasis and soil-transmitted helminthiasis control in Cameroon and Côte dʼIvoire: implementing control on a limited budget. Parasitology. 2009;136:1739–45.
Steinmann P, Keiser J, Bos R, Tanner M, Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect Dis. 2006;6:411–25.
Tchuem Tchuenté LA, Kamwa Ngassam RI, Sumo L, Ngassam P, Dongmo NC, Luogbou Nzu DD, et al. Mapping of schistosomiasis and soil-transmitted helminthiasis in the regions of Centre, East and West Cameroon. PLoS Negl Trop Dis. 2012;6:e1553.
Nkengazong L, Njiokou F, Teukeng F, Enyong P, Wanji S. Reassessment of endemicity level of urinary schistosomiasis in the Kotto-Barrombi focus (South-West Cameroon) and impact of mass drug administration (MDA) on the parasitic indices. J Cell Anim Biol. 2009;3:159–64.
Simões LF, Camargo EAF, Bastos LD, Neves MF, De Carvalho JF, et al. Susceptibility of Argentinean Biomphalaria tenagophila and Biomphalaria straminea to infection by Schistosoma mansoni and the possibility of geographic expansion of mansoni schistosomiasis. Rev Soc Bras Med Trop. 2013;46:611–6.
WHO. Schistosomiasis: progress report 2001–2011, strategic plan 2012–2020. Geneva: World Health Organisation. 2013. https://apps.who.int/iris/handle/10665/78074. Accessed 16 Mar 2020.
WHO. Field use of molluscicides in schistosomiasis control programmes. An operational manual for programme managers. Geneva: World Health Organisation. 2017. http://apps.who.int/iris. Accessed 16 Mar 2020.
Stensgaard AS, Utzinger J, Vounatsou P, Hürlimann E, Schur N, Saarnak CF, et al. Large-scale determinants of intestinal schistosomiasis and intermediate host snail distribution across Africa: does climate matter? Acta Trop. 2013;128:378–90.
Greer GJ, Mimpfoundi R, Malek AE, Joky A, Ngonseu E, Ratard RC. Human schistosomiasis in Cameroon. II. Distribution of the snail hosts. Am J Trop Med Hyg. 1990;42:573–80.
Kengne-Fokam AC, Nana-Djeunga HC, Bagayan M, Njiokou F. Biomphalaria camerunensis as a viable alternative intermediate host for Schistosoma mansoni in southern Cameroon. Parasit Vectors. 2018;11:181.
Boettger CR. Diagnose einer neuen Posthornschnecke aus Kamerun. Archiv Molluskenkunde. 1941;73:121–3.
Mandahl-barth G. Intermediate hosts of Schistosoma African Biomphalaria and Bulinus. 2. Bull World Health Organ. 1957;17:1–65.
Wright CA. The freshwater gastropod molluscs of west Cameroon. Bull Br Mus Nat Hist Zool. 1965;13:75–98.
Dung BT, Doanh PN, The DT, Loan HT, Losson B, Caron Y. Morphological and molecular characterization of lymnaeid snails and their potential role in transmission of Fasciola spp. in Vietnam. Korean J Parasitol. 2013;51:657–62.
Pesic V, Gloer P. A new freshwater snail genus (Hydrobiidae, Gastropoda) from Montenegro, with a discussion on gastropod diversity and endemism in Skadar Lake. ZooKeys. 2013;281:69–90.
Gasser RB, Stewart LE, Speare R. Genetic markers in ribosomal DNA from hookworm identification. Acta Trop. 1996;62:15–21.
Cunningham CO. Species variation within the internal transcribed spacer (ITS) region of Gyrodactylus (Monogenea: Gyrodactylidae) ribosomal RNA genes. J Parasitol. 1997;83:215–9.
Hope M, McManus DP. Genetic variations in geographically isolated populations and subspecies of Oncomelania hupensis determined by a PCR-based RFLP method. Acta Trop. 1994;57:75–82.
Stohard JR, Rollinson D. An evaluation of random amplified polymorphic DNA (RAPD) for the identification and phylogeny of freshwater snail of the genus Bulinus (Gastropoda: Planorbidae). J Moll Stud. 1996;62:165–76.
Stohard JR, Rollinson D. Molecular characterization of Bulinus globosus and B. nasutus on Zanzibar, and an investigation of their roles in the epidemiology of Schistosoma haematobium. Trans R Soc Trop Med Hyg. 1997;91:353–7.
Remigio EA, Blair D. Relationships among problematic North American stagnicoline snails (Pulmonata: Lymnaeidae) reinvestigated using nuclear ribosomal DNA internal transcribed spacer sequences. Can J Zool. 1997;75:1540–5.
Knight M, Brindley PJ, Richards CS, Lewis FA. Schistosoma mansoni: use of a cloned ribosomal RNA gene probe to detect restriction fragment length polymorphisms in the intermediate host Biomphalaria glabrata. Exp Parasitol. 1991;73:285–94.
Strahan K, Kane RA, Rollinson D. Development of cloned DNA probes for the identification of snail intermediate hosts within the genus Bulinus. Acta Trop. 1991;48:117–26.
Vidigal THDA, Neto ED, Simpson AJG, Carvalho OS. A low stringency polymerase chain reaction approach to identification of Biomphalaria glabrata and Biomphalaria tenagophila intermediate snail hosts of Schistosoma mansoni in Brazil. Mem Inst Oswaldo Cruz. 1996;91:739–44.
Pires ER, Vidigal THDA, Teles HMS, Simpson AJG, Carvalho OS. Specific identification of Biomphalaria tenagophila and Biomphalaria occidentalis populations by the low stringency polymerase chain reaction. Mem Inst Oswaldo Cruz. 1997;92:101–6.
Caldeira RL, Vidigal THDA, Martinela L, Simpson AJG, Carvalho OS. Identification of planorbids from Venezuela by polymerase chain reaction amplification and restriction fragment length polymorphism (PCR-RFLP). Mem Inst Oswaldo Cruz. 2000;95:171–7.
Caldeira RL, Vidigal THDA, Paulinelli ST, Simpson AJG, Carvalho OS. Molecular identification of similar species of the genus Biomphalaria (Mollusca: Planorbidae) determined by PCR-RFLP. Mem Inst Oswaldo Cruz. 1998;93:219–25.
Spatz L, Vidigal THDA, Caldeira RL, Dias Neto E, Cappa SMG, Carvalho OS. Study of Biomphalaria tenagophila tenagophila, B. t. guaibensis and B. occidentalis by polymerase chain reaction amplification and restriction enzyme digestion of the ribosomal RNA intergenic spacer regions. J Molluscan Stud. 1999;65:143–9.
Vidigal THDA, Caldeira RL, Simpson AJ, Carvalho OS. Further studies on the molecular systematics of Biomphalaria snails from Brazil. Mem Instituto Oswaldo Cruz. 2000;95:57–66.
Vidigal THDA, Montresor LC, Simpson AJG, Carvalho OS. Polymerase chain reaction and restriction fragment length polymorphism of cytochrome oxidase I used for differentiation Brazilian Biomphalaria species intermediate host of Schistosoma mansoni. Mem Inst Oswaldo Cruz. 2002;97:47–52.
Vidigal THDA, Spatz L, Nunes DN, Simpson AJG, Carvalho OS, Dias Neto E. Biomphalaria spp: identification of the intermediate snail hosts of Schistosoma mansoni by polymerase chain reaction amplification and restriction enzyme digestion of the ribosomal RNA gene intergenic spacer. Exp Parasitol. 1998;89:180–7.
Vidigal THDA, Magalhães KG, Carvalho ODS. Polymerase chain reaction and restriction fragment length polymorphism analysis of the ITS2 region for differentiation of Brazilian Biomphalaria intermediate hosts of the Schistosoma mansoni. Rev Soc Bras Med Trop. 2004;37:351–3.
Brown D. Freshwater snails of Africa and their medical importance. 2nd ed. London: Taylor & Francis Ltd.; 1994.
Maniatis T, Fritsch E, Sambrook J. Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Publications; 1982.
Kane RA, Rollinson D. Repetitive sequences in the ribosomal DNA internal transcribed spacer of Schistosoma haematobium, Schistosoma intercalatum and Schistosoma mattheei. Mol Biochem Parasitol. 1994;63:153–6.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;4:95–8.
Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian protein metabolism. New York: Academic Press; 1969. p. 21–132.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Librado P, Rozas J. DNASP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Nei M. Molecular evolutionary genetics. New York: Colombia University Press; 1987. p. 512.
Tajima F. Statistical methods for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Clement M, Posada D, Crandall K. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–60.
Okeke OC, Ubachukwu PO. Trematode infections of the freshwater snail Biomphalaria pfeifferi from a south-east Nigerian community with emphasis on cercariae of Schistosoma. J Helminthol. 2017;91:295–301.
DeJong RJ, Morgan JA, Paraense WL, Pointier JP, Amarista M, Ayeh-Kumi PF, et al. Evolutionary relationships and biogeography of Biomphalaria (Gastropoda: Planorbidae) with implications regarding its role as host of the human blood fluke, Schistosoma mansoni. Mol Biol Evol. 2001;18:2225–39.
Rollinson D, Knopp S, Levitz S, Stothard JR, TchuemTchuente LA, Garba A, et al. Time to set the agenda for schistosomiasis elimination. Acta Trop. 2013;128:423–40.
Fanello C, Santolamazza F, Della Torre A. Simultaneous identification of species and molecular forms of the Anopheles gambiae complex by PCR-RFLP. Med Vet Entomol. 2002;16:461–4.
Kengne-Fokam AC, Nana-Djeunga HC, Njiokou F. Mating behaviour in two Schistosoma mansoni intermediate hosts (Biomphalaria pfeifferi and Biomphalaria camerunensis) in Cameroon. Ann Aquac Res. 2016;3:1017.
Adamkewicz SL, Harasewych G, Blake J, Saudek D, Bult CJ. A molecular phylogeny of the Bivalve mollusks. Mol Biol Evol. 1997;14:619–29.
Salducci MD, Martin J, Pech N, Chappaz R, Costedoat C, Gilles A. Deciphering the evolutionary biology of freshwater fish using multiple approaches - insights for the biological conservation of the Vairone (Leuciscus souffia souffia). Conserv Genet. 2004;5:63–77.
Selander RK. Genic variation in natural populations. In: Ayala FJ, editor. Molecular evolution. Massachusetts: Sinauer Associates; 1976. p. 21–45.
Acknowledgements
The authors are thankful to Dr. Nana Djeunga Hugues Clotaire for his assistance during the preparation of the manuscript and the Liverpool school of tropical medicine for providing some laboratory reagents. The authors also thank the editor and the anonymous reviewers for their contribution that have significantly improved the quality of this paper.
Funding
This study was funded by the Parasitology and Ecology Laboratory of the Faculty of Science of the University of Yaoundé 1 and the Centre for Research in Infectious Diseases (CRID) Yaoundé Cameroon. Travel for sampling collection was supported by Mr. Foga Charles Patrick.
Author information
Authors and Affiliations
Contributions
MCTM conceived and designed the study, collected the field data, analyzed the data, and prepared the first draft of the manuscript. TTMT contributed in the study design, performed the data analyses and helped to draft the manuscript. ACKF participated in field data collection and helped to draft the manuscript. DNN analyzed the data. CSW contributed to the data analyses and helped to draft the manuscript. FN conceived and designed the study, provided general supervision at the various stages of the study process and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tchami Mbagnia, M.C., Melachio Tanekou, T.T., Kengne Fokam, A.C. et al. PCR-based molecular identification of two intermediate snail hosts of Schistosoma mansoni in Cameroon. Parasites Vectors 13, 158 (2020). https://doi.org/10.1186/s13071-020-04033-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-04033-1