
Abstract
Background
Two hemoplasma species, Mycoplasma suis and M. parvum, previously known as Eperythrozoon suis and E. parvum, respectively, have been identified in pigs. Swine hemoplasmosis is a global problem, and M. suis infection results in economic losses to pig producers worldwide. This study investigated the frequency and distribution of hemotropic mycoplasmas in pig farms of Korea. As hemoplasmas can be transmitted by ticks, we also analyzed the presence of the tick-borne pathogens Anaplasma spp. and Borrelia spp.
Methods
We screened 1867 samples from 464 pig farms located in four regions of Korea over the period from 2014 to 2018. PCR-positive samples were further analyzed by nucleotide sequencing and phylogenetic analysis of pathogen-specific markers for species identification.
Results
Of the 1867 pigs evaluated in the study, three (0.2%), 51 (2.7%), and one (0.1%) were found to be infected with M. suis, M. parvum, and the novel hemotropic M. haemosuis, respectively; Anaplasma spp. and Borrelia spp. were not detected. The 16S rRNA sequences of M. suis, M. parvum, and the novel hemotropic M. haemosuis were highly similar (99.3–100%, 99.6–100%, and 99.6–100%, respectively) to those of Mycoplasma spp. isolated from other countries. To the best of our knowledge, this is the first nationwide, large-scale study of the molecular detection of Mycoplasma spp. in domestic pigs in Korea.
Conclusions
Our results indicate that Mycoplasma infections are widespread in Korean domestic pigs, and that continuous monitoring and control strategies are required to prevent the spread of hemoplasmas, which, in addition to causing economic losses in the pig industry, pose a potential threat to public health. As transmission routes of hemoplasmas remain unelucidated, additional epidemiological studies are recommended to identify reservoirs and vectors of Mycoplasma spp. in Korea.
Similar content being viewed by others

Background
Hemoplasma refers to a group of hemotropic prokaryotes that lack a cell wall and are uncultivable in vitro [1]. Two hemoplasma species, Mycoplasma suis and M. parvum, previously known as Eperythrozoon suis and E. parvum, respectively, have been previously identified in pigs [2]; these two organisms can be distinguished based on morphology, pathogenicity [3], and mode of interaction with the host.
Mycoplasma suis, a small, rickettsia-like, rod-shaped, extracellular pleomorphic microorganism, which attacks erythrocytes [4] and is the causative agent of hemoplasmosis (previously called eperythrozoonosis) in domestic pigs [5]. This species was previously known as M. haemosuis (basonym E. suis) or “Candidatus M. haemosuis”, indicating the provisional status of an incompletely described taxon [6, 7]. Clinical signs of M. suis infection in pigs vary, but acute infection manifests as icterus and febrile hemolytic anemia and is characterized by low morbidity but high mortality rates [1]. Chronic M. suis infection in pigs causes growth delay and has been associated with low reproductive efficiency and an increased incidence of other infectious diseases [1, 8]. Pigs infected with M. suis are likely to be persistent chronic carriers of the pathogen even after the resolution of clinical signs [9].
Mycoplasma parvum is the last validly published species in the genus Eperythrozoon, E. parvum. This species is not available and there is no assurance that it can be found in a reasonable amount of time [6]. Since all the other known Eperythrozoon species have now been shown to be Mycoplasma species, it seems likely that E. parvum would prove to belong to the genus Mycoplasma. However, there is no direct proof and it cannot be decided when a sample of this bacterium can be found and studied [6]. Clinical signs of M. parvum infection are less well-known, and it is currently believed that this organism exerts relatively low pathogenicity in pigs. It has been shown that M. parvum was not associated with clinical signs even at the peak of bacteremia in intact piglets, and that severe anemia with pyrexia was detected only in splenectomized piglets [3, 10, 11].
Swine hemoplasmosis is a global disease, and M. suis infection causes economic losses for pig producers worldwide [1]. However, despite its evident economic importance, M. suis in pig herds is rarely reported, and its infection is regularly under-diagnosed or not diagnosed, which can lead to significant production losses [9]. To date, no information is available on hemotropic mycoplasmas specific to Korean pigs. Several Mycoplasma spp. have been detected by PCR and reported in other countries in pigs, such as M. suis/M. parvum and, the novel hemotropic M. haemosuis in China [12], M. suis and M. parvum in Japan [13], M. suis in China [14], M. suis in wild boars [15] and pigs [9] in Germany, and M. suis in Brazil [16]. This study thus investigated the frequency and distribution of hemotropic mycoplasma species in domestic pig farms of Korea. As M. suis can be transmitted by ticks, we also analyzed the presence of the tick-borne pathogens Anaplasma spp. and Borrelia spp., which has not been performed before in domestic pigs.
Methods
Sample size determination and sample collection
The total number of pigs raised in 6196 farms in Korea in 2018 was recorded at 11,640,677 [17]. In this study, we used simple random sampling, and sample size was determined by power analysis using an expected disease prevalence of 10%, accepted absolute error of 5%, and confidence level of 99% [18], as follows:
where n is the required sample size, pexp is the expected prevalence, and d is the desired absolute precision.
According to the formula, a minimum of 239 samples was required. We randomly selected 1867 samples from 464 pig farms located in four regions of Korea. Blood was collected from the jugular vein, and breed and region were recorded.
PCR
Genomic DNA was extracted from whole blood samples using the DNeasy Blood and Tissue Kit (Qiagen, Melbourne, Australia) according to the manufacturer’s protocol, and its quantity and quality were measured using a NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA) before storage at − 20 °C until analysis.
Screening was performed by nested PCR using the AccuPower HotStart PCR Premix Kit (Bioneer, Daejeon, Korea) and designated primer sets. Anaplasma spp. were detected based on amplification of the 16S rRNA gene using the primer sets EE1/EE2 and EE3/EE4 [19]. Borrelia spp. were identified based on the presence of the 5S (rrf)–23S (rrl) intergenic spacer using primer sets Bb23S3/Bb23Sa and Bb23SnF/Bb23SanR, and B. burgdorferi was detected by amplification of the outer surface protein A gene fragment using primer sets N1/C1c and N2/C2c [20]. Hemoplasmas were first identified based on the amplification of 16S rRNA with universal primers fHf1/rHf2 and M. suis-specific primers f2/r2 [16, 21]; positive results were then confirmed at the species level by PCR using cmsf2/cmsr2 and msf2/msf2 primer sets to amplify the 16S rRNA gene of M. suis, M. parvum, and the novel hemotropic M. haemosuis [12].
DNA cloning
Amplified 16S rRNA gene fragments were purified using the QIAquick Gel Extraction Kit (Qiagen) inserted into the pGEM-T Easy vector (Promega, Madison, WI, USA) following the manufacturer’s instructions, and the resulting constructs were used to transform Escherichia coli DH5α-competent cells (Thermo Fisher Scientific). Bacteria were incubated at 37 °C overnight, and plasmids were purified using a plasmid miniprep kit (Qiagen) according to the manufacturer’s instructions.
DNA sequencing and phylogenetic analysis
Recombinant plasmids were sequenced by Macrogen (Seoul, Korea), and 16S rRNA gene sequences were analyzed using the multiple sequence alignment program CLUSTAL Omega (ver. 1.2.1). Results of sequence alignment were corrected using BioEdit (ver. 7.2.5), and phylogenetic analysis was performed with MEGA (ver. 6.0) using the maximum likelihood method based on the Kimura 2-parameter distance model. The aligned sequences from the present study and those available from the GenBank database were analyzed using a similarity matrix. The stability of the obtained phylogenetic trees was estimated by bootstrap analysis with 1000 replicates.
Statistical analysis
The analytical software package GraphPad Prism version 5.04 (GraphPad Software Inc., La Jolla, CA, USA) was used for statistical analysis. The chi-square test was applied to assess significant differences between groups, and a P-value of less than 0.05 was considered statistically significant. Confidence intervals (95% CI) were calculated for all estimates.
Results
PCR detection
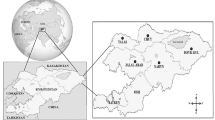
We randomly selected 1867 samples from 464 pig farms located in four regions of Korea (Fig. 1). PCR amplification of 16S rRNA gene fragments showed that 55 of the 1867 pigs tested (2.9%) were positive for Mycoplasma spp. Additional PCR analysis conducted on the 55 positive samples to identify M. suis, M. parvum and the novel hemotropic M. haemosuis species revealed incidences of 0.2% (3/1867; 95% CI: 0–0.3%), 2.7% (51/1867; 95% CI: 2.0–3.5%), and 0.1% (1/1867; 95% CI: 0–0.2%), respectively (Table 1). We did not detect any Anaplasma spp. or Borrelia spp.

The four regions of Korea (Northern, Central, Southern and Jeju Island) where pig blood samples were collected to detect hemoplasmas
No statistically significant differences in Mycoplasma prevalence were observed between breeding and fattening pigs; however, breeding pigs were more likely to be carriers than fattening pigs. Overall prevalences were as follows: M. suis (0.3%, 2/720; χ2 = 1.002, df = 1, P = 0.3169); M. parvum (2.9%, 21/720; χ2 = 0.1510, df = 1, P = 0.6976); and M. haemosuis (0.1%, 1/720; χ2 = 1.594, df = 1, P = 0.2068). Geographically, the prevalence of M. parvum was significantly increased in the south (χ2 = 19.432, df = 3, P = 0.0002), whereas M. suis was detected in both northern and southern regions (χ2 = 0.8050, df = 3, P = 0.8483). Mycoplasma haemosuis was only detected in the southern region (0.1%, 1/905; χ2 = 1.064, df = 3, P = 0.7859).
Molecular and phylogenetic analyses
Nucleotide alignment and phylogenetic analysis were performed using representative samples selected from different years and rearing regions. Sequences of the 16S rRNA gene from three M. suis (14-GN-32, 15-GN-22 and 16-GG-108; accession nos. MK492380, MK492381 and MK492382, respectively), eight M. parvum (14-GN-16, 14-GN-624, 15-GN-123, 16-GG-12, 16-CN-16, 18-JJ-43, 18-GB-16 and 18-JJ-19; accession nos. MK492383–MK492390, respectively) and one M. haemosuis (14-GN-19; accession no. MK492379) strains were analyzed. Sequence alignment revealed that three M. suis and eight M. parvum strains from the present study were 98.9–100% and 97.5–100% homologous to one another, respectively.
Phylogenetic analysis using the 16S rRNA (Fig. 2) gene demonstrated that the Mycoplasma species detected in this study clustered with those from the GenBank database. Results of nucleotide sequence alignment in the present study also showed high identity with those reported from other countries. The three M. suis 16S rRNA sequences shared high identity with those of M. suis isolated from pigs in China, Japan, Germany and the USA at 99.6% (GenBank: KC907396), 99.6% (GenBank: AB610847), 99.4% (GenBank: FQ790233) and 99.3% (GenBank: AF029394), respectively. The eight M. parvum 16S rRNA sequences shared 100% identity with those of M. parvum isolated from pigs in China (GenBank: JX489599) and Japan (GenBank: AB610846), and 99.6% identity with pig isolates from the USA (GenBank: CP006771). The single M. haemosuis 16S rRNA sequence shared 93.0–99.9% identity with sequences of Mycoplasma spp. isolated from pigs in China (99.8%, GenBank: JX489600; 99.9%, GenBank: JX489601), as well as cats in Australia (93.1%, GenBank: DQ464423) and the UK (93.0%, GenBank: DQ464420).

Phylogenetic tree of Mycoplasma spp. based on the 16S rRNA gene. This tree was constructed using the maximum likelihood method. Arrows indicate the sequences generated in the present study. GenBank accession numbers are shown in parentheses. Coxiella burnetii (GenBank: D89792) was used as the outgroup. Numbers at branches indicate bootstrap support levels (1000 replicates) and the scale bar indicates the number of substitutions per site
Discussion
Natural transmission routes of M. suis remain largely unknown [22]. It has been found, however, that the parasite can be shed in saliva, nasal and vaginal secretions and urine from infected animals, and can contaminate dust, water and food, favoring rapid spread of the pathogen [22]. The poor hygiene conditions in domestic pig farms create a favorable environment for the development of arthropods, which might be significant vectors of M. suis transmission among animals [14]. Mechanical transmission of M. suis by the stable fly Stomoxys calcitrans and the yellow fever mosquito Aedes aegypti has also been reported in pigs [23]. Other situations favorable to M. suis transmission between pigs, including intake of food contaminated with infected blood and reuse of needles by farm workers, may be additional risk factors associated with swine hemoplasmosis in China [14]. Importantly, infection with M. suis has also been reported among pig farm workers in China [14], suggesting that this hemoplasma species should be regarded as a potential zoonotic pathogen and a threat to public health.
In this study, three (0.2%) and 51 (2.7%) of the 1867 domestic pigs tested positive for M. suis and M. parvum, respectively. As 16S rRNA gene sequences of M. suis and M. parvum have high sequence identity [24], it is difficult to design PCR primers distinguishing the two species. In a previous study, comparative analysis of 16S rRNA sequences showed the existence of two different M. suis subtypes in wild boars: one (group A) was closely related to the known American and European M. suis isolates and the other (group B), which had only 96.9% identity with group A, formed an independent sub-cluster within M. suis isolates from China [15]. Based on these data, group B isolates that were morphologically identified as M. suis but were closer to M. parvum may need to be reclassified as M. parvum, and conclusions of previously published studies may need to be reevaluated when appropriate molecular data become available. The sequence of the M. suis 16S rRNA gene was first determined in the Illinois strain (GenBank: U88565) in 1997 [25], whereas the complete genome sequence of this strain (GenBank: CP002525) was determined in 2011 [26]. However, M. parvum cannot be maintained in vivo, which delayed the identification of this species, and 16S rRNA and RNase P RNA genes in M. parvum Morioka 1, 4 and 9 strains were sequenced much later [24]. The complete genome sequence of the M. parvum Indiana strain (GenBank: CP006771) was determined in 2014 [11].
Interestingly, in the present study, comparative analysis of 16S rRNA sequences revealed the existence of Mycoplasma species other than M. suis or M. parvum in only one (0.1%) pig. This novel hemotropic M. haemosuis, which was recently detected in pigs from China [12], is not an officially recognized species and is not in the List of Prokaryotic Names with Standing in Nomenclature [6]. Results from our 16S rRNA-based phylogenetic analysis suggest that this putative novel porcine hemoplasma species is a member of a new cluster genetically related to “Candidatus M. turicensis” isolates from the cat, which, together with M. haemocanis and M. haemofelis, belong to the Haemofelis subgroup, whereas M. suis and M. parvum, together with M. ovis, M. wenyonii, “Candidatus M. haemominutum” and “Candidatus M. haematoparvum”, belong to the Haemominutum subgroup. This division of hemoplasma species into these two subgroups has previously been confirmed by 16S rRNA sequencing data [12, 27, 28].
Results of this study indicated that breeding pigs are more likely to be infected with Mycoplasma than fattening pigs, which is consistent with a previous report that sows presented with higher rates of M. suis, M. parvum and the novel hemotropic M. haemosuis than growing pigs [12]. These observations may reflect the fact that the risk for infection increases with herd age, as the chances of contact with M. suis-contaminated sources grow with time [29]. Moreover, M. suis may cause immunosuppression in adult pigs, which favors co-infection and secondary infection [29]. Regarding geographical distribution, in this study, M. suis was detected in both northern and southern regions, whereas the prevalence of M. parvum was significantly higher in the southern region, and the novel hemotropic M. haemosuis was exclusively detected in the southern region. These data suggest the prevalence of Mycoplasma spp. differs among geographical locations, and is likely associated with the habitat and distribution of their vectors. Of note, the prevalence of M. suis-infected pigs was also higher in the southern region of Germany [9].
Conclusions
To the best of our knowledge, this is the first nationwide, large-scale study on the molecular detection of Mycoplasma infection among Korean domestic pigs. Our results indicate that infections with M. suis, M. parvum and the novel hemotropic M. haemosuis are widespread in Korea. Unfortunately, a lack of information concerning Mycoplasma spp. carriage by domestic pigs may delay the administration of effective measures to limit the spread of hemoplasma infection. Therefore, continuous monitoring and control strategies should be implemented in pig farming to prevent the spread of hemoplasmas through herds. Additional epidemiological studies are required to identify the reservoirs and vectors of Mycoplasma spp., to reduce or halt the spread of these pathogens among domestic pigs.
Availability of data and materials
Data supporting the conclusions of this article are included within the article. The newly generated sequences were submitted to the GenBank database under the accession numbers MK492379-MK492390. The datasets used and/or analyzed during the present study are available from the corresponding author upon reasonable request.
Abbreviations
- 95% CI:
-
95% confidence interval
- df:
-
degrees of freedom
- IACUC:
-
Institutional Animal Care and Use Committee
- KNU:
-
Kyungpook National University
References
Messick JB. Hemotrophic mycoplasmas (hemoplasmas): a review and new insights into pathogenic potential. Vet Clin Pathol. 2004;33:2–13.
Splitter EJ. Eperythrozoon suis n. sp. and Eperythrozoon parvum n. sp., 2 new blood parasites of swine. Science. 1950;111:513–4.
Seamer J. Studies with Eperythrozoon parvum Splitter, 1950. Parasitology. 1960;50:67–80.
Zachary JF, Basgall EJ. Erythrocyte membrane alterations associated with the attachment and replication of Eperythrozoon suis: a light and electron microscopic study. Vet Pathol. 1985;22:164–70.
Neimark H, Johansson KE, Rikihisa Y, Tully JG. Proposal to transfer some members of the genera Haemobartonella and Eperythrozoon to the genus Mycoplasma with descriptions of “Candidatus Mycoplasma haemofelis”, “Candidatus Mycoplasma haemomuris”, “Candidatus Mycoplasma haemosuis” and “Candidatus Mycoplasma wenyonii”. Int J Syst Evol Microbiol. 2001;51:891–9.
Euzeby JP. List of prokaryotic names with standing in nomenclature. 2015. http://www.bacterio.net/index.html/. Accessed 20 Dec 2018.
Neimark H, Johansson KE, Rikihisa Y, Tully JG. Revision of haemotrophic Mycoplasma species names. Int J Syst Evol Microbiol. 2002;52:683.
Heinritzi K. Eperythrozoon infection in swine as a disease factor. Berl Munch Tierarztl Wochenschr. 1989;102:337–42.
Ritzmann M, Grimm J, Heinritzi K, Hoelzle K, Hoelzle LE. Prevalence of Mycoplasma suis in slaughter pigs, with correlation of PCR results to hematological findings. Vet Microbiol. 2009;133:84–91.
Barnett SF. Eperythrozoon parvum in pigs in Kenya. Bull Epizoot Dis Afr. 1963;11:185–95.
do Nascimento NC, dos Santos AP, Chu Y, Guimaraes AM, Baird AN, Weil AB, et al. Microscopy and genomic analysis of Mycoplasma parvum strain Indiana. Vet Res. 2014;45:86.
Fu Y, Shi T, Xu L, Wei W, Lu F, Zhang X, et al. Identification of a novel Hemoplasma species from pigs in Zhejiang Province, China. J Vet Med Sci. 2017;79:864–70.
Watanabe Y, Fujihara M, Suzuki J, Sasaoka F, Nagai K, Harasawa R. Prevalence of swine hemoplasmas revealed by real-time PCR using 16S rRNA gene primers. J Vet Med Sci. 2012;74:1315–8.
Yuan CL, Liang AB, Yao CB, Yang ZB, Zhu JG, Cui L, et al. Prevalence of Mycoplasma suis (Eperythrozoon suis) infection in swine and swine-farm workers in Shanghai, China. Am J Vet Res. 2009;70:890–4.
Hoelzle K, Engels M, Kramer MM, Wittenbrink MM, Dieckmann SM, Hoelzle LE. Occurrence of Mycoplasma suis in wild boars (Sus scrofa L.). Vet Microbiol. 2010;143:405–9.
Guimaraes AM, Biondo AW, Lara AC, Messick JB. Exploratory study of Mycoplasma suis (Eperythrozoon suis) on four commercial pig farms in southern Brazil. Vet Rec. 2007;160:50–3.
Korean Statistical Information Service. Number of pigs and pig farms by city and province/Herd size. 2018. http://kosis.kr/statHtml/statHtml.do?orgId=101&tblId=DT_1EO311&conn_path=I2&language=en. Accessed 1 Nov 2018.
Thrusfield M. Veterinary epidemiology. 3rd ed. Oxford: Blackwell Science; 2005.
Seo MG, Ouh IO, Lee H, Geraldinoc PJL, Rhee MH, Kwon OD, et al. Differential identification of Anaplasma in cattle and potential of cattle to serve as reservoirs of Anaplasma capra, an emerging tick-borne zoonotic pathogen. Vet Microbiol. 2018;226:15–22.
VanBik D, Lee SH, Seo MG, Jeon BR, Goo YK, Park SJ, et al. Borrelia species detected in ticks feeding on wild Korean water deer (Hydropotes inermis) using molecular and genotypic analyses. J Med Entomol. 2017;54:1397–402.
Messick JB, Cooper SK, Huntley M. Development and evaluation of a polymerase chain reaction assay using the 16S rRNA gene for detection of Eperythrozoon suis infection. J Vet Diagn Invest. 1999;11:229–36.
Dietz S, Mack SL, Hoelzle K, Becker K, Jannasch C, Stadler J, et al. Quantitative PCR analysis of Mycoplasma suis shedding patterns during experimental infection. Vet Microbiol. 2014;172:581–5.
Prullage JB, Williams RE, Gaafar SM. On the transmissibility of Eperythrozoon suis by Stomoxys calcitrans and Aedes aegypti. Vet Parasitol. 1993;50:125–35.
Watanabe Y, Fujihara M, Obara H, Nagai K, Harasawa R. Two genetic clusters in swine hemoplasmas revealed by analyses of the 16S rRNA and RNase P RNA genes. J Vet Med Sci. 2011;73:1657–61.
Rikihisa Y, Kawahara M, Wen B, Kociba G, Fuerst P, Kawamori F, et al. Western immunoblot analysis of Haemobartonella muris and comparison of 16S rRNA gene sequences of H. muris, H. felis, and Eperythrozoon suis. J Clin Microbiol. 1997;35:823–9.
Messick JB, Santos AP, Guimaraes AM. Complete genome sequences of two hemotropic mycoplasmas, Mycoplasma haemofelis strain Ohio2 and Mycoplasma suis Strain Illinois. J Bacteriol. 2011;193:2068–9.
Tasker S, Helps CR, Day MJ, Harbour DA, Shaw SE, Harrus S, et al. Phylogenetic analysis of hemoplasma species: an international study. J Clin Microbiol. 2003;41:3877–80.
Willi B, Tasker S, Boretti FS, Doherr MG, Cattori V, Meli ML, et al. Phylogenetic analysis of “Candidatus Mycoplasma turicensis” isolates from pet cats in the United Kingdom, Australia, and South Africa, with analysis of risk factors for infection. J Clin Microbiol. 2006;44:4430–5.
Song Q, Zhang W, Song W, Liu Z, Khan MK, He L, et al. Seroprevalence and risk factors of Mycoplasma suis infection in pig farms in central China. Prev Vet Med. 2014;117:215–21.
Acknowledgements
Not applicable.
Funding
This work was supported by a grant from the Basic Science Research Programme through the National Research Foundation (NRF) of Korea funded by the Ministry of Education (grant number NRF-2016R1D1A1B02015366).
Author information
Authors and Affiliations
Contributions
All authors approved the manuscript. Contributions were as follows: conception, design and conducting experiments, MGS, DK; interpretation, data analysis and writing, all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study, conducted from 2014 to 2018, was beyond the purview of the Institutional Animal Care and Use Committee (IACUC) at Kyungpook National University (KNU), as the IACUC at KNU only evaluates those involving laboratory animals maintained in indoor facilities and does not regulate research involving outdoor animals. However, under the regulatory “Act on the Prevention of Contagious Animal Disease (Amendment Act 2016)” national and local veterinary institutes in Korea regularly conduct control measures in accordance with annual infectious animal disease programs. Blood samples were collected from pigs by practicing veterinarians at local, government-run veterinary institutes during monitoring, surveillance, treatment, or regular check-ups after receipt of verbal consent from pig owners, in accordance with the administrative rules of the Ministry of Agriculture, Food and Rural Affairs, Korea.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Seo, MG., Kwon, OD. & Kwak, D. Prevalence and phylogenetic analysis of hemoplasma species in domestic pigs in Korea. Parasites Vectors 12, 378 (2019). https://doi.org/10.1186/s13071-019-3638-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3638-x