
Abstract
Background
In recent years, human infection by the simian malaria parasite Plasmodium knowlesi has increased in Southeast Asia, leading to growing concerns regarding the cross-species spread of the parasite. Consequently, a deeper understanding of the biology of P. knowlesi is necessary in order to develop tools for control of the emerging disease. TatD-like DNase expressed at the surface of P. falciparum has recently been shown to counteract host innate immunity and is thus a potential malaria vaccine candidate.
Methods
The expression of the TatD DNase of P. knowlesi (PkTatD) was confirmed by both Western-blot and immunofluorescent assay. The DNA catalytic function of the PkTatD was confirmed by digestion of DNA with the recombinant PkTatD protein in the presence of various irons.
Results
In the present study, we investigated the expression of the homologous DNase in P. knowlesi. The expression of TatD-like DNase in P. knowslesi (PkTatD) was verified by Western blot and indirect immunofluorescence assays. Like that of the P. falciparum parasite, PkTatD was also found to be located on the surface of erythrocytes infected by the parasites. Biochemical analysis indicated that PkTatD can hydrolyze DNA and this activity is magnesium-dependent.
Conclusions
We identified that PkTatD expressed on the surface of P. knowlesi-infected RBCs is a Mg2+-dependent DNase and exhibits a stronger hydrolytic capacity than TatD from P. falciparum. The data support our previous findings that TatD-like DNase is a unanimously expressed virulence factor of Plasmodium parasites.
Similar content being viewed by others


Background
The simian malaria parasite Plasmodium knowlesi is the pathogen of a neglected tropical disease. P. knowlesi infection progresses rapidly and causes high parasitemia, which has severe consequences [1]. However, although it can cause severe and occasionally even fatal diseases in humans, it is seldom classified as a regular infectious agent by public health bodies [2]. Plasmosium knowlesi was first described in 1932 by Das Gupta and Knowles. Until 1965, it was considered as a zoonosis. Furthermore, until 2004, a proportion of natural P. knowlesi infections acquired by human patients in Sarawak, Malaysia, were likely misdiagnosed as being caused by the morphologically similar parasite P. malariae [3, 4]. Since then, the development of nested polymerase chain reaction [5] and dual-color fluorescence in situ hybridization assay [6] techniques as new diagnostic tools has significantly increased the accuracy of P. knowlesi detection. This has led to an increase in the identification of human P. knowlesi malaria in other parts of Malaysia, revealing that P. knowlesi is now the main cause of malaria in Malaysia [7]. Extensive study has revealed that P. knowlesi is widespread not only in Malaysia but in other countries in Southeast Asia, such as Singapore [8], Cambodia [9], Indonesia [10], Thailand [11], the Philippines [12] and Vietnam [2, 13]. Furthermore, imported P. knowlesi infections due to forest ecotourism in Southeast Asia have been reported in Europe and Japan [14]. Consequently, P. knowlesi is now recognized as the fifth Plasmodium parasite that infects humans, the others being P. falciparum, P. malariae, P. vivax and P. ovale [15].
Natural immunity mediated by immune cells such as macrophages, neutrophils and natural killer cells plays an important role in the elimination of invading pathogens from a host. For example, neutrophils provide an early response to inflammatory reactions, whereupon they are activated by chemotactic signals and then migrate quickly to infection sites [16]. However, clinical pathological investigations have revealed that the infiltration of innate immune cells to sites where Plasmodium parasites accumulate is not very significant. This is largely due to countermeasures employed antagonistically by such parasites against the host’s immune system, as outlined below.
During pathogen infection, activated neutrophils and macrophages release fibrous elements composed of proteases and DNA to restrict the invading pathogens [17, 18]. The fibrous elements released by neutrophils are termed as neutrophil extracellular traps (NETs) and they facilitate the innate immune response by capturing pathogens [19, 20]. However, P. falciparum and Streptococcus release DNase to counteract these NETs [21].
Thus, inhibition of pathogen-derived DNases can impede the propagation of pathogens in the host and help the immune system to control the infection. Furthermore, the TatD-like DNase of P. falciparum has been characterized as an important virulence factor and a potential malaria vaccine candidate [22, 23].
Herein, we reveal that P. knowlesi also expresses a TatD-like DNase (PkTatD), homologous to that of P. falciparum, and that it has a similar molecular structure and functionality.
Results and discussion
Sequence characteristics of the P. knowlesi TatD-like DNase
Sequence analysis of Plasmodium TatD proteins showed that TatD proteins are conserved in Plasmodium spp. As shown in Fig. 1, sequence similarity of TatD sequences among Plasmodium spp. was up to 73.01% and all TatD sequences contain a conserved motif with 17 amino acids typical to the TatD deoxyribonuclease family; this suggests that the TatD family proteins of Plasmodium parasites perform a similar DNA catalytic function in the interaction with their hosts. Furthermore, a signal peptide consisting of 25 amino acids was located at the N-terminus, which indicates, unlike that of other organisms [24], that the Plasmodium TatD proteins are likely secreted outside the infected cells.

Sequence alignment of Plasmodium TatD proteins. The alignments of TatD sequences of Escherichia coli and Plasmodium species show that the plasmodial TatD sequences contain a 25–27AAs signal peptide (black box). All Plasmodium species have a conserved motif of 17 AAs (yellow box). The sequence similarity among Plasmodium species is up to 73.01%. The red asterisks indicate conserved active site residues
Preparation of PkTatD recombinant proteins and anti-PkTatD polyclonal antibodies
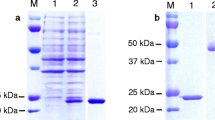
For the functional characterization of the PkTatD, recombinant proteins and specific antibodies were essentially generated. The optimized expression conditions for the recombinant proteins were found to be 22 °C, 16 h, after induction with 0.1 mM IPTG. The molecular weights of the His-tagged and GST-tagged recombinant proteins, without the putative signal peptide, were 51.8 and 76.8 kDa, respectively. The recombinant proteins were verified by Western blot using anti-His-tagged and anti-GST-tagged monoclonal antibodies, respectively. The soluble His-tagged (Fig. 2a) and GST-tagged (Fig. 2b) recombinant proteins were purified by affinity chromatography and verified by Western blot. The purified His-tagged recombinant protein was used to prepare the polyclonal antibodies. Both recombinant PkTatD proteins and specific antibodies are essential tools for further characterization of the molecule.

Generation and purification of soluble recombinant proteins. a The purified His-tagged recombinant protein (51.8 kDa). b The purified GST-tagged recombinant protein (76.8 kDa). Lane M: protein molecular weight markers; Lanes 1 and 2: SDS-PAGE and Western blot analyses, respectively
Identification of endogenous TatD-like DNase in P. knowlesi
The expression of PkTatD in P. knowlesi was initially verified by Western blot using an anti-PkTatD antibody. The anti-PkTatD antibody reacted with a single 54 kDa protein, which is consistent with the molecular weight of PkTatD, and the PkTatD-specific antibody did not react with macaque RBCs (Fig. 3a). Furthermore, the results of IFA indicated that PkTatD is mainly located on the surfaces of the parasite-infected RBCs and in the cytoplasm of the infected erythrocytes (Fig. 3b, c), which was in a similar pattern to that of erythrocytic stage P. falciparum [22].

Identification of endogenous TatD protein in P. knowlesi. a Western blot analysis of the expression of native PkTatD. Lanes 1 and 2 represent infected RBCs and healthy macaque RBCs (negative control), respectively. A single band of approximately 54 kDa was detected in infected RBCs. b The different stages of P. knowlesi were stained with Giemsa. c Indirect immunofluorescence assay with anti-PkTatD polyclonal antibodies. The green fluorescence indicates that the PkTatD is located on the surface of the infected RBCs and in the cytoplasm of the parasite. The nuclei were stained blue with DAPI. Scale-bar: 5 μm
Even though the sequence similarity between PfTatD and PkTatD is 63.70%, the P. knowlesi TatD-like DNase specific antibody did not cross-react with P. falciparum TatD (PfTatD) in both Western blot (Fig. 4a) and IFA (Fig. 4b), suggesting that the immune epitopes of the two molecules are different and the antibodies to either species are likely not cross-protective.

Anti-PkTatD-like DNase polyclonal antibodies did not cross react with that of P. falciparum. The cross reactivity of anti-PkTatD-like DNase antibodies with PfTatD-like DNase was tested with Western blot (a) and immunofluorescence (b), and no cross reactivity was observed. The parasite nuclei were stained blue with DAPI. Negative means a serum from a healthy rat was used as a negative control. Blank means no primary antibody was added in the first step incubation. Scale-bar: 5 μm
Divalent-metal dependence of PkTatD activity
The DNA hydrolysis activity of P. falciparum TatD-like DNase has been previously characterized [22]. Here, we demonstrated that by incubating PkTatD GST-tagged recombinant protein with DNA and different concentrations of various divalent metal ions, PkTatD also hydrolyzed DNA in a concentration-dependent manner (Fig. 5a). Similar to that of PfTatD, the catalytic activity of PkTatD increased at higher temperatures (Fig. 5b). However, we found that the DNA hydrolytic activity of PkTatD is Mg2+-dependent (Fig. 5c, d), and higher concentrations of Cu2+ was inhibitory to the enzyme (Fig. 5c).

Ion-dependent DNase activity of PkTatD. a PkTatD-GST recombinant protein at concentrations from 0 to 5 μM was incubated with 200 ng of DNA in PBS (pH 7.4) at 37 °C for 1 h. DNA was completely hydrolyzed at a concentration of 3 μM. GST protein was used as a negative control. b DNA was completely hydrolyzed at 35 °C and the enzyme remained active at 41 °C. c PkTatD-GST recombinant protein and DNA were incubated with divalent metal ions at concentrations from 0 to 10 mM. The enzyme activity was enhanced by Mg2+ but inhibited by Cu2+. d Mg2+ (10 mM) and DNA were incubated with recombinant PkTatD-GST protein at concentrations from 0 to 5 μM
Deoxyribonucleases generally fall into two categories: divalent metal ion-dependent DNase I and divalent metal ion repressed DNase II [25, 26]. It has been previously reported that the activity of Yeast TatD is Mg2+-dependent [27], but the activity of P. falciparum TatD is inhibited by Mg2+ [22]. In this study, the hydrolysis activity was strongly enhanced when Mg2+ was added (Fig. 5c, d), but inhibited by Cu2+ (Fig. 5c). Thus, PfTatD showed a biochemical feature of DNase II, while PkTatD likely belonged to the DNase I group.
Conclusions
We have identified a TatD-like DNase in P. knowlesi, PkTatD, which shared a conserved sequence feature with other Plasmodium species and was expressed both on the surface of P. knowlesi-infected RBCs and inside the cells. PkTatD is a Mg2+-dependent DNase and exhibits a stronger hydrolytic capacity that PfTatD. The data reported here further demonstrate that TatD DNases are essential factors for the plasmodial parasites in their interaction with the host immune system.
Methods
Bioinformatic analysis of P. knowlesi TatD-like DNase
The putative TatD sequence alignments of P. knowlesi, P. berghei, P. falciparum, P. ovale, P. vivax, P. cynomolgi, P. inui, P. reichenowi, P. yoelii, P. chabaudi, P. malariae and E. coli were retrieved from the PlasmoDB database (www.plasmodb.org) and signatures of the sequences were bioinformatically analyzed using DNAMAN 7 (Lynnon Corporation, San Ramon, USA).
Parasite culture
The P. falciparum strain 3D7 was cultured using human O+ erythrocytes in malaria culture medium (MCM) according to standard methods [28]. The parasites were synchronized with 5% sorbitol.
The P. knowlesi A1-H1 strain was cultivated as previously described [29]. It was originally isolated from a human traveler returning from Malaysia in 1965, most likely from a zoonotic infection, and has since been passaged through rhesus macaque monkeys and subsequently adapted in vitro in rhesus macaque RBCs. The RBCs were provided by the Zoological Research Center of Chinese Academy of Sciences. Briefly, parasites were proliferated in macaque erythrocytes and MCM and then synchronized by centrifugation through a cushion of Nycodenz (Axis-Shield, Oslo, Norway) [30].
Expression and purification of PkTatD recombinant proteins
The coding sequence of the PkTatD gene (PKNH_0201600) was optimized for expression in E. coli by synthesis and cloned into the pGEX4T-1 and pET28a vectors. His-tagged and GST-tagged recombinant proteins were expressed in E.coli (TransGen Biotech, Beijing, China) and the soluble proteins were purified by affinity chromatography using glutathione sepharose and His GraviTrap (GE Healthcare, Uppsala, Sweden), respectively [31].
Preparation of anti-PkTatD polyclonal antibodies
Five female Wister rats were immunized with a total of 200 μg/rat His-tagged recombinant protein emulsified in complete Freund’s adjuvant (Sigma, Missouri, USA) (for the first immunization) and in incomplete Freund’s adjuvant (for the next three immunizations). The immune sera were collected after the antibody titer reached 1:16,000.
Detection of PkTatD by Western blot assay
The expression of PkTatD protein of synchronized P. knowlesi trophozoites was analyzed through SDS-PAGE and a Western blot assay. The trophozoites and schizonts of P. falciparum were also analyzed using the same sera to assess the cross-reactivity of the anti-PkTatD antibodies. Briefly, the denatured proteins were electrophoresed in a 10% acrylamide gel then transferred onto PVDF membranes. The membranes were blocked for 2 h with 3% BSA and incubated overnight at 4 °C with the serum (1:200) from an infected rat as the primary antibody (the serum from a healthy rat was used as a control). After washing in 1× PBS buffer, the membranes were incubated with an alkaline-phosphatase-labeled goat anti-rat IgG antibody (1:20,000) for 1 h. Thereafter, the membranes were developed in a buffer containing BCIP/NBT (5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium chloride).
Indirect immunofluorescence assay
The indirect immunofluorescence assay (IFA) technique was used to further localize the PkTatD protein in the infected erythrocytes. Thin smears of P. knowlesi-infected and P. falciparum-infected erythrocytes were respectively fixed in cold methanol for 10 s and blocked with 3% BSA at 37 °C for 30 min. The samples were then incubated for 1 h at 37 °C with the immune sera (1:50) as primary antibodies (serum from a healthy rat was used as a control), then incubated with Alexa Fluor 488 goat anti-rat IgG (1:200) for 1 h at 37 °C. The parasite nuclei were stained with DAPI. The images were captured with a fluorescence microscope (Leica Camera AG, Wetzlar, Germany).
Analysis of the DNA hydrolytic activity of the PkTatD recombinant protein
The genomic DNA of a mouse liver was extracted by phenol/chloroform extraction. The DNA was incubated with GST-tagged recombinant PkTatD protein at a concentration of 0.1, 0.5, 1.5, 3, 4 or 5 μM in PBS buffer (pH 7.4) and a total volume of 20 μl at 37 °C. The GST protein and PBS were used as negative control and blank control, respectively. To identify the optimum enzymatic temperature, the hydrolysis reaction was carried out at 16 °C, 28 °C, 35 °C, 37 °C and 41 °C. To investigate the effects of divalent metal ions on the enzyme activity, different concentrations of Mg2+, Cu2+, and other divalent metal ions were added to the reactions. The results were analyzed using 1.2% agarose gel.
Abbreviations
- BCIP/NBT:
-
5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium chloride
- IPTG:
-
isopropyl-β-D-thiogalactoside
- MCM:
-
malaria culture medium
- PBS:
-
phosphate-buffered saline
- PfTatD:
-
Plasmodium falciparum TatD-like DNase
- PkTatD:
-
Plasmodium knowlesi TatD-like DNase
- SDS-PAGE:
-
sodium dodecyl sulfate polyacrylamide gel electrophoresis
References
Barber BE, Grigg MJ, William T, Yeo TW, Anstey NM. The treatment of Plasmodium knowlesi Malaria. Trends Parasitol. 2017;33:242–53.
Moyes CL, Henry AJ, Golding N, Huang Z, Singh B, Baird JK, et al. Defining the geographical range of the Plasmodium knowlesi reservoir. PLoS Neglect Trop Dis. 2014;8:e2780.
Antinori S, Galimberti L, Milazzo L, Corbellino M. Plasmodium knowlesi: the emerging zoonotic malaria parasite. Acta Trop. 2013;125:191–201.
Knowles R. Gupta BMD. A study of monkey-malaria, and its experimental transmission to man. Ind Med Gaz. 1932;67:301–20.
Miguel-Oteo M, Jiram AI, Ta-Tang TH, Lanza M, Hisam S, Rubio JM. Nested multiplex PCR for identification and detection of human Plasmodium species including Plasmodium knowlesi. Asian Pac J Trop Med. 2017;10:299–304.
Shah J, Poruri A, Mark O, Khadilka U, Mohring F, Moon RW, et al. A dual colour fluorescence in situ hybridization (FISH) assay for identifying the zoonotic malaria parasite Plasmodium knowlesi with a potential application for the specific diagnosis of knowlesi malaria in peripheral-level laboratories of Southeast Asia. Parasit Vectors. 2017;10:342.
Muller M, Schlagenhauf P. Plasmodium knowlesi in travellers, update 2014. Int J Infect Dis. 2014;22:55–64.
Ng OT, Ooi EE, Lee CC, Lee PJ, Ng LC, Pei SW, et al. Naturally acquired human Plasmodium knowlesi infection, Singapore. Emerg Infect Dis. 2008;14:814–6.
Khim N, Siv S, Kim S, Mueller T, Fleischmann E, Singh B, et al. Plasmodium knowlesi infection in humans, Cambodia, 2007–2010. Emerg Infect Dis. 2011;17:1900–2.
Lubis IND, Wijaya H, Lubis M, Lubis CP, Divis PCS, Beshir KB, et al. Contribution of Plasmodium knowlesi to multispecies human malaria infections in North Sumatera, Indonesia. J Infect Dis. 2017;215:1148–55.
Jongwutiwes S, Putaporntip C, Iwasaki T, Sata T, Kanbara H. Naturally acquired Plasmodium knowlesi malaria in human, Thailand. Emerg Infect Dis. 2004;10:2211–3.
Luchavez J, Espino F, Curameng P, Espina R, Bell D, Chiodini P, et al. Human infections with Plasmodium knowlesi, the Philippines. Emerg Infect Dis. 2008;14:811–3.
Van den Eede P, Van HN, Van Overmeir C, Vythilingam I, Duc TN, Hung LX, et al. Human Plasmodium knowlesi infections in young children in central Vietnam. Malar J. 2009;8:249.
Fong MY, Lau YL, Chang PY, Anthony CN. Genetic diversity, haplotypes and allele groups of Duffy binding protein (PkDBPα II) of Plasmodium knowlesi clinical isolates from Peninsular Malaysia. Parasit Vectors. 2014;7:161.
Singh B, Kim Sung L, Matusop A, Radhakrishnan A, Shamsul SS, Cox-Singh J, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004;363:1017–24.
Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol. 2006;16:396–400.
Von Kockritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood. 2008;111:3070–80.
Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14:949–53.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5.
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5:e1000639.
Sumby P, Barbian KD, Gardner DJ, Whitney AR, Welty DM, Long RD, et al. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc Natl Acad Sci USA. 2005;102:1679–84.
Chang Z, Jiang N, Zhang Y, Lu H, Yin J, Wahlgren M, et al. The TatD-like DNase of Plasmodium is a virulence factor and a potential malaria vaccine candidate. Nat Commun. 2016;7:11537.
Wang W, Liu F, Jiang N, Lu H, Yang N, Feng Y, et al. Plasmodium TatD-Like DNase antibodies blocked parasite development in the mosquito gut. Front Microbiol. 2018;9:1023.
Chen L, Shen D, Sun N, Xu J, Wang W, Dou D. Phytophthora sojae TatD nuclease positively regulates sporulation and negatively regulates pathogenesis. Mol Plant Microbe Interact. 2014;27:1070–80.
MacLea KS, Krieser RJ, Eastman A. Revised structure of the active form of human deoxyribonuclease IIalpha. Biochem Biophys Res Commun. 2002;292:415–21.
Abe A, Liao TH. The immunological and structural comparisons of deoxyribonucleases I. Glycosylation differences between bovine pancreatic and parotid deoxyribonucleases. J Biol Chem. 1983;258:10283–8.
Qiu J, Yoon JH, Shen B. Search for apoptotic nucleases in Yeast: role of Tat-D nuclease in apoptotic DNA degradation. J Biol Chem. 2005;280:15370–9.
Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–5.
Zeeman AM, der Wel AV, Kocken CH. Ex vivo culture of Plasmodium vivax and Plasmodium cynomolgi and in vitro culture of Plasmodium knowlesi blood stages. Methods Mol Biol. 2013;923:35–49.
Moon RW, Hall J, Rangkuti F, Ho YS, Almond N, Mitchell GH, et al. Adaptation of the genetically tractable malaria pathogen Plasmodium knowlesi to continuous culture in human erythrocytes. Proc Natl Acad Sci USA. 2013;110:531–6.
Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40.
Acknowledgements
Not applicable.
Funding
This study was supported by grants of the National Key Research and Development Programme of China (grant nos. 2017YFD0500400, 2017YFD0501200) and the National Natural Science Foundation of China (grant nos. 81420108023, 81772219).
Availability of data and materials
All relevant data supporting the conclusions of this article are included within the article.
Author information
Authors and Affiliations
Contributions
All authors contributed to the critical review of the final manuscript. QJC conceived and designed the study, and critically revised the paper. YPZ, XYS, BX, LBJ and NJ performed the experiments. YPZ, NJ, NY, YF and QJC performed the data analysis. YPZ and QJC drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All laboratory animal protocols and procedures were performed following the regulations of the Animal Ethics Committee of Shenyang Agricultural University (permit no. SYXK<Liao>2011-0001).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, Y., Xiao, B., Jiang, N. et al. Expression and functional analysis of the TatD-like DNase of Plasmodium knowlesi. Parasites Vectors 11, 629 (2018). https://doi.org/10.1186/s13071-018-3251-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-018-3251-4