
Abstract
It is over 100 years since the life-cycle of the malaria parasite Plasmodium was discovered, yet its intricacies remain incompletely understood - a knowledge gap that may prove crucial for our efforts to control the disease. Phenotypic screens have partially filled the void in the antimalarial drug market, but as compound libraries eventually become exhausted, new medicines will only come from directed drug development based on a better understanding of fundamental parasite biology. This review focusses on the unusual cell cycles of Plasmodium, which may present a rich source of novel drug targets as well as a topic of fundamental biological interest. Plasmodium does not grow by conventional binary fission, but rather by several syncytial modes of replication including schizogony and sporogony. Here, we collate what is known about the various cell cycle events and their regulators throughout the Plasmodium life-cycle, highlighting the differences between Plasmodium, model organisms and other apicomplexan parasites and identifying areas where further study is required. The possibility of DNA replication and the cell cycle as a drug target is also explored. Finally the use of existing tools, emerging technologies, their limitations and future directions to elucidate the peculiarities of the Plasmodium cell cycle are discussed.
Similar content being viewed by others

Background
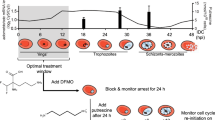
The malaria parasite owes its success in part to its ability to ‘divide and conquer’ [1]. It pursues a complex, two-host life-cycle involving both mosquito and human hosts, in which each bottleneck is followed by a replication phase (Fig. 1). There are four periods of mitotic DNA synthesis and one period of meiosis during the course of the Plasmodium life-cycle [2]. The properties of cell division at these replication phases differ fundamentally from conventional models of eukaryotic cell division: rather than binary fission, the parasite opts primarily for schizogony whereby a multinucleate syncytium is formed, prior to budding and cytokinesis [3]. Equally intriguing is the remarkably rapid process of gamete formation, where male gametocytes undergo three rounds of DNA replication in a matter of minutes, producing eight male gametes [4, 5].

Schematic showing the life-cycle of P. falciparum. Each replicative stage of the life-cycle, together with the approximate fold-replication, is highlighted in purple. Approximate parasite numbers within each host at each stage are also shown to highlight the severe bottlenecks and massive expansions that occur throughout the life-cycle
Such a complex life-cycle presumably requires sophisticated global and local regulators, involving refined checkpoint and DNA repair mechanisms [3], yet these are currently only poorly understood. Along with the unusual spatial and temporal dynamics of DNA replication, cell cycle regulators have been shown to be distinct from human counterparts [6]. Thus, replication in Plasmodium appears to be an excellent drug target: its mechanisms and regulators are distinct from those of the host organisms, the scale of reproductive output is directly crucial to pathogenicity, and it offers the possibility of interfering with the transmissibility of the parasite. Furthermore, the parasite possesses two organelles of bacterial origin, the apicoplast and mitochondrion, both of which carry their own genomes and may harbour distinct drug targets in the form of prokaryotic-type replication proteins (apicoplast replication was recently well-reviewed [7], so this article focusses only on nuclear replication). Finally, in the current era of artemisinin resistance, which appears to involve parasite ‘dormancy’, understanding cell cycle arrest and checkpoints is of utmost importance.
The Plasmodium cell cycle
The standard eukaryotic cell cycle follows a clearly defined series of stages during which the cell grows (interphase), replicates its chromosomes (S phase) and divides (M phase), with S phase often being flanked by two gap phases called G1 and G2 (Fig. 2). The process is tightly regulated by an extensive regulatory network that ensures the cell is ready to progress onwards through the cycle after each phase [8, 9]. Apicomplexans, including Plasmodium spp., deviate significantly from this classical model, allowing each cell to produce dozens or hundreds of new daughter cells during a single replicative cycle. At the heart of these adaptations is schizogony: a syncytial and yet asynchronous form of replication and cell division, which occurs in the mammalian host during hepatic and erythrocytic infections, while a largely analogous process called sporogony occurs in the mosquito vector during oocyst formation (Fig. 1).

Schematic of the conventional eukaryotic cell cycle, highlighting the points at which cycle checkpoints operate
For the most virulent human malaria parasite, P. falciparum, the parasites in the erythrocytic cycle, when grown in vitro, produce an average of 16 daughter merozoites from a single infected erythrocyte. Of these, about two-thirds can successfully invade new erythrocytes, resulting in growth rates of up to 11-fold per 48-hour cycle [10, 11]. Infected hepatocytes and oocysts have been less closely studied, but each of these can produce many thousands of daughter cells during a single replicative cycle, taking ~ 7–10 days. Finally, the extremely rapid replicative process of male gametogenesis takes just 10–15 minutes (Table 1). Thus, there are huge variations of both speed and magnitude between the various replicative stages of the Plasmodium life-cycle.
Cell-cycle mechanics at the cellular level
The Plasmodium cell cycle is best characterised during erythrocytic schizogony, with the start of each cycle being associated with the invasion of a new erythrocyte by a merozoite. During the first 24 hours post-invasion (hpi), ring and early-trophozoite parasites possesses a single haploid nucleus in interphase or G1. The centriolar plaque (CP), which is functionally equivalent to the centrosomes in higher eukaryotes, begins to assemble and duplicate 20–24 hpi, marking the shift from growth to replication. Replication of the chromosomes initiates at estimated times of 29 hpi [12] or 24–26 hpi [13], overlapping with the semi-conservative duplication of the CP. It was initially proposed that several rounds of continual chromosome replication would then occur, followed by a single co-ordinated mass chromosome segregation, nuclear division and formation of daughter merozoites [14]; however, an alternative model entails repeated cycles of replication, segregation and nuclear envelope division, based upon the observed segregation patterns of the CP [13]. In this model the characteristic asynchronicity of schizogony correlates with the inheritance of the CP, with nuclei that inherit the larger mother CP being ready to divide sooner than those nuclei with the daughter CP.
It is not known what signal induces a schizont to stop its asynchronous nuclear divisions and undergo a final - and apparently synchronous [13] - division, followed by budding into membrane-bound daughter cells. Space or nutrients within the host erythrocyte may become limiting, or the timed production of a global cell cycle regulator may be hard-wired into Plasmodium’s transcriptional cascade [15]. The latter model could potentially enforce synchronicity in the final nuclear division, but it would presumably run the risk of ‘catching’ existing nuclei at different stages of S phase. Streipen and co-workers, working largely in the related apicomplexan parasite Toxoplasma gondii, have proposed that ‘local’ versus ‘global’ control is centrally coordinated by the CPs [3, 16], while a remnant flagellum is the organising principle for daughter cell budding [17]. This may also be true for Plasmodium, although schizogony is numerically more complex than endodyogeny in T. gondii and the challenges of its coordination may in fact be more analogous to the replication of hyphal yeasts [18]. Overall, significant gaps remain in our understanding of the cell biology underlying schizogony, and even less is known about the other phases of Plasmodium replication (Fig. 1), for which erythrocytic schizogony may yet prove an inadequate model.
Mechanics of DNA replication at the molecular level
Many early-diverging eukaryotes, such as Trypanosoma and Oxytricha, organise their genomes in extremely unusual ways [19, 20]; by comparison, the basic genome structure of Plasmodium is quite conventional. There are 14 linear chromosomes with telomeres and centromeres, plus two small organellar genomes in the mitochondrion and apicoplast [21]. This conventional genome is replicated, however, within the framework of fundamentally unusual cell biology. Although much remains to be studied, some of the proteins and parameters involved in this process have now begun to be elucidated.
Plasmodium encodes the basic replicative machinery that is found in all eukaryotes, including DNA polymerases [21, 22], proliferating cell nuclear antigen (PCNA) [23, 24] and minichromosome maintenance proteins (MCMs) [25, 26]. Components of the Origin Recognition Complex (ORC) are also present although only two ORC components have been characterised thus far in P. falciparum, PfORC1 and PfORC5 [14, 27], while a putative ORC2 has been shown to be upregulated during gametocyte activation in P. berghei [28]. The remaining members of the complex are either absent or lack sufficient homology with characterised members to allow clear identification, although in P. berghei putative homologues of ORC4 and ORC3 have recently been proposed [28]. The ORC-associated protein Cdc6 also lacks a clear homologue in P. falciparum but a candidate has been proposed in P. berghei [28]. Cdc6 is required for the recruitment of Cdt1 and the loading of MCM proteins (to date only a putative Cdt1-like gene has been identified in Plasmodium). Interestingly, PfORC1 shares a homology region with Cdc6 at the C-terminus and it may, therefore, be regulated in a Cdc6-like manner. In S. cerevisiae and humans, the onset of Sphase results in the recruitment of two further components to activate the MCM helicase: Cdc45 and the GINS complex (consisting of Sld5, Psf1, Psf2 and Psf3). Sequence analysis has failed to identify a clear homologue of Cdc45 in Plasmodium, while the members of the GINS complex have been putatively identified, based on low sequence homology, but it remains to be determined whether they are functional.
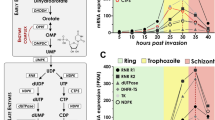
ORC binds DNA and facilitates the initiation of replication. It recognises a conserved consensus sequence in the yeast Saccharomyces cerevisiae, but in other eukaryotes there is no consensus sequence and the preferred composition of DNA bound by ORC varies from organism to organism [29]. Putative ORC-binding sequences in P. falciparum have recently been identified in silico, based on the AT-rich ORC-binding site of S. cerevisiae (Fig. 3a): they are spaced only ~ 2500 bp apart in this highly AT-rich genome and although selected sites do show significant enrichment of ORC1 in ChIP assays [30] (Fig. 3b), only a small proportion of these sites appear to be used for intra-erythrocytic replication. DNA ‘combing’ of labelled DNA fibres was recently used to investigate the actual distribution of active replication forks during erythrocytic schizogony (Fig. 3c) and this revealed a mean distance of 65 kb between individual origins, with replication forks moving at a mean rate of 1.19 kb/min [31]. The rate of replication was not constant, but decreased as the cells neared completion of schizogony, coinciding with a reduction in the mean distance between individual origins. Interestingly, this is the opposite of the pattern seen in human cells, where replication speeds up and origins become more widely spaced as S phase proceeds. In a schizont, which is replicating not 2-fold but ~ 16-fold, the availability of nucleotides, the physical space available or the increasing compaction of chromatin may all limit the speed of replication as S phase advances [31].

Development of techniques to examine the replication of the Plasmodium genome. a Bioinformatic analysis of conserved sequences at and surrounding replication origins in the S. cerevisiae genome has led to identification of common motifs for searching the Plasmodium genome [30]. Origins in S. cerevisiae consist of compact autonomously replicating sequences (ARS) with ‘A domain’ motifs (orange) and surrounding ‘B domains’ (green). The Plasmodium genome has a high concentration of individual A and B domains (~ every 2500 bp) but a much lower concentration when the requirement for closely associated domains is imposed (grey boxes). Bioinformatics approaches can only identify putative origin sites and may fail to identify true origins (*) or identify sequences which are incapable of functioning as origins. b Chromatin immuno-precipitation (ChIP) of the proteins required before and during replication, such as members of the Origin Recognition Complex (ORC), allows experimental characterisation of origin sequences [30]. Following reversible DNA-protein cross linking, the genome is fragmented and the proteins of interest are purified along with the associated DNA fragments, which are then sequenced. This may include origins that would never be activated, and may miss those where the protein complex has dissociated from the chromosome. c Synthetic nucleoside labelling and DNA combing techniques allow the labelling and fluorescent immunodetection of de novo DNA synthesis [31]. Parasites expressing viral thymidine kinase can incorporate the synthetic nucleosides IdU (red) and CldU (green) which can be visualised in individual nuclei or on combed DNA fibres, allowing the calculation of inter-origin distances and replication rates. The synthetic nucleosides will only be incorporated around active origins (*) while inactive origins will remain unlabelled and therefore undetected
During erythrocytic schizogony the first replication (and possible nuclear division) requires 4–6 hours [13]. This, however, is not the maximal rate of replication for the parasite. After ingestion by a mosquito, male gametocytes undergo a 3-fold replication of their genome in less than 15 minutes, resulting in the production of 8 motile microgametes with 1N genome content [4, 5]. (Female gametocytes also mature and exit their host cells upon entering a mosquito but no replication or cell division occurs.) In the model rodent malaria species P. berghei, in which much of the work on gametocytes has been conducted, the first round of replication is usually completed within a minute of gametocyte activation [32]. Such extreme speed is unprecedented in eukaryotic gametogenesis, and may reflect strong pressure to complete the sexual cycle and exit the midgut before the parasite cells are digested along with the blood meal. It has yet to be determined whether the replication rate, the number of simultaneously active origins or both are significantly increased during gametocyte replication but the question of replication fidelity clearly becomes particularly pressing at this unique point in the parasite’s life-cycle. If the replication speed remains the same as it is in erythrocytic schizogony, then almost all of the suggested ORC-binding sites [30] must be used as origins simultaneously. Precedents do exist for such extremely flexible origin usage: in the earliest replications of Xenopus embryos, origins occur every 5–15 kb, spacing out only after the mid-blastula transition [33]. Plasmodium genome replication may be under similarly flexible control, although nothing is yet known about how this might be differentially enforced in gametogenesis, sporogony, hepatic and erythrocytic schizogony.
Regulation of the Plasmodium cell cycle
As described above, Plasmodium undergoes multiple unconventional cell cycles, in a variety of host cell types and for varying durations. The intricacy and temporal/spatial accuracy with which these cell cycles are governed requires global and local regulators that must be fine-tuned and potentially equally unconventional. Although the genomic revolution for Plasmodium has permitted some investigation of these regulators, our understanding at present is patchy and incomplete.
Cyclins and CDKs
In eukaryotic cells, cell cycle progression is governed by cyclins and cyclin-dependent protein kinases (CDKs), along with other proteins such the anaphase promoting complex (APC), which promotes waves of cyclin degradation. The interplay between these regulatory and catalytic components and their timely upregulation, inhibition and degradation prompts sequential progression through G1, S, G2 and M phases [34] (Fig. 2).
The peculiarities of Plasmodium schizogony begin with the lack of a G2 phase as the syncytial nuclei appear to alternate asynchronously between S and M phases prior to the orchestrated event of cytokinesis [35] (Fig. 4a). This raises questions about whether control of replicative cycles through diffusible cytoplasmic factors is feasible [2, 12]. Although such factors may exist, the Apicomplexa have a very unusual repertoire of cyclins, CDKs and CDK-related kinases (CRKs): in total, three cyclins, Cyc1, Cyc3 and Cyc4 [6], and seven CDK or CDK-related kinases, PK5, PK6, Mrk1, Crk-1, Crk-3, Crk-5 [36] and Crk-4 [12], have been identified in Plasmodium (Table 2).

Illustration of cell cycle phases in Plasmodium erythrocytic schizogony (a) and Plasmodium male gametogenesis (b). The predicted involvement of cyclins, CDKs and other kinases is shown at each phase. Placement of such components is only loosely chronological since most details are unknown. CDKs/Crks with a dashed outline indicate cyclin independence. Crks or CDKs predicted to be involved in transcriptional regulation are transparent (without a white background). Interactions identified in vitro between cyclins and CDKs are indicated by a dashed orange arrow. Table 2 identifies all sources used to construct the figure. The cell cycles at sporogony and hepatic schizogony are omitted due to the lack of information about these stages
None of the cyclins are homologs of canonical cell-cycle cyclins (e.g. mammalian Cyc D, E and A) and they tend to be ubiquitously expressed at many stages and not to ‘cycle’ as seen in mammalian cells [6, 37]. Amongst the CDKs, PK5 is the only enzyme that clusters with mammalian cell-cycle CDKs; of the others, Mrk1 is the putative functional homologue of CDK7, which is both a CDK-activating kinase and a component of the general transcription factor TFIIH [38,39,40], Crk1 and Crk3 are homologous to transcriptional CDKs, and PK6 and Crk5 are ‘atypical’ CDKs specific to Apicomplexa [41,42,43].
Although PK5 is the putative homologue of mammalian CDK1, Plasmodium encodes no cognate cyclins for such an enzyme and the activator for PK5 remains unknown: in vitro, it is unusually promiscuous and can be activated by all three Plasmodium cyclins as well as mammalian p25, cyclin H and RINGO [37, 44, 45]. The partnership with Cyc1 is questionable because recent immunoprecipitation studies failed to identify it [46]. Nevertheless, PK5 has been shown to be involved in ORC1 phosphorylation, implicating it in DNA replication in erythrocytic stages [45]. Mrk1 is not apparently a CDK-activating kinase in Plasmodium and although it can interact in vitro with the replication factor complex (PfRFC-5) and PfMCM6 [40], it actually appears to be crucial for cytokinesis rather than replication, as indeed is Cyc1 [46], with Mrk1 acting in a complex with Cyc1 and MAT1 [46]. Crk5 can be activated in vitro by Cyc1 and Cyc4 but its in vivo partner is again unknown; it is involved in, but not essential for, erythrocytic schizogony because its absence results in viable parasites with fewer merozoites per schizont [43]. PK6 is proposed to be involved in the onset of S phase in erythrocytic stages but in vivo characterisation is lacking, and recombinant PK6 is cyclin-independent in vitro [47]. The remaining CDKs, Crk-1 and Crk-3, are predicted to have roles in transcriptional regulation and thus in cell growth and proliferation [42, 48]. Overall, it is clear that Plasmodium cell cycles are not regulated by conventional waves of cyclin/CDK activity, although schizogony and sporogony do require Cyc1 and Cyc3, respectively [6, 46], and several CDKs are evidently involved.
Non-cyclin/CDK cell cycle regulators
A host of cell cycle regulators beside the cyclins/CDKs have been identified in Plasmodium (Fig. 4). Some of these are homologs of conventional eukaryotic regulators, including the NIMA kinases (Nek1-4) and the Aurora kinases (Ark1-3): Nek1 plays an essential roles in mitosis [43] and Nek2-4, in gametogenesis/meiosis [49,50,51], while the Arks are involved in mitotic spindle formation and cytokinesis [52]. Another - perhaps more interesting - group of regulators are specific to the unusual cell cycle modes of apicomplexans and there is considerable interest in the plant-like calcium-dependent protein kinases (CDPKs) as possible parasite-specific drug targets, with CDPK4 playing multiple roles in male gametogenesis [28] and CDPK7, in erythrocytic schizogony [53]. Another Plasmodium-specific kinase, PfCRK4, was recently identified as essential for DNA replication in erythrocytic schizogony, although the pathway in which it acts remains to be elucidated [12]. The regulation of Plasmodium’s atypical cell cycles could clearly be rich source of novel drug targets, but much work is still required to piece together the molecular signalling pathways involved.
Cell cycle checkpoints in Plasmodium
In addition to the cyclin-CDK regulatory network, there are also defined checkpoints in yeast and mammalian systems that control cell cycle advancement. These serve as quality control for cell growth (G1 checkpoint), successful DNA replication or DNA damage (S and G2 checkpoints) and chromosome attachment to the spindle (M checkpoint) [54]. Checkpoints are particularly important for avoiding re-replication and preventing the propagation of incompletely replicated or damaged daughter genomes.
The existence of cell cycle checkpoints in Plasmodium remains, in general, uncertain, and genes encoding key checkpoint proteins such as Rb, p53, ATM and ATR have not been identified. There is, however, some evidence of a G1 checkpoint in the related parasite T. gondii [55], and blood-stage Plasmodium parasites can induce a comparable, reversible rest state/dormancy at the prereplicative ring stage in response to nutrient starvation [56] and drug pressure [57,58,59]. What induces these states in the absence of a conventional G1 checkpoint pathway is unclear.
DNA repair machinery is largely conserved in the parasite genome, as described below, and parasites respond to DNA damage by upregulating repair machinery and altering chromosome structure [60]. However, there is no apparent G2, offering no opportunity for a G2 checkpoint [13, 35] and the feasibility of intra-S and M checkpoints is challenged by the striking variation in the speed of genome replication at different life-cycle stages, particularly the unprecedented rate in male gametocytes [61], sharply contrasting with a more conventional rate during erythrocytic schizogony [13, 31]. Checkpoint regulation may be temporally possible during erythrocytic schizogony - and perhaps also oocyst sporogony and hepatic schizogony - but not gametogenesis. Spatially, schizogony also poses challenges to checkpoint control. Although chromosomes do appear to align with the hemispindle, which is anchored to a CP, they remain uncondensed: it is thought that the centromeres remain constantly attached to CPs and that this may help to separate the uncondensed chromosomes accurately [3]. Finally, the syncytial nature of Plasmodium replication raises questions about diffusible checkpoint factors and the how the replication of individual genomes could be stalled within a shared cytoplasm [2]. Nevertheless, the observation that schizonts produce very varied numbers of merozoites while the erythrocytic cycle is always ~ 48 hours [62] does imply that intra-S or M checkpoints may delay some nuclei, and that the time span of schizogony is not imposed by the number of merozoites to be produced.
Variations in cell cycle speed also raise questions about replicative fidelity and tolerance of karyotypic variation. Indeed, this parasite’s relentless ability to develop resistance against all antimalarials is undoubtedly linked to genomic plasticity, but it remains debatable whether plasticity is inherent in the P. falciparum genome, or is actually promoted by unusually relaxed cell-cycle control. Under drug pressure, P. falciparum erythrocytic stages can initiate AT-track mediated, random duplication of segments of the genome, followed by the establishment of pseudopolyploids around a high priority locus. This permits fine-tuning of amplicon numbers, relevant to drug pressure, while avoiding genome damage and any deleterious mutations in off-target loci [63]. In fact, 20 years ago the concept of ‘hypermutator’ strains specific to certain global regions was proposed to explain the fact that drug-resistant parasites regularly arise in Southeast Asia [64], but this has proved difficult to evidence, or to link it to any molecular defect in DNA repair or checkpoints [65, 66]. Two recent studies have suggested that the mitotic mutation rate does not vary between P. falciparum strains and is not increased by drug pressure; rather, beneficial resistance mutants are simply selected under drug pressure [67, 68]. In this regard, P. falciparum’s active maintenance of a highly AT-biased genome (81% A/T), with high insertion/deletion rates and an overrepresentation of homopolymeric A/T tracts [69], may be the key to rapid adaptive evolution [70].
The extremely fast replication of male gametes raises a particular conundrum in terms of checkpoints: does this phase require especially stringent regulation, or conversely, more relaxed control to favour speed over fidelity? The observation that some male gametes are produced with apparently partial or absent nuclei (unpublished observations) and the fact that male-expressed genes display fast rates of evolution [71] may suggest the latter. Proteins involved in DNA replication and mitosis are simultaneously phosphorylated within the first 20 seconds of gametocyte activation, contrary to the traditional view of sequential progression through the cell cycle, and this may facilitate the rapidity of gametogenesis [32]. Indeed, the relatively limited repertoire of cell cycle kinases in Plasmodium may also imply that some have dual functions: CDPK4 has been implicated in assembly of the pre-replicative complex, mitotic spindle formation, cytokinesis and axoneme motility [28, 32, 72]. Extending this concept, highly divergent kinases may also operate ‘incognito’ as checkpoint kinases in Plasmodium.
Regardless of cell cycle speed, the parasite is clearly able to promote genomic diversity during mitosis, as well as more conventionally at meiosis. It seems unlikely that the intricacy and precision of the Plasmodium cell cycles would proceed unchecked, but evidence is currently lacking for clearly defined checkpoints and there may be great flexibility in which checkpoints are enforced during different types of replication.
DNA repair in Plasmodium
DNA damage can originate from a range of sources, the most common in Plasmodium being reactive oxygen species generated by metabolism, free radicals, which are often produced after uptake of antimalarial drugs such as chloroquine or artemisinin, and errors made during DNA replication. Damage may affect individual bases or may lead to the generation of potentially deadly double strand breaks (DSBs). The mutational spectrum observed in P. falciparum is highly unusual: the SNP mutation rate is 2.45 × 10-10 mutations per base pair per life-cycle but this is dwarfed by a 10-fold higher indel mutation rate, driven by low complexity AT-rich sequences and a significant G:C -> A:T transition bias [70]. This can promote the formation of pseudopolyploid loci, as described above, but the core genome nevertheless remains intact, due to the presence of an effective DNA repair system including most - although notably not all - of the pathways commonly found in model eukaryotes.
Damage to individual bases is resolved by the excision repair pathways that include nucleotide excision repair (NER), base excision repair (BER) and mismatch repair (MMR). Orthologs of the majority of genes involved in the NER pathway have been identified bioinformatically, with the exception of p62 and XPC [73]. Similarly, the majority of the MMR pathway is present but there are notable differences from other eukaryotes, with RecJ exonucleases appearing to be absent while a UvrD helicase homolog, found in E. coli but absent in humans, is present [74, 75]. Plasmodium falciparum lacks homologs for short-patch BER but a long-path BER pathway is present [76].
The majority of eukaryotes rely upon two major pathways for the repair of double-strand breaks, homologous recombination (HR) and non-homologous end joining (NHEJ). The Plasmodium genome encodes a functional HR pathway but the core genes of the NHEJ pathway appear to be absent across the genus [21, 77], supported by the inability to detect NHEJ products in vitro after the experimental generation of DSBs [78, 79]. Accordingly, HR is the prime source of DSB repair in the parasite [79,80,81] (by contrast, T. gondii possesses a functional NHEJ pathway, indicating loss of this pathway in Plasmodium [82, 83]). HR, indeed, appears to be essential for the completion of the parasite life-cycle because the knockout of a zinc finger protein, PbZfp, in P. berghei leads to a loss of transmission competence in mosquitoes due to a failure to recruit the topisomerase-like enzyme Spo11 to recombination hotspots [80]. During all haploid growth phases the parasite must therefore rely upon alternative end joining pathways such as microhomology-mediated end joining (MMEJ) to repair DSBs within the core genome, because no repair template exists to allow HR [84]. Bioinformatic comparisons with the S. cerevisiae and human genomes have identified Plasmodium orthologs of the MMEJ components, but studies of irradiated parasites suggest that the process is inefficient or used infrequently, and that parasites with a 1N genome content are unable to repair damage as efficiently as trophozoites with genome content of 2N or more [85,86,87].
Notably, this restriction does not apply to multigene families, such as the var family of key virulence genes in P. falciparum. There are ~ 60 hypervariable var gene homologs in every parasite genome so these can be repaired via HR even in haploid parasites, using homologs within the same family. This leads to important diversification of these gene families during mitotic growth [68] (as well as during meiosis), generating new antigenic variants that can facilitate immune evasion during chronic human infections. Var gene recombination does not require substantial stretches of high sequence homology and the genes do not necessarily recombine with their closest homologues [68]; the physical clustering of var genes at the nuclear periphery may favour sequence pairing even in the absence of extensive homology.
DNA replication in Plasmodium as a potential drug target
Historically, DNA replication has been an excellent drug target in malaria parasites, as demonstrated by Fansidar: an anti-folate drug combination which proved vital after the emergence of chloroquine resistance in the late 1950s [88]. Fansidar is a synergistic combination of two drugs that block the pathway to production of reduced folate cofactors (essential for nucleotide production and DNA synthesis), but resistance to the combination arose fairly rapidly [89, 90]. However, directly targeting the regulatory machinery of the parasite, such as cell-cycle checkpoint control, or eliciting DNA damage as a route to parasite killing, may provide a greater hurdle to resistance development. Indeed, DNA damage, together with protein damage, is thought to be a mode of action for the frontline antimalarial drug artemisinin, mediated through free radicals [91]. Plasmodium falciparum responds to artemisinin by inducing dormancy at G0/G1, downregulating DNA synthesis-related cyclins/CDKs, upregulating putative ‘negative-regulatory’ cyclins/CDK’s [35] and upregulating DNA repair machinery - specifically, PfRad51, PfRad54, PfRPAIL and PfRPAIS [60, 92]. Artemisinin has also been shown to induce G0/G1 arrest in Leishmania donovani promastigotes [93] and in multiple cancer cell lines [35]. For Plasmodium, understanding the cell cycle arrest phenotype takes on new urgency because it is considered a key mechanism of artemisinin resistance [94]. This resistance is not yet fully understood in molecular terms, but it correlates with mutations in the Kelch-13 protein, which in turn correlate with elevated levels of the phosphoinositide-3-kinase enzyme PfPI3K [95]. PfPI3K, a lipid kinase, is distantly related to protein kinases that are key checkpoint proteins in most eukaryotes but are missing in Plasmodium - an intriguing similarity that is currently under investigation in our laboratory.
The ability of resistant parasites to survive in a dormant state and recrudesce weeks later may be exacerbated by the recent finding that erythrocytic schizogony is remarkably flexible in resistant parasites. Resistant clones from Southeast Asia have a prolonged ring stage and considerably shortened trophozoite stage, presumably reducing drug exposure to the more vulnerable trophozoite [57]. This phenotype was stable without artemisinin drug pressure, which is particularly worrying because it could reduce the available window for antimalarials that target the trophozoite stage and that may be used as partner drugs in artemisinin combination therapies, or as alternative drugs if such therapies fail.
Artemisinin resistance first arose at the Thai/Cambodian border, the historic epicentre of drug resistance development, and Cambodian isolates have been shown to have a mild mutator phenotype and mutations in a number of DNA repair genes, including members of the mismatch repair pathway Mlh1, Pms1 and Exo1 [66]. (This is in addition to mutations in the Kelch-13 propeller domain [96], which serves as a molecular marker for resistance). In bacterial systems, mild mutators acquire mutations at a lower rate than hypermutator lines but remain able to purge deleterious mutations efficiently from the genome and thus they can outcompete hypermutators in the long term [97,98,99,100]. A fine balance is clearly required between the need for accuracy and adaptability, and this balance has probably shifted in P. falciparum in response to continual pressure from antimalarial drug programmes. By analogy with cancer cells - which frequently have deficient checkpoint and DNA repair pathways - artemisinin resistant parasites may be on a knife edge between efficient growth and the potential disaster of an under-regulated cell cycle, so it may be possible to target them with drugs that exacerbate their defects. Proteasome inhibitors can synergise with artemisinin by exacerbating the effects of protein damage [101], so perhaps a similar strategy to inhibit a crucial checkpoint or DNA repair pathway could exacerbate the effects of artemisinin-induced DNA damage.
A better understanding of cell cycle regulation, checkpoints and repair mechanisms in P. falciparum is needed to aid the discovery of compounds that specifically inhibit checkpoints or DNA repair. There may also be scope for drugs that target the cell cycle itself, since components such as the cyc-CDK machinery are distinct from human counterparts. Indeed, like many Plasmodium proteins, some conserved replication proteins also have distinct N-terminal extensions or inserted domains that could serve as highly specific drug targets. Such discoveries could not only circumvent artemisinin resistance but could also identify a much-needed novel class of antimalarial agents. Recently, the CDK inhibitors olomoucine (PfCRK1, PfPK5 and PfPK6 inhibitor), roscovitine (PfPK5 and PfPK6 inhibitor) and WR636638 (chalcone inhibitor of PfMRK) were shown to have differential effects on artemisinin induced dormancy [35]. The DNA synthesis inhibitor aphidicolin and PfPK5 inhibitor flavopiridol also decrease DNA replication in malaria parasites [102]. Overall, the very fact that artemisinin is effective validates DNA damage as a drug target, but the looming catastrophe of widespread artemisinin resistance urgently demands a better understanding of the resistance mechanism, its relationship with DNA repair machinery, and with the control of the Plasmodium cycle cell as a whole.
State-of-the-art focus: Experimental techniques to study the Plasmodium cell cycle
Investigation of the Plasmodium cell cycle has been hampered a lack of transferable techniques from other well-studied systems. Erythrocytic schizogony has been most thoroughly investigated because the blood stages of P. falciparum are relatively easy to culture. However, during schizogony the small size of nuclei (diameter > 1 μm) limits the resolution of immunofluorescence microscopy and the multiple asynchronous rounds of replication complicate flow cytometric definition of S phase parasites based upon DNA content. Furthermore, synchronization of blood-stage parasites, beyond age-range categories or morphological classification via microscopy, has been problematic because the most commonly used agents for chemically synchronising cells at a precise cell-cycle boundary are ineffective in Plasmodium [103].
Fortunately, in recent years significant technological advances have occurred that will facilitate the study of the Plasmodium cell cycle. The advent of bromodeoxyuridine (BrdU) labelling for Plasmodium [104] means that accurate detection of S phase entry and tracing of de novo DNA synthesis is now possible (Fig. 3c). CRK4 depletion in P. falciparum has been shown halt DNA replication [12] and might be exploited as a valuable synchronisation tool. Progress in reverse-genetic techniques such as Crispr/Cas9 [105], Selection linked integration (SLI) [106], and the DiCre system for conditional knockdown [107] have improved the genetic tractability and speed of genetic experiments in the parasite. Such techniques can undoubtedly help to further characterise the cyclin/CDK machinery and the elusive checkpoint systems in Plasmodium. Along with advances in imaging methods, such as improved deconvolution of widefield microscopy, super resolution techniques and spinning disk confocal microscopy, we are now better equipped than ever to study the cell cycles of Plasmodium.
Conclusion
This review summarises our current knowledge about the events of the Plasmodium cell cycles at both the cellular and molecular levels, emphasising many areas of incomplete understanding. The cell cycles pursued by this parasite vary enormously at different life-cycle stages, and even the most accessible of these, which can be grown readily in vitro in human erythrocytes, urgently demands further study. This is especially so because of the great potential for novel drug targets in this area of Plasmodium biology. The increasing availability of experimental tools for such work means that there has never been a better time to advance our understanding of the cell cycle in this globally important human parasite.
Abbreviations
- APC:
-
anaphase promoting complex
- Ark:
-
Aurora kinase
- ARS:
-
autonomously replicating sequence
- BER:
-
base excision repair
- BrdU/CldU/IdU:
-
bromo/chloro/iodo-deoxyuridine
- CDK:
-
cyclin dependent kinase
- CDPK:
-
calcium-dependent protein kinase
- ChIP:
-
chromatin immuno-precipitation
- CP:
-
centriolar plaque
- CRK:
-
CDK-related kinase
- DSB:
-
DNA double strand break
- hpi:
-
hours post-invasion
- HR:
-
homologous recombination
- MCM:
-
minichromosome maintenance protein
- MMEJ:
-
microhomology-mediated end joining
- MMR:
-
mismatch repair
- Nek:
-
NIMA kinase
- NER:
-
nucleotide excision repair
- NHEJ:
-
non-homologous end joining
- ORC:
-
Origin Recognition Complex
- Pb :
-
Plasmodium berghei
- PCNA:
-
proliferating cell nuclear antigen
- Pf :
-
Plasmodium falciparum
- RFC:
-
replication factor complex
- SLI:
-
Selection linked integration
References
Striepen B, Jordan CN, Reiff S, van Dooren GG. Building the perfect parasite: cell division in apicomplexa. PLoS Pathog. 2007;3:e78.
Arnot DE, Gull K. The Plasmodium cell-cycle: facts and questions. Ann Trop Med Parasitol. 1998;92:361–5.
Francia ME, Striepen B. Cell division in apicomplexan parasites. Nat Rev Microbiol. 2014;12:125–36.
Janse CJ, Van der Klooster PF, Van der Kaay HJ, Van der Ploeg M, Overdulve JP. Rapid repeated DNA replication during microgametogenesis and DNA synthesis in young zygotes of Plasmodium berghei. Trans R Soc Trop Med Hyg. 1986;80:154–7.
Kawamoto F, Alejo-Blanco R, Fleck SL, Sinden RE. Plasmodium berghei: ionic regulation and the induction of gametogenesis. Exp Parasitol. 1991;72:33–42.
Roques M, Wall RJ, Douglass AP, Ramaprasad A, Ferguson DJ, Kaindama ML, et al. Plasmodium P-type cyclin CYC3 modulates endomitotic growth during oocyst development in mosquitoes. PLoS Pathog. 2015;11:e1005273.
Milton ME, Replication NSW. maintenance of the Plasmodium falciparum apicoplast genome. Mol Biochem Parasitol. 2016;208:56–64.
Trovesi C, Manfrini N, Falcettoni M, Longhese MP. Regulation of the DNA damage response by cyclin-dependent kinases. J Mol Biol. 2013;425:4756–66.
Diffley JF. Regulation of early events in chromosome replication. Curr Biol. 2004;14:R778–86.
Reilly HB, Wang H, Steuter JA, Marx AM, Ferdig MT. Quantitative dissection of clone-specific growth rates in cultured malaria parasites. Int J Parasitol. 2007;37:1599–607.
Murray L, Stewart LB, Tarr SJ, Ahouidi AD, Diakite M, Amambua-Ngwa A, et al. Multiplication rate variation in the human malaria parasite Plasmodium falciparum. Sci Rep. 2017;7:6436.
Ganter M, Goldberg JM, Dvorin JD, Paulo JA, King JG, Tripathi AK, et al. Plasmodium falciparum CRK4 directs continuous rounds of DNA replication during schizogony. Nat Microbiol. 2017;2:17017.
Arnot DE, Ronander E, Bengtsson DC. The progression of the intra-erythrocytic cell cycle of Plasmodium falciparum and the role of the centriolar plaques in asynchronous mitotic division during schizogony. Int J Parasitol. 2011;41:71–80.
Gupta A, Mehra P, Dhar SK. Plasmodium falciparum origin recognition complex subunit 5: functional characterization and role in DNA replication foci formation. Mol Microbiol. 2008;69:646–65.
Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J, DeRisi JL. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003;1:E5.
Suvorova ES, Francia M, Striepen B, White MW. A novel bipartite centrosome coordinates the apicomplexan cell cycle. PLoS Biol. 2015;13:e1002093.
Francia ME, Jordan CN, Patel JD, Sheiner L, Demerly JL, Fellows JD, et al. Cell division in apicomplexan parasites is organized by a homolog of the striated rootlet fiber of algal flagella. PLoS Biol. 2012;10:e1001444.
Gladfelter AS. Nuclear anarchy: asynchronous mitosis in multinucleated fungal hyphae. Curr Opin Microbiol. 2006;9:547–52.
Kissinger JC. A tale of three genomes: the kinetoplastids have arrived. Trends Parasitol. 2006;22:240–3.
Chen X, Jung S, Beh LY, Eddy SR, Landweber LF. Combinatorial DNA rearrangement facilitates the origin of new genes in Ciliates. Genome Biol Evol. 2015;7:2859–70.
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511.
White JH, Kilbey BJ. DNA replication in the malaria parasite. Parasitol Today. 1996;12:151–5.
Mitra P, Banu K, Deshmukh AS, Subbarao N, Dhar SK. Functional dissection of proliferating-cell nuclear antigens (1 and 2) in human malarial parasite Plasmodium falciparum: possible involvement in DNA replication and DNA damage response. Biochem J. 2015;470:115–29.
Kilbey BJ, Fraser I, McAleese S, Goman M, Ridley RG. Molecular characterisation and stage-specific expression of proliferating cell nuclear antigen (PCNA) from the malarial parasite, Plasmodium falciparum. Nucleic Acids Res. 1993;21:239–43.
Ansari A, Tuteja R. Genome wide comparative comprehensive analysis of Plasmodium falciparum MCM family with human host. Commun Integr Biol. 2012;5:607–15.
Patterson S, Robert C, Whittle C, Chakrabarti R, Doerig C, Chakrabarti D. Pre-replication complex organization in the atypical DNA replication cycle of Plasmodium falciparum: characterization of the mini-chromosome maintenance (MCM) complex formation. Mol Biochem Parasitol. 2006;145:50–9.
Deshmukh AS, Srivastava S, Herrmann S, Gupta A, Mitra P, Gilberger TW, et al. The role of N-terminus of Plasmodium falciparum ORC1 in telomeric localization and var gene silencing. Nucleic Acids Res. 2012;40:5313–31.
Fang H, Klages N, Baechler B, Hillner E, Yu L, Pardo M, et al. Multiple short windows of calcium-dependent protein kinase 4 activity coordinate distinct cell cycle events during Plasmodium gametogenesis. eLife. 2017;6:e26524.
Leonard AC, Méchali M. DNA replication origins. Cold Spring Harb Perspect Biol. 2013;5:a010116.
Agarwal M, Bhowmick K, Shah K, Krishnamachari A, Dhar SK. Identification and characterization of ARS-like sequences as putative origin(s) of replication in human malaria parasite Plasmodium falciparum. FEBS J. 2017;284:2674–95.
Stanojcic S, Kuk N, Ullah I, Sterkers Y, Merrick CJ. Single-molecule analysis reveals that DNA replication dynamics vary across the course of schizogony in the malaria parasite Plasmodium falciparum. Sci Rep. 2017;7:4003.
Invergo BM, Brochet M, Yu L, Choudhary J, Beltrao P, Billker O. Sub-minute phosphoregulation of cell cycle systems during Plasmodium gamete formation. Cell Rep. 2017;21:2017–29.
Blow JJ, Gillespie PJ, Francis D, Jackson DA. Replication origins in Xenopus egg extract are 5–15 kilobases apart and are activated in clusters that fire at different times. J Cell Biol. 2001;152:15–25.
Malumbres M, Harlow E, Hunt T, Hunter T, Lahti JM, Manning G, et al. Cyclin-dependent kinases: a family portrait. Nat Cell Biol. 2009;11:1275–6.
Gray KA, Gresty KJ, Chen N, Zhang V, Gutteridge CE, Peatey CL, et al. Correlation between cyclin dependent kinases and artemisinin-induced dormancy in Plasmodium falciparum in vitro. PLoS One. 2016;11:e0157906.
Deshmukh AS, Agarwal M, Dhar SK. Regulation of DNA replication proteins in parasitic protozoans: possible role of CDK-like kinases. Curr Genet. 2016;62:481–6.
Merckx A, Le Roch K, Nivez MP, Dorin D, Alano P, Gutierrez GJ, et al. Identification and initial characterization of three novel cyclin-related proteins of the human malaria parasite Plasmodium falciparum. J Biol Chem. 2003;278:39839–50.
Chen Y, Jirage D, Caridha D, Kathcart AK, Cortes EA, Dennull RA, et al. Identification of an effector protein and gain-of-function mutants that activate Pfmrk, a malarial cyclin-dependent protein kinase. Mol Biochem Parasitol. 2006;149:48–57.
Peng Y, Keenan SM, Welsh WJ. Structural model of the Plasmodium CDK, Pfmrk, a novel target for malaria therapeutics. J Mol Graph Model. 2005;24:72–80.
Jirage D, Chen Y, Caridha D, O'Neil MT, Eyase F, Witola WH, et al. The malarial CDK Pfmrk and its effector PfMAT1 phosphorylate DNA replication proteins and co-localize in the nucleus. Mol Biochem Parasitol. 2010;172:9–18.
Ward P, Equinet L, Packer J, Doerig C. Protein kinases of the human malaria parasite Plasmodium falciparum: the kinome of a divergent eukaryote. BMC Genomics. 2004;5:79.
Halbert J, Ayong L, Equinet L, Le Roch K, Hardy M, Goldring D, et al. A Plasmodium falciparum transcriptional cyclin-dependent kinase-related kinase with a crucial role in parasite proliferation associates with histone deacetylase activity. Eukaryot Cell. 2010;9:952–9.
Dorin-Semblat D, Bottrill AR, Solyakov L, Tobin A, Doerig C. Experimental tools for the study of protein phosphorylation in Plasmodium. Methods Mol Biol. 2013;923:241–57.
Le Roch K, Sestier C, Dorin D, Waters N, Kappes B, Chakrabarti D, et al. Activation of a Plasmodium falciparum cdc2-related kinase by heterologous p25 and cyclin H. Functional characterization of a P. falciparum cyclin homologue. J Biol Chem. 2000;275:8952–8.
Deshmukh AS, Agarwal M, Mehra P, Gupta A, Gupta N, Doerig CD, et al. Regulation of Plasmodium falciparum Origin Recognition Complex subunit 1 (PfORC1) function through phosphorylation mediated by CDK-like kinase PK5. Mol Microbiol. 2015;98:17–33.
Robbins JA, Absalon S, Streva VA, Dvorin JD. The malaria parasite cyclin H homolog PfCyc1 is required for efficient cytokinesis in blood-stage Plasmodium falciparum. MBio. 2017;8(3):e00605-17.
Bracchi-Ricard V, Barik S, Delvecchio C, Doerig C, Chakrabarti R, Chakrabarti D. PfPK6, a novel cyclin-dependent kinase/mitogen-activated protein kinase-related protein kinase from Plasmodium falciparum. Biochem J. 2000;347:255–63.
Iwanaga T, Sugi T, Kobayashi K, Takemae H, Gong H, Ishiwa A, et al. Characterization of Plasmodium falciparum cdc2-related kinase and the effects of a CDK inhibitor on the parasites in erythrocytic schizogony. Parasitol Int. 2013;62:423–30.
Reininger L, Billker O, Tewari R, Mukhopadhyay A, Fennell C, Dorin-Semblat D, et al. A NIMA-related protein kinase is essential for completion of the sexual cycle of malaria parasites. J Biol Chem. 2005;280:31957–64.
Reininger L, Tewari R, Fennell C, Holland Z, Goldring D, Ranford-Cartwright L, et al. An essential role for the Plasmodium Nek-2 Nima-related protein kinase in the sexual development of malaria parasites. J Biol Chem. 2009;284:20858–68.
Reininger L, Garcia M, Tomlins A, Müller S, Doerig C. The Plasmodium falciparum, Nima-related kinase Pfnek-4: a marker for asexual parasites committed to sexual differentiation. Malar J. 2012;11:250.
Carvalho TG, Doerig C, Reininger L. Nima- and Aurora-related kinases of malaria parasites. Biochim Biophys Acta. 2013;1834:1336–45.
Kumar P, Tripathi A, Ranjan R, Halbert J, Gilberger T, Doerig C, et al. Regulation of Plasmodium falciparum development by calcium-dependent protein kinase 7 (PfCDPK7). J Biol Chem. 2014;289:20386–95.
Shaltiel IA, Krenning L, Bruinsma W, Medema RH. The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J Cell Sci. 2015;128:607–20.
Suvorova ES, Radke JB, Ting LM, Vinayak S, Alvarez CA, Kratzer S, et al. A nucleolar AAA-NTPase is required for parasite division. Mol Microbiol. 2013;90:338–55.
Babbitt SE, Altenhofen L, Cobbold SA, Istvan ES, Fennell C, Doerig C, et al. Plasmodium falciparum responds to amino acid starvation by entering into a hibernatory state. Proc Natl Acad Sci USA. 2012;109:E3278–87.
Hott A, Casandra D, Sparks KN, Morton LC, Castanares GG, Rutter A, et al. Artemisinin-resistant Plasmodium falciparum parasites exhibit altered patterns of development in infected erythrocytes. Antimicrob Agents Chemother. 2015;59:3156–67.
Bohórquez EB, Juliano JJ, Kim HS, Meshnick SR. Mefloquine exposure induces cell cycle delay and reveals stage-specific expression of the pfmdr1 gene. Antimicrob Agents Chemother. 2013;57:833–9.
Gray AM, Arguin PM, Hamed K. Surveillance for the safety and effectiveness of artemether-lumefantrine in patients with uncomplicated Plasmodium falciparum malaria in the USA: a descriptive analysis. Malar J. 2015;14:349.
Gupta DK, Patra AT, Zhu L, Gupta AP, Bozdech Z. DNA damage regulation and its role in drug-related phenotypes in the malaria parasites. Sci Rep. 2016;6:23603.
Sinden RE. The cell biology of malaria infection of mosquito: advances and opportunities. Cell Microbiol. 2015;17:451–66.
Read M, Sherwin T, Holloway SP, Gull K, Hyde JE. Microtubular organization visualized by immunofluorescence microscopy during erythrocytic schizogony in Plasmodium falciparum and investigation of post-translational modifications of parasite tubulin. Parasitology. 1993;106:223–32.
Guler JL, Freeman DL, Ahyong V, Patrapuvich R, White J, Gujjar R, et al. Asexual populations of the human malaria parasite, Plasmodium falciparum, use a two-step genomic strategy to acquire accurate, beneficial DNA amplifications. PLoS Pathog. 2013;9:e1003375.
Rathod PK, McErlean T, Lee PC. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc Natl Acad Sci USA. 1997;94:9389–93.
Brown TS, Jacob CG, Silva JC, Takala-Harrison S, Djimdé A, Dondorp AM, et al. Plasmodium falciparum field isolates from areas of repeated emergence of drug resistant malaria show no evidence of hypermutator phenotype. Infect Genet Evol. 2015;30:318–322.
Lee AH, Fidock DA. Evidence of a mild mutator phenotype in Cambodian Plasmodium falciparum malaria parasites. PLoS One. 2016;11:e0154166.
Bopp SE, Manary MJ, Bright AT, Johnston GL, Dharia NV, Luna FL, et al. Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Genet. 2013;9:e1003293.
Claessens A, Hamilton WL, Kekre M, Otto TD, Faizullabhoy A, Rayner JC, et al. Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of Var genes during mitosis. PLoS Genet. 2014;10:e1004812.
Russell K, Cheng CH, Bizzaro JW, Ponts N, Emes RD, Le Roch K, et al. Homopolymer tract organization in the human malarial parasite Plasmodium falciparum and related apicomplexan parasites. BMC Genomics. 2014;15:848.
Hamilton WL, Claessens A, Otto TD, Kekre M, Fairhurst RM, Rayner JC, et al. Extreme mutation bias and high AT content in Plasmodium falciparum. Nucleic Acids Res. 2017;45:1889–901.
Khan SM, Reece SE, Waters AP, Janse CJ, Kaczanowski S. Why are male malaria parasites in such a rush?: Sex-specific evolution and host-parasite interactions. Evol Med Public Health. 2013;2013:3–13.
Billker O, Dechamps S, Tewari R, Wenig G, Franke-Fayard B, Brinkmann V. Calcium and a calcium-dependent protein kinase regulate gamete formation and mosquito transmission in a malaria parasite. Cell. 2004;117:503–14.
Tajedin L, Anwar M, Gupta D, Tuteja R. Comparative insight into nucleotide excision repair components of Plasmodium falciparum. DNA Repair (Amst). 2015;28:60–72.
Shankar J, Tuteja R. UvrD helicase of Plasmodium falciparum. Gene. 2008;410:223–33.
Ahmad M, Tuteja R. Emerging importance of mismatch repair components including UvrD helicase and their cross-talk with the development of drug resistance in malaria parasite. Mutat Res. 2014;770:54–60.
Haltiwanger BM, Matsumoto Y, Nicolas E, Dianov GL, Bohr VA, Taraschi TF. DNA base excision repair in human malaria parasites is predominantly by a long-patch pathway. Biochemistry. 2000;39:763–72.
Aravind L, Iyer LM, Wellems TE, Miller LH. Plasmodium biology: genomic gleanings. Cell. 2003;115:771–85.
Straimer J, Lee MC, Lee AH, Zeitler B, Williams AE, Pearl JR, et al. Site-specific genome editing in Plasmodium falciparum using engineered zinc-finger nucleases. Nat Methods. 2012;9:993–8.
Kirkman LA, Lawrence EA, Deitsch KW. Malaria parasites utilize both homologous recombination and alternative end joining pathways to maintain genome integrity. Nucleic Acids Res. 2014;42:370–9.
Gopalakrishnan AM, Aly ASI, Aravind L, Kumar N. Multifunctional involvement of a C2H2 zinc finger protein (PbZfp) in malaria transmission, histone modification, and susceptibility to DNA damage response. MBio. 2017;8(4):e01298-17.
Lee AH, Symington LS, Fidock DA. DNA repair mechanisms and their biological roles in the malaria parasite Plasmodium falciparum. Microbiol Mol Biol Rev. 2014;78:469–86.
Fox BA, Ristuccia JG, Gigley JP, Bzik DJ. Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot Cell. 2009;8:520–9.
Fox BA, Falla A, Rommereim LM, Tomita T, Gigley JP, Mercier C, et al. Type II Toxoplasma gondii KU80 knockout strains enable functional analysis of genes required for cyst development and latent infection. Eukaryot Cell. 2011;10:1193–206.
Singer M, Marshall J, Heiss K, Mair GR, Grimm D, Mueller AK, et al. Zinc finger nuclease-based double-strand breaks attenuate malaria parasites and reveal rare microhomology-mediated end joining. Genome Biol. 2015;16:249.
Oakley MS, Gerald N, Anantharaman V, Gao Y, Majam V, Mahajan B, et al. Radiation-induced cellular and molecular alterations in asexual intraerythrocytic Plasmodium falciparum. J Infect Dis. 2013;207:164–74.
Waki S, Yonome I, Suzuki M. Plasmodium falciparum: attenuation by irradiation. Exp Parasitol. 1983;56:339–45.
Waki S, Yonome I, Suzuki M. X-ray sensitivity and DNA synthesis in synchronous culture of Plasmodium falciparum. Z Parasitenkd. 1985;71:213–8.
Hyde JE. Exploring the folate pathway in Plasmodium falciparum. Acta Trop. 2005;94:191–206.
White NJ. Antimalarial drug resistance. J Clin Invest. 2004;113:1084–92.
Ekland EH, Fidock DA. In vitro evaluations of antimalarial drugs and their relevance to clinical outcomes. Int J Parasitol. 2008;38(7):743.
Gopalakrishnan AM, Kumar N. Antimalarial action of artesunate involves DNA damage mediated by reactive oxygen species. Antimicrob Agents Chemother. 2015;59:317–25.
Gopalakrishnan AM, Kumar N. Opposing roles for two molecular forms of replication protein A in Rad51-Rad54-mediated DNA recombination in Plasmodium falciparum. MBio. 2013;4(3):e00252–13.
Sen R, Bandyopadhyay S, Dutta A, Mandal G, Ganguly S, Saha P, et al. Artemisinin triggers induction of cell-cycle arrest and apoptosis in Leishmania donovani promastigotes. J Med Microbiol. 2007;56:1213–8.
Witkowski B, Lelièvre J, Barragán MJ, Laurent V, Su XZ, Berry A, et al. Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob Agents Chemother. 2010;54:1872–7.
Mbengue A, Bhattacharjee S, Pandharkar T, Liu H, Estiu G, Stahelin RV, et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature. 2015;520:683–7.
Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–5.
Denamur E, Matic I. Evolution of mutation rates in bacteria. Mol Microbiol. 2006;60:820–7.
Orlén H, Hughes D. Weak mutators can drive the evolution of fluoroquinolone resistance in Escherichia coli. Antimicrob Agents Chemother. 2006;50:3454–6.
Baquero MR, Nilsson AI, Turrientes MC, Sandvang D, Galán JC, Martínez JL, et al. Polymorphic mutation frequencies in Escherichia coli: emergence of weak mutators in clinical isolates. J Bacteriol. 2004;186:5538–42.
Denamur E, Tenaillon O, Deschamps C, Skurnik D, Ronco E, Gaillard JL, et al. Intermediate mutation frequencies favor evolution of multidrug resistance in Escherichia coli. Genetics. 2005;171:825–7.
Dogovski C, Xie SC, Burgio G, Bridgford J, Mok S, McCaw JM, et al. Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol. 2015;13:e1002132.
Graeser R, Wernli B, Franklin RM, Kappes B. Plasmodium falciparum protein kinase 5 and the malarial nuclear division cycles. Mol Biochem Parasitol. 1996;82:37–49.
Naughton JA, Bell A. Studies on cell-cycle synchronization in the asexual erythrocytic stages of Plasmodium falciparum. Parasitology. 2007;134:331–7.
Merrick CJ. Transfection with thymidine kinase permits bromodeoxyuridine labelling of DNA replication in the human malaria parasite Plasmodium falciparum. Malar J. 2015;14:490.
Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio JJ. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol. 2014;32:819–21.
Birnbaum J, Flemming S, Reichard N, Soares AB, Mesén-Ramírez P, Jonscher E, et al. A genetic system to study Plasmodium falciparum protein function. Nat Methods. 2017;14:450–6.
Jones ML, Das S, Belda H, Collins CR, Blackman MJ, Treeck M. A versatile strategy for rapid conditional genome engineering using loxP sites in a small synthetic intron in Plasmodium falciparum. Sci Rep. 2016;6:21800.
Prudêncio M, Rodriguez A, Mota MM. The silent path to thousands of merozoites: the Plasmodium liver stage. Nat Rev Microbiol. 2006;4:849–56.
Mikolajczak SA, Kappe SH. A clash to conquer: the malaria parasite liver infection. Mol Microbiol. 2006;62:1499–506.
Khan SM, Waters AP. Malaria parasite transmission stages: an update. Trends Parasitol. 2004;20:575–80.
Baton LA, Ranford-Cartwright LC. Spreading the seeds of million-murdering death: metamorphoses of malaria in the mosquito. Trends Parasitol. 2005;21:573–80.
Beier JC. Malaria parasite development in mosquitoes. Annu Rev Entomol. 1998;43:519–43.
Doerig C, Endicott J, Chakrabarti D. Cyclin-dependent kinase homologues of Plasmodium falciparum. Int J Parasitol. 2002;32:1575–85.
Doerig C, Horrocks P, Coyle J, Carlton J, Sultan A, Arnot D, et al. Pfcrk-1, a developmentally regulated cdc2-related protein kinase of Plasmodium falciparum. Mol Biochem Parasitol. 1995;70:167–74.
Rangarajan R, Bei A, Henry N, Madamet M, Parzy D, Nivez MP, et al. Pbcrk-1, the Plasmodium berghei orthologue of P. falciparum cdc-2 related kinase-1 (Pfcrk-1), is essential for completion of the intraerythrocytic asexual cycle. Exp Parasitol. 2006;112:202–7.
Dorin-Semblat D, Schmitt S, Semblat JP, Sicard A, Reininger L, Goldring D, et al. Plasmodium falciparum NIMA-related kinase Pfnek-1: sex specificity and assessment of essentiality for the erythrocytic asexual cycle. Microbiology. 2011;157:2785–94.
Tewari R, Straschil U, Bateman A, Böhme U, Cherevach I, Gong P, et al. The systematic functional analysis of Plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell Host Microbe. 2010;8:377–87.
Acknowledgements
We acknowledge K. Monsell for early development of some of the topics in this article and F. Noulin and P. Horrocks for critical reading.
Funding
This work was supported by an ERC research grant to CJM, ‘Plasmocycle’.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
CJM conceived and planned the review. HM, CWD and CJM researched and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Matthews, H., Duffy, C.W. & Merrick, C.J. Checks and balances? DNA replication and the cell cycle in Plasmodium. Parasites Vectors 11, 216 (2018). https://doi.org/10.1186/s13071-018-2800-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-018-2800-1