
Abstract
Background
Liver fibrosis is an excessive wound-healing reaction that requires the participation of inflammatory cells and hepatic stellate cells (HSCs). The pathogenesis of liver fibrosis caused by viruses and alcohol has been well characterized, but the molecular mechanisms underlying liver fibrosis induced by the liver fluke Clonorchis sinensis are poorly understood. Lysophospholipase A (LysoPLA), which deacylates lysophospholipids, plays a critical role in mediating the virulence and pathogenesis of parasites and fungi; however, the roles of C. sinensis lysophospholipase A (CsLysoPLA) in C. sinensis-induced liver fibrosis remain unknown.
Methods
A mouse macrophage cell line (RAW264.7) was cultured and treated with CsLysoPLA. IL-25 and members of its associated signaling pathway were detected by performing quantitative real-time PCR, Western blotting and immunofluorescent staining. A human hepatic stellate cell line (LX-2) was cultured and exposed to IL-25. LX-2 cell activation markers were examined via quantitative real-time PCR, Western blotting and immunofluorescent staining. Migration was analyzed in transwell plates.
Results
Treating RAW264.7 cells with CsLysoPLA significantly induced IL-25 expression. Elevated PKA, B-Raf, and ERK1/2 mRNA levels and phosphorylated B-Raf and ERK1/2 were detected in CsLysoPLA-stimulated RAW264.7 cells. The PKA inhibitor H-89 weakened B-Raf and ERK1/2 phosphorylation whereas the AKT activator SC79 attenuated ERK1/2 phosphorylation in RAW264.7 cells. Both H-89 and SC79 inhibited CsLysoPLA-induced IL-25 upregulation. In addition, stimulation of LX-2 cells with IL-25 upregulated the expression of mesenchymal cell markers, including α-smooth muscle actin (α-SMA) and collagen type I (Collagen-I), and promoted cell migration.
Conclusions
CsLysoPLA activates HSCs by upregulating IL-25 in macrophages through the PKA-dependent B-Raf/ERK1/2 pathway and potentially promotes hepatic fibrosis during C. sinensis infection.
Similar content being viewed by others


Background
Clonorchiasis, a food-borne zoonosis, is caused by Clonorchis sinensis infection [1,2,3]. Adults of C. sinensis parasitize the intra-hepatic bile ducts of their hosts. Long-term infection by C. sinensis results in chronic liver injury leading to liver fibrosis [4, 5]. Mechanical damage caused by the adult C. sinensis worm and excretory/secretory proteins (ESPs) as well as the interplay between worms and the host immune system are responsible for pathological changes [6, 7]. However, the exact molecular mechanisms involved in C. sinensis-induced liver fibrosis remain unclear.
IL-25 (also known as IL-17E) is a member of the IL-17 cytokine family and is considered a T helper type 2 (Th2) cell-derived cytokine [8]. IL-25 is also expressed in alveolar macrophages, mast cells and eosinophils [9,10,11]. Unlike the proinflammatory effects exerted by other members of the IL-17 family, IL-25 promotes type 2 inflammation by locally upregulating IL-4, IL-5 and IL-13 [8, 9]. In mice, the intranasal administration or forced expression of IL-25 induces pulmonary inflammation similar to asthma [12, 13]. Administration of an IL-25 blocking antibody in allergen-exposed mice results in a moderate reduction in airway inflammation [14]. IL-25 also has the ability to modulate tumor pathogenesis. IL-25 administration in mouse xenograft models of human melanoma, breast, lung, colon, and pancreatic cancers induces antitumor activity that requires the presence of B cells and eosinophil infiltration [15]. In addition, IL-25, which is essential for host defense, is induced at high levels following helminth infection [16, 17].
Liver fibrosis is an excessive wound-healing reaction associated with chronic injury to the liver, such as that caused by virus and parasite infections, alcohol abuse, and metabolic and autoimmune diseases [18, 19]. When the liver is subjected to chronic injury, hepatic stellate cells (HSCs) are exposed to autocrine or paracrine signals, including oxidative stress, apoptotic bodies, and cytokines such as TGF-β1 and PDGF, and transform into activated myofibroblast-like cells [20]. Activated HSCs not only generate extracellular matrix (ECM) but also secrete cytokines and growth factors to promote the development of liver fibrosis [21]. Liver fibrosis is a sequela of various inflammatory processes comprising both innate and adaptive immune responses [22, 23]. Infection with C. sinensis is characterized by a Th2-dominant immune response, which is vital for the development of liver fibrosis [24,25,26], and hepatic macrophages also reportedly play a critical role [27]. Supporting this link, macrophages were shown to produce IL-25 in a rat model of particle-induced airway inflammation [9]. IL-25 is a Th2 cytokine, and according to the same study investigating rat airway inflammation, hepatic macrophages overexpress IL-25 and may contribute to liver fibrosis caused by C. sinensis.
Lysophospholipase A (LysoPLA) is a member of the phospholipase family and has been identified in many mammalian tissues and cells. This enzyme deacylates lysophospholipids and likely plays a pivotal role in the virulence and pathogenesis of parasites and fungi [28,29,30]. Previously, we expressed and characterized C. sinensis lysophospholipase A (CsLysoPLA) and observed that it upregulated the expression of pro-fibrotic genes in a hepatic stellate cell line (LX-2) [30, 31]. In the present study, we detected IL-25 levels in a macrophage cell line (RAW264.7) treated with CsLysoPLA in vitro and analyzed levels of signaling molecules. Furthermore, we evaluated cell migration and mRNA expression levels in LX-2 cells after IL-25 administration.
Methods
Expression and purification of recombinant CsLysoPLA
As previously described [30, 31], the CsLysoPLA coding region was amplified by polymerase chain reaction (PCR) using a cDNA plasmid library derived from adult C. sinensis worms as a template. The PCR product was cloned into the His6-tagged expression vector pET-28a(+) after digestion with BamH I/Xho I (Thermo Fisher Scientific, Waltham, MA, USA). The recombinant plasmid was then transformed into Escherichia coli BL21 (DE3) for overexpression induced by isopropyl-β-D-thiogalactoside (IPTG). Escherichia coli were harvested by centrifugation and resuspended in phosphate-buffered saline (PBS), sonicated on ice, and centrifuged to collect the supernatant. The recombinant protein was purified using a His Bind Purification Kit (Novagen, Darmstadt, Germany), eluted with 150 mM imidazole and dialyzed in PBS to remove the imidazole.
Culture and treatment of RAW264.7 and LX-2 cells
RAW264.7 cells (2.5 × 105 cells/well) were seeded in 24-well plates in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Carlsbad, USA) containing 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were cultured at 37 °C in an atmosphere containing 5% CO2 until they reached 80% confluence. The cells were then exposed to CsLysoPLA (1, 5, 10 or 20 μg/ml) for 12 h, 24 h and 48 h, C. sinensis fructose-1,6-bisphosphatase (CsFBPase) (20 μg/ml) and mouse serum albumin (MSA, 20 μg/ml) (Fitzgerald, MA, USA) as control proteins, or equal volumes of PBS as blank controls. To investigate the effects of signaling inhibitors or activators, RAW264.7 cells were treated with CsLysoPLA (10 μg/ml) in the presence or absence of H-89 (a PKA inhibitor) (20 μM) (Beyotime, Shanghai, China) or SC79 (an AKT activator) (4 μg/ml) (Sigma-Aldrich, Steinheim, Germany) for 15 min, 30 min, 60 min and 90 min as previously described [32,33,34]. LX-2 cells (2 × 105 cells/well) were seeded in 6-well plates and grown to 70% confluence at 37 °C in an atmosphere containing 5% CO2 followed by serum starvation for 24 h. The cells were then stimulated with IL-25 (20 ng/ml) (R&D, Minneapolis, USA) with or without BAY 11–7083 (an NF-κB inhibitor) (0.1 μg/ml) (Beyotime, Shanghai, China) for 24 h; TGF-β1 (5 ng/ml) (Peprotech, Rocky Hill, USA) was used as a positive control.
Reverse transcription and quantitative real-time PCR
Total cellular RNA was extracted from RAW264.7 or LX-2 cells using TRIzol reagent according to the manufacturer’s protocol. cDNA was synthesized using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA) and amplified on a Bio-Rad CFX96 Real-Time system (Bio-Rad, Hercules, CA, USA) with SYBR Green I (Takara, Dalian, China) and specific primers for quantitative analysis. Briefly, the cDNA was pre-denatured at 95 °C for 30 s, followed by 40 cycles at 95 °C for 5 s and 60 °C for 30 s. β-actin was amplified as a house-keeping gene for each sample. Relative fold-changes in mRNA expression were determined by calculating 2-ΔΔCt. Primer sequences are listed in Table 1.
Immunofluorescent staining
RAW264.7 or LX-2 cells were cultured on slides. The cells were fixed with 4% paraformaldehyde for 20 min at room temperature (RT), then washed with PBS and permeabilized in PBS containing 0.3% Triton X-100 for 10 min. The slides were blocked with PBS containing 1% bovine serum albumin for 30 min and incubated with a primary monoclonal antibody against IL-25 (R&D, Minneapolis, USA) or collagen type I (Collagen-I) (Abcam, London, UK) overnight at 4 °C. The slides were then incubated with a Cy3-conjugated or FITC-conjugated secondary antibody (Proteintech, Chicago, USA) for 1 h in darkness. Finally, the slides were stained with 4′,6-diamidino-2-phenylindole (DAPI) and mounted with antifade reagent (Beyotime, Shanghai, China). Images were obtained using an Olympus BX63 microscope and cellSens Dimension (Version1.8) software (Olympus, Tokyo, Japan). The intensity of IL-25 and collagen type I staining was analyzed with Image-Pro Plus v6.0 software.
Western blotting
Protein lysates from cells receiving different treatments were prepared using RIPA buffer (Beyotime, Shanghai, China) containing protease and phosphatase inhibitors (KeyGEN, Nanjing, China). Equal amounts of total protein (30 μg of protein per lane) were loaded onto gels for electrophoresis and then transferred onto PVDF membranes (Millipore, Billerica, USA), followed by incubation in blocking buffer (25 mM Tris, pH 7.4, 0.15 M NaCl, 0.1% Tween-20, 5% nonfat milk) for 1 h at RT. The membranes were incubated with primary antibodies against α-smooth muscle actin (α-SMA) (Proteintech, Chicago, USA), IL-25 (R&D, Minneapolis, USA), total ERK1/2, phospho-ERK1/2, total AKT, phospho-AKT, phospho-B-Raf (Cell Signaling Technology, Boston, USA) or GADPH (Proteintech, Chicago, USA) at 4 °C overnight. After washing, the membranes were incubated with HRP-conjugated secondary antibodies for 1 h at RT. Proteins were visualized with an ECL kit (Advansta, CA, USA). GADPH was used as a loading control. The intensity of Western blotting bands was analyzed with Quantity One v4.6.2 software.
Cell migration assays
Cell migration assays were performed as previously described [35]. Briefly, the upper wells of transwells with 8.0-μm pore polycarbonate membrane inserts in a 24-well plate (Corning, NY, USA) were filled with 100 μl of serum-free medium containing 5 × 104 LX-2 cells, and the lower wells contained IL-25 (20 ng/ml) in 600 μl of serum-free medium. The plate was incubated for 12 h at 37 °C. Migrated cells adhering to the undersides of inserts were fixed with 100% methanol for 30 min and then stained with 0.1% crystal violet (Leagene, Beijing, China) for 15 min. Migrated cells were counted by enumerating the number of stained cells in five independent fields for each experiment under a light microscope (Leica, Wetzlar, Germany).
Statistical analysis
All data are presented as the mean ± SEM. Data were analyzed by performing independent Student’s t-tests and ANOVA followed by Bonferroni’s post-hoc multiple comparisons test using SPSS software for Windows (version 16.0; SPSS, Inc., IL, USA). A P value <0.05 was considered statistically significant.
Results
CsLysoPLA upregulated IL-25 expression in the RAW264.7 macrophage cell line
CsLysoPLA stimulation increased IL-25 transcription levels in a time-dependent manner in RAW264.7 cells (F (238) = 27.805, P < 0.0001) (Fig. 1a). To confirm the specificity of this CsLysoPLA-induced IL-25 transcription, CsFBPase, a member of the C. sinensis excretory/secretory proteins (CsESPs), and MSA were applied as controls. Unlike CsLysoPLA, incubation with CsFBPase or MSA did not elicit significant changes in IL-25 mRNA levels (Fig. 1a). Furthermore, the levels of other cytokines associated with macrophage function, such as TNF-α, iNOS, IL-6, IL-4, IL-13, IL-10 and IL-33, did not change significantly in RAW264.7 cells (Fig. 1b). Western blotting also revealed an increase in IL-25 protein expression compared with the expression in PBS-treated RAW264.7 cells (Fig. 1c, d). Similarly, stronger green fluorescence emitted by an anti-IL-25 monoclonal antibody was observed in RAW264.7 cells incubated with CsLysoPLA compared with the PBS group (Fig. 1e, f).

IL-25 is highly expressed in CsLysoPLA-stimulated RAW264.7 cells. a Quantitative real-time PCR analysis of IL-25 in RAW264.7 cells treated with CsLysoPLA (1, 5, 10 and 20 μg/ml) and PBS (0 μg/ml, negative control) for 12, 24 and 48 h. CsFBPase (20 μg/ml) and MSA (20 μg/ml) were used as control proteins. Data are shown as mean ± SEM. b Quantitative real-time PCR analysis of TNF-α, iNOS, IL-6, IL-13, IL-10 and IL-33 in RAW264.7 cells treated with CsLysoPLA (1, 5, 10 and 20 μg/ml) and PBS (0 μg/ml, negative control) for 24 h. c Western blot analysis of IL-25 in RAW264.7 cells treated with CsLysoPLA (10 μg/ml) and the equal volume of PBS for 24 h. GADPH was used as a loading control. d Quantification of western blot data in Fig. 1c. Data are shown as mean ± SEM. ** P < 0.01, ***P < 0.001. e Immunofluorescence staining analysis of IL-25 (green) in RAW264.7 cells treated with CsLysoPLA (10 μg/ml) and the equal volume of PBS for 24 h. Nuclei were stained with DAPI (blue). Original magnification ×100. Scale-bars: 200 μm. f Quantification of immunofluorescence data in Fig. 1e. Data are shown as mean ± SEM. ***P < 0.001
CsLysoPLA stimulated IL-25 expression in RAW264.7 cells via the PKA-dependent B-Raf-ERK1/2 signaling pathway
After 24 h, increased mRNA levels of protein kinase A (PKA) (F (2,6) = 19.815, P = 0.002), B-Raf (F (2,6) = 17.593, P = 0.003), extracellular signal-regulated kinase 1 (ERK1) (F (2,6) = 61.151, P < 0.0001) and extracellular signal-regulated kinase 2 (ERK2) (F (2,6) = 14.275, P = 0.005) were detected in CsLysoPLA-stimulated RAW264.7 cells (Fig. 2a). Western blotting showed that CsLysoPLA induced both B-Raf and downstream ERK1/2 phosphorylation 15 min after stimulation, and phosphorylation gradually increased until 90 min after stimulation (Fig. 2b, c). By contrast, CsLysoPLA inhibited AKT phosphorylation. The PKA inhibitor H-89 attenuated B-Raf and ERK1/2 phosphorylation from 15 min to 90 min (Fig. 2b, c). The AKT activator SC79 also reduced the levels of phosphorylated ERK1/2 in RAW264.7 cells (Fig. 2d, e). Moreover, both H-89 (t (4) = 3.933, P = 0.017) and SC79 (t (4) = 4.480, P = 0.011) inhibited IL-25 expression induced by CsLysoPLA (Fig. 2f).

CsLysoPLA facilitates IL-25 expression in RAW264.7 cells via PKA-dependent B-Raf-ERK1/2 pathway. a RAW264.7 cells were stimulated with CsLysoPLA (5, 10 μg/ml) for 24 h. Relative expressions of PKA, B-Raf, and ERK1/2 genes were examined by Quantitative real-time PCR. b RAW264.7 cells were stimulated with CsLysoPLA (10 μg/ml) in the absence or presence H-89 (20 μM) for 15, 30, 60 and 90 min. The protein levels of phospho-B-Raf, total ERK1/2, phospho-ERK1/2, total AKT, phospho-AKT were detected by western blotting with their respective antibodies. GADPH was used as a loading control. c Quantification of western blot data in (b). “−/−” PBS control, “+/−” cells treated with CsLysoPLA and “+/+” cells treated with CsLysoPLA and H-89. d RAW264.7 cells were stimulated with CsLysoPLA (10 μg/ml) with or without SC79 (4 μg/ml) for 15 and 30 min. The protein levels of total ERK1/2, phospho-ERK1/2, total AKT, phospho-AKT were detected by western blotting with their respective antibodies. PBS was used as negative control. e Quantification of western blot data in (d). “−/−” PBS control, “+/−” cells treated with CsLysoPLA and “+/+” cells treated with CsLysoPLA and SC79. f RAW264.7 cells were stimulated by CsLysoPLA (10 μg/ml) in the presence of H-89 (20 μM) or SC79 (4 μg/ml) for 12 h. The level of IL-25 mRNA was analyzed by quantitative real-time PCR. Data are shown as mean ± SEM. *P < 0.05
IL-25-induced activation and migration of LX-2 cells
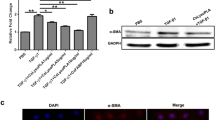
Both α-SMA and Collagen-I are markers of activated HSCs [19]. α-SMA mRNA levels in LX-2 cells were significantly upregulated (t (4) = 4.634, P = 0.010) following stimulation with IL-25 for 24 h (Fig. 3a), and α-SMA protein levels exhibited the same change (Fig. 3b, c). Furthermore, LX-2 cells were pretreated with an NF-κB inhibitor (BAY 11–7083) before stimulation with IL-25. BAY 11–7083 blockade resulted in a reduction in α-SMA protein expression compared with IL-25 treatment (Fig. 3d, e). Red fluorescence from a Cy3-conjugated anti-collagen-I monoclonal antibody was markedly stronger in IL-25-treated LX-2 cells compared with cells that underwent PBS treatment (Fig. 3f, g). In addition, much greater amounts of LX-2 cells migrated to the lower well following IL-25 treatment compared with PBS-treated cells (t (4) = 2.984, P = 0.017) (Fig. 3h, i).

IL-25 facilitates LX-2 cells activation and migration. a LX-2 cells were stimulated with IL-25 (20 ng/ml) and TGF-β1 (5 ng/ml) for 24 h. Relative α-SMA expression was examined by quantitative real-time PCR. b LX-2 cells were stimulated with IL-25 (20 ng/ml) and TGF-β1 (5 ng/ml) for 24 h. α-SMA protein expression was detected by western blotting. GADPH was used as loading control. c Quantification of western blot data in (b). d LX-2 cells were stimulated with IL-25 (20 ng/ml) in the absence or presence of BAY 11–7083 (0.1 μg/ml) for 24 h. α-SMA protein expression was detected by western blotting. GADPH was used as loading control. e Quantification of western blot data in (d). f LX-2 cells were stimulated with IL-25 (20 ng/ml) for 24 h. Collagen-I protein expression was determined by immunofluorescence staining using Collagen-I antibody (red). Nuclei were stained with DAPI (blue). Original magnification ×200. g Quantification of immunofluorescence data in Fig. 3f. h LX-2 cells were stimulated with IL-25 (20 ng/ml) for 24 h. The migration was analyzed using a Transwell plate. Light microscopy was used to collect images of migrated cells. Original magnification ×100. The number of migration cells was counted as a mean of five independent fields for each experiment. PBS was used as negative control and TGF-β1 as the positive control. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, *** P < 0.001. Scale-bars: f, 100 μm; h, 200 μm
Discussion
Liver fibrosis caused by C. sinensis infection affects patient quality of life, but the underlying mechanisms have yet to be clarified. Liver fibrosis is a well-known repair response during liver injury, and HSCs and many cytokines take part in its progression [19,20,21]. In the current study, treatment with CsLysoPLA induced IL-25 expression in RAW264.7 cells. PKA, B-Raf, and ERK1/2 mRNA levels in CsLysoPLA-stimulated RAW264.7 cells increased. CsLysoPLA induced the phosphorylation of both B-Raf and ERK1/2, whereas the PKA inhibitor H-89 attenuated B-Raf and ERK1/2 phosphorylation, confirming the role of CsLysoPLA. The AKT activator SC79 reduced the levels of phosphorylated ERK1/2 in RAW264.7 cells. Both H-89 and SC79 inhibited IL-25 upregulation induced by CsLysoPLA. In addition, IL-25 upregulated the expression of α-SMA and Collagen-I in LX-2 cells and promoted cell migration.
The host immune response to C. sinensis infection tends to be Th2-dominant [24, 25]. IL-25 is a Th2 cytokine regulator, and previous reports revealed an association between IL-25 and pulmonary disorders such as pulmonary fibrosis and airway remodeling [14, 36]. According to a previous study investigating a rat model of particle-induced airway inflammation, macrophages are potential sources of IL-25 [9]. Additionally, CsLysoPLA was proposed to play a role in C. sinensis-induced liver fibrosis. Therefore, we stimulated RAW264.7 cells with CsLysoPLA and observed that CsLysoPLA significantly promoted the expression of IL-25 but not TNF-α, iNOS, IL-6, IL-4, IL-13, IL-10 or IL-33, which mediate macrophage functions [37, 38]. Thus, CsLysoPLA may interfere with macrophage function by upregulating IL-25 expression. An excretory/secretory protein from C. sinensis, CsFBPase, did not elevate IL-25 expression in RAW264.7 cells when applied as a control, which suggests a specific function for CsLysoPLA in RAW264.7 cells.
Rat lysophospholipase removes palmitate from Gα subunits, accelerating the cycling of Gα subunits between palmitoylation and depalmitoylation and resulting in increased G protein signaling efficacy [39]. The ERK signaling cascade plays an important role in regulating gene expression, cell proliferation and differentiation, and apoptosis [40,41,42]. ERK1/2 activation is modulated by Gα via the cAMP/PKA signaling cascade, which ultimately activates the B-Raf-MEK-ERK module [43,44,45]. In addition, the AKT and ERK pathways undergo negative crosstalk to induce AKT-mediated ERK signaling pathway inactivation [46,47,48]. In the present study, we observed PKA, B-Raf and ERK1/2 activation and AKT inhibition. Blocking PKA and activating AKT with the chemical inhibitors H-89 and SC79, respectively, further confirmed the signaling pathways involved in CsLysoPLA-induced IL-25 expression. Enhanced AKT activation, inhibited ERK1/2 excitation and attenuated CsLysoPLA-induced IL-25 overexpression were present in RAW264.7 cells. These results suggested that the ERK1/2 signaling pathway is involved in CsLysoPLA-induced IL-25 elevation in macrophages; additionally, there may be other transcriptional mediators contributing to IL-25 production. In summary, CsLysoPLA-mediated IL-25 production is partially dependent on the PKA-dependent B-Raf/ERK1/2 pathway.
Macrophages play a major functional role in liver fibrosis. Both macrophage depletion in Cd11b-DTR transgenic mice and macrophage blockade in mice via liposomal clodronate injection in response to CCL4 resulted in prominently reduced HSC activation and numbers as well as attenuated fibrosis [49, 50]. To our knowledge, macrophages take part in the development of liver fibrosis by secreting a diverse range of cytokines, chemokines and other soluble regulators that directly act on HSCs [23].
HSC activation represents a pivotal event in liver fibrosis [51]. Activated HSCs convert to a myofibroblast-like phenotype, upregulate mesenchymal cell markers such as α-SMA and Collagen-I, and migrate to sites of damage [19, 23]. IL-25 significantly increases collagen secretion by normal human lung fibroblasts [14]. In the present study, we first investigated the direct interaction between IL-25 and HSCs. IL-25 enhanced the expression of α-SMA and Collagen-I and promoted the migration of LX-2 cells. Both of these effects likely result in the secretion and accumulation of excessive ECM proteins and facilitate the pathogenesis of liver fibrosis [51]. Thus, the present findings suggest IL-25 may be a profibrotic cytokine that regulates fibrogenesis by directly activating HSCs and promoting their migration. Recent studies indicate NF-κB is essential for IL-25-mediated inflammation and hyper-responsiveness [52, 53]. We observed inhibited α-SMA expression in LX-2 cells following the blockade of NF-κB with a chemical inhibitor (BAY 11–7083), suggesting that IL-25 may stimulate the expression of profibrotic genes in HSCs via NF-κB signaling pathway activation. The precise mechanism underlying IL-25 function requires further investigation.
Conclusions
Our previous work showed that IL-25 is significantly elevated in the serum of C. sinensis-infected mice, and this trend correlated with the degree of liver fibrosis during infection. Based on these results, we speculate that CsLysoPLA infiltrates blood capillaries broken following mechanical damage and chemical injury induced by the adult worm and its excretory/secretory products, then activates HSCs by upregulating IL-25 in macrophages through the PKA-dependent B-Raf/ERK1/2 pathway, thus promoting hepatic fibrosis during infection (Fig. 4). This hypothesis needs further verification in vivo in future studies. Nonetheless, our work may provide valuable information for the development of liver fibrosis therapies.

Schematic chart showing the potential role of CsLysoPLA in hepatic fibrosis. CsLysoPLA activates PKA, B-Raf and ERK1/2, and inhibits AKT phosphorylation, upregulating IL-25 in macrophages. IL-25 enhances the expression of α-SMA and Collagen-I in LX-2 cells, promoting hepatic fibrosis
Abbreviations
- Collagen-I:
-
Collagen type I
- CsESPs:
-
C. sinensis excretory/secretory proteins
- CsFBPase:
-
C. sinensis Fructose-1, 6-bisphosphatase
- CsLysoPLA:
-
C. sinensis lysophospholipase A
- DAPI:
-
4′,6-diamidino-2-phenylindole
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- ECM:
-
Extracellular matrix
- FBS:
-
Fetal bovine serum
- HSCs:
-
Hepatic stellate cells
- MSA:
-
Mouse serum albumin
- PBS:
-
Phosphate-buffered saline
- Th2:
-
Helper T cells.
- α-SMA:
-
α-smooth muscle actin
References
Lin J, Qu H, Chen G, He L, Xu Y, Xie Z, et al. Clonorchis sinensis acetoacetyl-CoA thiolase: identification and characterization of its potential role in surviving in the bile duct. Parasit Vectors. 2015;8:125.
Kim JG, Ahn CS, Kim SH, Bae YA, Kwon NY, Kang I, et al. Clonorchis sinensis omega-class glutathione transferases play major roles in the protection of the reproductive system during maturation and the response to oxidative stress. Parasit Vectors. 2016;9(1):337.
Tang Z, Shang M, Chen T, Ren P, Sun H, Qu H, et al. The immunological characteristics and probiotic function of recombinant Bacillus subtilis spore expressing Clonorchis sinensis cysteine protease. Parasit Vectors. 2016;9(1):648.
Sripa B, Kaewkes S, Sithithaworn P, Mairiang E, Laha T, Smout M, et al. Liver fluke induces cholangiocarcinoma. PLoS Med. 2007;4(7):e201.
Choi BI, Han JK, Hong ST, Lee KH. Clonorchiasis and cholangiocarcinoma: etiologic relationship and imaging diagnosis. Clin Microbiol Rev. 2004;17(3):540–52.
Yan C, Wang YH, Yu Q, Cheng XD, Zhang BB, Li B, et al. Clonorchis sinensis excretory/secretory products promote the secretion of TNF-alpha in the mouse intrahepatic biliary epithelial cells via Toll-like receptor 4. Parasit Vectors. 2015;8:559.
Xu Y, Lin J, Bian M, Chen W, Liang P, Wang X, et al. CsRNASET2 is an important component of Clonorchis sinensis responsible for eliciting Th2 immune response. Parasitol Res. 2015;114(6):2371–9.
Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15(6):985–95.
Kang CM, Jang AS, Ahn MH, Shin JA, Kim JH, Choi YS, et al. Interleukin-25 and interleukin-13 production by alveolar macrophages in response to particles. Am J Respir Cell Mol Biol. 2005;33(3):290–6.
Ikeda K, Nakajima H, Suzuki K, Kagami S, Hirose K, Suto A, et al. Mast cells produce interleukin-25 upon Fc epsilon RI-mediated activation. Blood. 2003;101(9):3594–6.
Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J Exp Med. 2007;204(8):1837–47.
Rickel EA, Siegel LA, Yoon BR, Rottman JB, Kugler DG, Swart DA, et al. Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. J Immunol. 2008;181(6):4299–310.
Pan G, French D, Mao W, Maruoka M, Risser P, Lee J, et al. Forced expression of murine IL-17E induces growth retardation, jaundice, a Th2-biased response, and multiorgan inflammation in mice. J Immunol. 2001;167(11):6559–67.
Gregory LG, Jones CP, Walker SA, Sawant D, Gowers KH, Campbell GA, et al. IL-25 drives remodelling in allergic airways disease induced by house dust mite. Thorax. 2013;68(1):82–90.
Benatar T, Cao MY, Lee Y, Lightfoot J, Feng N, Gu X, et al. IL-17E, a proinflammatory cytokine, has antitumor efficacy against several tumor types in vivo. Cancer Immunol Immunother. 2010;59(6):805–17.
Hurst SD, Muchamuel T, Gorman DM, Gilbert JM, Clifford T, Kwan S, et al. New IL-17 family members promote Th1 or Th2 responses in the lung: In vivo function of the novel cytokine IL-25. J Immunol. 2002;169(1):443–53.
Zhao A, Urban JF Jr, Sun R, Stiltz J, Morimoto M, Notari L, et al. Critical Role of IL-25 in nematode infection-induced alterations in intestinal function. J Immunol. 2010;185(11):6921–9.
Anthony B, Allen JT, Li YS, McManus DP. Hepatic stellate cells and parasite-induced liver fibrosis. Parasit Vectors. 2010;3(1):60.
Iwaisako K, Brenner DA, Kisseleva T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J Gastroenterol Hepatol. 2012;27(Suppl 2):65–8.
Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25(2):195–206.
Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123(5):1902–10.
Wick G, Grundtman C, Mayerl C, Wimpissinger TF, Feichtinger J, Zelger B, et al. The immunology of fibrosis. Annu Rev Immunol. 2013;31:107–35.
Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14(3):181–94.
Xu Y, Liang P, Bian M, Chen W, Wang X, Lin J, et al. Interleukin-13 is involved in the formation of liver fibrosis in Clonorchis sinensis-infected mice. Parasitol Res. 2016;115(7):2653–60.
Kim EM, Yu HS, Jin Y, Choi MH, Bae YM, Hong ST. Local immune response to primary infection and reinfection by Clonorchis sinensis in FVB mice. Parasitol Int 2016. doi:10.1016/j.parint.2016.11.006.
Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;104(6):777–85.
Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60(5):1090–6.
Wang A, Deems RA, Dennis EA. Cloning, expression, and catalytic mechanism of murine lysophospholipase I. J Biol Chem. 1997;272(19):12723–9.
Wang A, Dennis EA. Mammalian lysophospholipases. Biochim Biophys Acta. 1999;1439(1):1–16.
Zhang F, Liang P, Chen W, Wang X, Hu Y, Liang C, et al. Stage-specific expression, immunolocalization of Clonorchis sinensis lysophospholipase and its potential role in hepatic fibrosis. Parasitol Res. 2013;112(2):737–49.
Ma C, Hu X, Hu F, Li Y, Chen X, Zhou Z, et al. Molecular characterization and serodiagnosis analysis of a novel lysophospholipase from Clonorchis sinensis. Parasitol Res. 2007;101(2):419–25.
Feng X, Sun T, Bei Y, Ding S, Zheng W, Lu Y, et al. S-nitrosylation of ERK inhibits ERK phosphorylation and induces apoptosis. Sci Rep. 2013;3:1814.
Yang R, Piperdi S, Gorlick R. Activation of the RAF/mitogen-activated protein/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase pathway mediates apoptosis induced by chelerythrine in osteosarcoma. Clin Cancer Res. 2008;14(20):6396–404.
Jo H, Mondal S, Tan D, Nagata E, Takizawa S, Sharma AK, et al. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc Natl Acad Sci USA. 2012;109(26):10581–6.
Zhou L, Shang M, Shi M, Zhao L, Lin Z, Chen T, et al. Clonorchis sinensis lysophospholipase inhibits TGF-β1-induced expression of pro-fibrogenic genes through attenuating the activations of Smad3, JNK2, and ERK1/2 in hepatic stellate cell line LX-2. Parasitol Res. 2016;115(2):643–50.
Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci USA. 2014;111(1):367–72.
Juhas U, Ryba-Stanisławowska M, Szargiej P, Myśliwska J. Different pathways of macrophage activation and polarization. Postepy Hig Med Dosw. 2015;69:496–502.
Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604.
Duncan JA, Gilman AG. A Cytoplasmic acyl-protein thioesterase that removes palmitate from G protein a subunits and p21 (RAS). J Biol Chem. 1998;273(25):15830–7.
Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773(8):1213–26.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–84.
Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351 Pt 2:289–305.
Thomas CM, Hong T, van Pijkeren JP, Hemarajata P, Trinh DV, Hu W, et al. Histamine derived from probiotic Lactobacillus reuteri suppresses TNF via modulation of PKA and ERK signaling. PLoS One. 2012;7(2):e31951.
Goldsmith ZG, Dhanasekaran DN. G Protein regulation of MAPK networks. Oncogene. 2007;26(22):3122–42.
Sachs K, Perez O, Pe'er D, Lauffenburger DA, Nolan GP. Causal protein-signaling networks derived from multiparameter single-cell data. Science. 2005;308(5721):523–9.
Rommel C, Clarke BA, Zimmermann S, Nuñez L, Rossman R, Reid K, et al. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286(5445):1738–41.
Dai R, Chen R, Li H. Cross-talk between PI3K/Akt and MEK/ERK pathways mediates endoplasmic reticulum stress-induced cell cycle progression and cell death in human hepatocellular carcinoma cells. Int J Oncol. 2009;34:1749–57.
Guan KL, Figueroa C, Brtva TR, Zhu T, Taylor J, Barber TD, et al. Negative regulation of the serine/threonine kinase B-Raf by Akt. J Biol Chem. 2000;275:27354–9.
Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58(4):1461–73.
Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115(1):56–65.
Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–56.
Wong CK, Li PW, Lam CW. Intracellular JNK, p38 MAPK and NF-κB regulate IL-25 induced release of cytokines and chemokines from costimulated T helper lymphocytes. Immunol Lett 2007;112(2):82-91.
Cheung PF, Wong CK, Ip WK, Lam CW. IL-25 regulates the expression of adhesion molecules on eosinophils: mechanism of eosinophilia in allergic inflammation. Allergy. 2006;61(7):878–85.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Key Basic Research and Development Project (973 project, No. 2010CB530000), the Science and Technology Planning Project of Guangdong Province (No. 2016A050502008), the National Important Sci-Tech Special Projects (No. 2012ZX10004220), and the National Natural Science Foundation of China (No. 81101270).
Availability of data and materials
All data generated or analysed during this study are included in this article.
Authors’ contributions
Conceived and designed the experiments: LNZ, JX, YH and XBY. Performed the experiments: LNZ. Analyzed the data: LNZ, MCS and LZ. Contributed reagents/materials/analysis tools: LNZ, ZPL, ZLT, HCS, TJC and ZYL. Wrote the paper: LNZ, YH and XBY. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, L., Shi, M., Zhao, L. et al. Clonorchis sinensis lysophospholipase A upregulates IL-25 expression in macrophages as a potential pathway to liver fibrosis. Parasites Vectors 10, 295 (2017). https://doi.org/10.1186/s13071-017-2228-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-017-2228-z