
Abstract
Background
African animal trypanosomosis, transmitted cyclically by tsetse flies or mechanically by other biting flies, causes serious inflictions to livestock health. This study investigates the extent of non-tsetse transmitted animal trypanosomosis (NTTAT) by Trypanosoma (T.) evansi and T. vivax in domestic animals in the tsetse-free regions of Northern Ethiopia, Afar and Tigray.
Methods
A cross sectional study was conducted on 754 dromedary camels, 493 cattle, 264 goats, 181 sheep, 84 donkeys, 25 horses and 10 mules. The microhaematocrit centrifugation technique was used as parasitological test. Plasma was collected for serodiagnosis with CATT/T.evansi and RoTat 1.2 immune trypanolysis (ITL) while buffy coat specimens were collected for molecular diagnosis with T. evansi type A specific RoTat 1.2 PCR, T. evansi type B specific EVAB PCR and T. vivax specific TvPRAC PCR.
Results
The parasitological prevalence was 4.7% in Tigray and 2.7% in Afar and significantly higher (z = 2.53, p = 0.011) in cattle (7.3%) than in the other hosts. Seroprevalence in CATT/T.evansi was 24.6% in Tigray and 13.9% in Afar and was significantly higher (z = 9.39, p < 0.001) in cattle (37.3%) than in the other hosts. On the other hand, seroprevalence assessed by ITL was only 1.9% suggesting cross reaction of CATT/T.evansi with T. vivax or other trypanosome infections. Molecular prevalence of T. evansi type A was 8.0% in Tigray and in Afar and varied from 28.0% in horses to 2.2% in sheep. It was also significantly higher (p < 0.001) in camel (11.7%) than in cattle (6.1%), donkey (6%), goat (3.8%), and sheep (2.2%). Four camels were positive for T. evansi type B. Molecular prevalence of T. vivax was 3.0% and was similar in Tigray and Afar. It didn’t differ significantly among the host species except that it was not detected in horses and mules.
Conclusions
NTTAT caused by T. vivax and T. evansi, is an important threat to animal health in Tigray and Afar. For the first time, we confirm the presence of T. evansi type B in Ethiopian camels. Unexplained results obtained with the current diagnostic tests in bovines warrant particular efforts to isolate and characterise trypanosome strains that circulate in Northern Ethiopia.
Similar content being viewed by others


Background
Ethiopia is the richest country in livestock population in Africa with more than 52 million heads of cattle, 46 million small ruminants, about 9 million equines (donkeys, horses and mules) and 1 million camels [1]. The livestock resource contributes to 12% of the total gross domestic product (GDP) and over 45% of the agricultural GDP of Ethiopia. However, the benefit derived from livestock is far below its potential. Inadequate food supply, high disease prevalence, poor genetic resources and poor marketing are the main bottlenecks for the development of the livestock sector [2].
African trypanosomosis is one of the most important animal diseases encountered in all agro-ecological zones of the country and hinders the efforts made for food self-sufficiency [3]. African trypanosomosis is a general term for infections in many different hosts (man and his domestic animals and wild animals) caused by various trypanosome species with Trypanosoma (T.) brucei, T. congolense, T. vivax, T. evansi and T. equiperdum as the most important ones [4]. African animal trypanosomoses (AAT) cause serious inflictions to the health of livestock ranging from anaemia, loss of condition and emaciation, abortion, death etc. [5-10]. The trypanosomes responsible for AAT in Ethiopia are T. vivax, T. congolense, T. brucei, T. evansi and T. equiperdum [11].
T. congolense and T. brucei are exclusively found in the tsetse-infested areas of Ethiopia while T. evansi and T. equiperdum occur in the tsetse-free areas. T. vivax can be found in both tsetse-infested and tsetse-free areas except in the highlands, which are >2500 meter above sea level [11,12].
In Africa, T. vivax is transmitted both cyclically by Glossina spp. and mechanically by horse flies (Tabanidae) and stable flies (Stomoxys sp.). It circulates in several species of ungulates including cattle, small ruminants, equids, camelids and wild animals such as antelopes [4]. Wild ungulates, especially buffaloes and antelopes, as well as trypanotolerant cattle are generally symptomless carriers [13]. T. vivax is also endemic in Latin America where its transmission is exclusively mechanical through biting flies [14-17].
T. evansi has multiple means of transmission of which mechanical transmission by biting insects is the most important in camels and other large animals. Other transmission routes such as the bite of vampire bats and oral transmission in carnivores has been documented [4,18,19].
In Ethiopia, T. evansi is widely distributed across the six agro-climatic zones and mainly coincides with the distribution of camels [20]. Trypanosomosis due to T. evansi (surra) is the number one protozoan disease of camels. Horses are also very susceptible. Infected camels and equines may die within 3 months. Moreover, cattle, buffalo, pigs, goat and sheep infected with T. evansi suffer from immunosuppression, resulting in increased susceptibility to other diseases or in vaccination failure [21-23]. For example, experimental infections in buffalo and pigs have shown reduced cellular and humoral responses after vaccination against classical swine fever and Pasteurella multicoda in T. evansi infected animals compared to uninfected animals [24-26].
T. evansi strains with kDNA minicircle type A are the most abundant and found in Africa, South America and Asia [27-29]. They are also characterised by the presence of the gene for the Variant Surface Glycoprotein (VSG) RoTat 1.2. This RoTat 1.2 VSG is expressed early during infections resulting in the detectability of anti-RoTat 1.2 antibodies in animals infected with T. evansi type A [30]. In contrast, T. evansi strains with type B minicircle are far less common and have so far been isolated only from camels in Kenya [31-35]. Ngaira et al. showed that T. evansi type B typically lacks the RoTat 1.2 gene and as a consequence, infections with this type are not detected with serological and molecular tests based on RoTat 1.2 VSG, like CATT/T.evansi and RoTat 1.2 PCR [32,34,36,37].
Despite the considerable number of epidemiological studies carried out in Ethiopia on cattle and camel trypanosomosis in parts of Southern Nations, Nationalities, and Peoples’ Region (SNNPR), and in Oromiya and Amhara regions, information from Tigray and pastoral areas of Afar, belonging to the tsetse-free areas of Ethiopia, is scanty [38-45]. In addition, due to limited logistic resources and poor diagnostic facilities, the exact burden and socioeconomic impact of AAT is probably underestimated and information on prevailing trypanosome species and affected hosts remains inaccurate and fragmented [44,46,47]. Therefore, this study was designed to investigate the distribution of T. evansi and T. vivax in selected districts of Tigray and in pastoral areas of Afar.
Diagnosis of AAT is often based on clinical suspicion. Parasite detection is cumbersome in many cases where only low numbers of trypanosomes circulate in the host body fluids [47]. Techniques for concentration of the trypanosomes by centrifugation of a blood specimen can be applied. After centrifugation of some blood in a capillary tube, the trypanosomes can be detected directly under the microscope at the level of the white blood cell layer (the buffy coat) [48]. Where differential diagnosis is needed, the capillary tube can be broken and the buffy coat spread on a microscope slide for examination according to Murray et al. [49]. A more sensitive technique is the mini Anion Exchange Centrifugation Technique (mAECT) but the technique works best with T. brucei and T. evansi and has poor diagnostic potential for T. congolense and T. vivax [50-53].
As an alternative to parasitological diagnosis, molecular diagnostic tests have been developed. For the diagnosis of surra, the PCR RoTat 1.2 and Q-PCR RoTat 1.2 are specific for T. evansi type A and PCR EVAB is specific for T. evansi type B [34,37,54]. For the molecular diagnosis of T. vivax, the ITS-1 PCR and proline racemase PCR (TvPRAC PCR) can be employed [55,56]. Neither parasitological nor molecular tests are 100% sensitive, due to the often low number of circulating parasites.
Serological tests are able to reveal ongoing or past trypanosome infections based on antibody detection. For surra, the most specific antibody detection tests make use of the T. evansi specific variant surface glycoprotein (VSG) RoTat 1.2 as antigen. The CATT/T.evansi is such a test in the form of a direct agglutination test and is the only rapid diagnostic test for surra that is recommended by the World Organization for Animal Health [57,58]. By virtue of its format as a direct agglutination test, CATT/T.evansi can be applied on any host species. Knowledge about the antigenic repertoires of T. vivax is almost non-existent. Most antibody detection tests for T. vivax make use of more or less purified native antigens leaving room for non-specific reactions. In regions where T. vivax and T. brucei or T. evansi occur together in the same host species, it is almost impossible to identify the infecting trypanosome species at the level of circulating antibodies in the host [47,59-61]. Only recently, recombinant T. vivax specific antigens are being investigated for their diagnostic potential [62].
The present study provides data on the epidemiology of AAT in domestic animals in two tsetse-free regions of Ethiopia.
Methods
Study areas
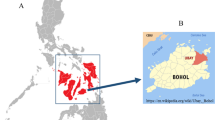
The study was conducted in selected districts (weredas) of Tigray and pastoral areas of Afar, representing tsetse-free areas of Ethiopia. Tigray region is located in the northern part of Ethiopia between longitudes 36°27′ E and 39°59′ E and latitudes 12°15′ N and 14°57′ N (Figure 1). It shares international boundaries with Eritrea and Sudan and regional boundaries with Amhara and Afar regions of Ethiopia. Tigray is divided into four zones and 35 weredas [63]. Selected “tabias” or peasant associations from the districts of Raya-Azebo (southern zone), Tselemti (northwestern zone) and Kafta-Humera and Tsegede (western zone), were included.

Map of Ethiopia showing study districts in Tigray and Afar regions and tsetse belt areas.
Afar region, one of the four major pastoral regions in Ethiopia, occupies an area of about 270,000 km2 and is situated between longitudes 39°34′E and 42°28′E and latitudes 8°49′N and 14°30′N [64]. The region shares international boundaries with the State of Eritrea and Djibouti, as well as regional boundaries with the regions of Tigray, Amhara, Oromia and Somali (Figure 1). The Afar region consists of 5 administrative zones (sub-regions) [65]. Taking into account the accessibility to the pastoral communities, “kebeles” or sampling stations were selected in the districts of Megale (zone 2), Awash Fentale and Amibara (zone 3) and Gulina and Yalo (zone 4).
Ethics statement
The study protocol was approved by the Veterinary Ethics Committee of the Institute of Tropical Medicine (EXT2012-1).
Study design, study animals and specimen collection
Considering 95% confidence level and average prevalence of 30% [44], the number of specimens to collect was planned according to Thrusfield [66] as n = (1.96)2 × Pexp(1-Pexp)/d2; where: n = required sample size, d = absolute precision required (d = 0.05), Pexp = expected prevalence of the disease. A cross sectional study was conducted from February till July 2013 on 1811 domestic animals comprising 754 dromedary camels, 493 cattle, 264 goats, 181 sheep, 84 donkeys, 25 horses and 10 mules. The animals were sampled at watering and grazing points and at veterinary clinics where they were brought for accaricide spraying and vaccination. Individual study subjects from willing owners were randomly selected regardless of age, gender and body condition [66]. From each animal, 9 mL of jugular vein blood was collected in a heparinised Venosafe tube (Terumo, Leuven, Belgium), labelled with a unique code, placed in a coolbox at 4°C and processed as described below.
Packed cell volume (PCV) and microhaematocrit centrifugation technique (mHCT)
The microhaematocrit (mHCT) was performed as described by Woo [67]. Briefly, 4 microhaematocrit capillary tubes were filled with approximately 50 μL of blood from the Venosafe tube and stoppered with sealant. After centrifugation at 12,000 rpm for 5 min, the PCV was recorded and the tubes were mounted in a specially designed viewing chamber and examined under the microscope at 10x16 magnification for the presence of motile trypanosomes at the level of the buffy coat as described by Fikru et al. [44]. Animals with confirmed presence of trypanosomes were treated free of charge with 0.25 mg/kg melarsamine hydrochloride (cymelarsan) in the case of camels or with 0.5 mg/kg isometamidium chloride (samorin, trypamidium) or 7 mg/kg diminazeneaceturate (berenil) in the case of ruminants.
Preparation of plasma and buffy coat specimens
The blood collected in the heparinised Venosafe tubes was centrifuged for 10 min at 1,000 rpm and plasma was collected with a single use plastic transfer pipette into 2 mL tubes with screwcaps (Sarstedt, Nümbrecht, Germany). Plasma was stored at 4°C until testing for specific antibodies with CATT/T.evansi and subsequently frozen at −20°C. From the remaining blood specimen, 500 μL of buffy coat layer were collected by means of a micropipette with a filter tip and mixed with an equal volume of guanidium EDTA buffer (GEB; 6 M guanidium chloride, 0.2 M EDTA, pH 8.0) and stored at ambient temperature until DNA extraction [68]. Of those animals that were parasitologically positive, part of the buffy coat was collected for cryopreservation in liquid nitrogen for later isolation of the parasite according to Pyana et al. [69].
CATT/T. evansi
Detection of T. evansi specific antibodies was carried out by CATT/T.evansi on plasma that was prediluted 1:4 in CATT diluent, according to the instructions of the manufacturer (Institute of Tropical Medicine, Antwerp, Belgium).
Immune trypanolysis test for surra
From each plasma specimen, 30 μL were spotted on Whatman 4 filter paper (Whatman, Maidstone, UK) in Ethiopia and shipped to the Institute of Tropical Medicine, Antwerp, Belgium. For elution of plasma and test procedures, the protocol developed by Camara and co-workers, with modifications, was employed [70]. Briefly, from each filter paper, two 6 mm diameter disks were punched and placed in a well of a flat bottom microlon microtitre plate (Greiner Bio-One, Wemmel, Belgium). Antibodies were eluted overnight at 4°C in 40 μL of fetal bovine serum (FBS) followed by 1 hour on a plate shaker at ambient temperature. Twenty μL of the eluted fraction were transferred into a well of a U-bottom polystyrene microtitre plate (Sterilin, Newport, UK). Next, T. evansi RoTat 1.2 trypanosomes, grown in a mouse, were diluted in ice-cold guinea pig serum (GPS) and kept on ice to obtain a suspension of 5 trypanosomes per microscopic field according to the matching method [71]. Twenty μL of this suspension were added to each well of the microtiter plate with the eluted specimens and incubated at ambient temperature for 1 hour. Antibody mediated complement lysis was assessed by dispensing 5 μL of the reaction mixture on a microscope slide, covered by a 18×18 mm cover slip and examination at 25×10 magnification under a phase-contrast microscope. Trypanolysis was considered positive when at least 50% of the trypanosomes were lysed [70].
DNA extraction
DNA extraction was performed with the High Pure PCR Product Purification Kit (Roche Diagnostics, Mannheim, Germany). Since unexpected clotting of the buffy coat specimens preserved in GEB was observed, 200 μL of tissue lysis buffer and 50 μL of proteinase K (Roche Diagnostics, Mannheim, Germany) were added to the 1 mL buffy coat-GEB mixture followed by digestion for 90 min at 56°C under constant shaking at 1,000 rpm. Eventually, DNA was extracted from 240 μL of this mixture and stored at −20°C until use. DNA concentrations were measured in the Nanodrop ND-1000 UV–vis spectrophotometer (NanoDrop Technologies, Wilmington, USA) or the Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, USA).
PCR
All PCR amplifications were carried out in 200 μL thin-wall PCR tubes (ABgene, Epsom, UK) in a T3 thermocycler 48 (Biometra, Göttingen, Germany). Amplified products were visualised under UV after electrophoresis in a 2% agarose gel at 135 V for 30 minutes and staining with ethidium bromide. To check the quality of DNA, a PCR targeting vertebrate cytochrome b was performed [72,73]. To detect T. evansi type A, the RoTat 1.2 PCR was conducted [37] while the EVAB PCR was used for the detection of T. evansi type B [34]. Detection of T. vivax was performed by means of TvPRAC PCR [56]. ITS1-PCR was used to test part of the specimen collection for T. congolense, T. theileri and Trypanozoon [55]. Each PCR assay was done in 25 μL reaction volumes with 12.5 μL HotStarTaq polymerase master mix (Qiagen, Leipzig, Germany) containing 2.5 units HotStarTaq DNA polymerase, 1 × PCR buffer with 1.5 mM MgCl2 and 200 μM of each dNTP, 0.8 μM of each primer (Biolegio, Amsterdam, Netherlands), 8 μL accugene water (Lonza, Verviers, Belgium) and 2.5 μL of template DNA.
The target genes, primer names and sequences and expected amplicon lengths are represented in Table 1. Compared to the references cited in the table, some minor changes were made at the level of the polymerase and master mix, the initial denaturation step and the numbers of cycles. Cycling conditions for the different PCRs were as follows. Cytochrome B PCR: 94°C for 15 min and 35 cycles of 30 sec at 94°C, 30 sec at 52°C, 30 sec at 72°C and final extension for 5 min at 72°C. RoTat 1.2 PCR: 94°C for 15 min and 40 cycles of 30 sec at 94°C, 30 sec at 59°C, 30 sec at 72°C and final extension for 5 min at 72°C. EVAB PCR: 94°C for 15 min and 30 cycles of 30 sec at 94°C, 30 sec at 60°C, 60 sec at 72°C and final extension for 1 min at 72°C. TvPRAC PCR: 94°C for 15 min and 30 cycles of 30 sec at 94°C, 30 sec at 63°C, 30 sec at 72°C and final extension for 5 min at 72°C. ITS-1 PCR: 94°C for 15 min and 40 cycles of 30 sec at 94°C, 30 sec at 60°C, 30 sec 72°C and final extension for 5 min at 72°C.
Data analysis
All data were recorded in Microsoft Excel. STATA/MP 13.1 [74] was used for statistical analysis. Percentages with 95% confidence interval (CI) were used to express prevalence. Logistic regression was applied for assessing differences in prevalence of AAT between domestic animal species and evaluating the effect of infection (test positive) on PCV values. To assess agreement between the diagnostic tests, Cohen’s kappa coefficient was calculated and interpreted according to Landis and Koch [75]. P-values <0.05 were considered as significant.
Results
In total, 1811 animals were sampled of which 959 (53%) in Tigray and 852 (47%) in Afar. In general, there was statistically significant interaction (X 2 = 330.12, p < 0.001) between regions and sampled domestic animal species, i.e. more cattle and camels were sampled in Tigray than in Afar, while more sheep and goats were sampled in Afar than in Tigray.
Parasite detection
In 68 animals, trypanosomes were detected (Table 2). Thus, the overall parasitologically confirmed prevalence of trypanosomosis was 3.8% (CI 2.9-4.6%) with 4.7% (CI 3.4-6.0%) in Tigray and 2.7% (CI 1.6-3.8%) in Afar. No trypanosomes were detected in equines. The parasitological prevalence in cattle (7.3%, CI 5.0-9.5%) was significantly higher (z = 2.53, p = 0.011) than in camels (4.0%, CI 2.6-5.4%), sheep (0.6%, CI 0–1.7%) and goats (0.4%, CI 0–1.2%).
Serology
With CATT/T.evansi, antibodies were detected in 354 animals (Table 2). Thus, the overall seroprevalence was 19.6% (CI 17.7-21.4%) with 24.6% (CI 21.9-27.3%) in Tigray and 13.9% (CI 11.5-16.2%) in Afar. Among the equines, CATT/T.evansi was only positive in donkeys (10.7%, CI 4.0-17.4%). The overall seroprevalence was significantly higher (z = 9.39, p < 0.001) in cattle (37.3%, CI 33.1-41.6%) than in camels (13.7%, CI 11.2-16.1%), in goats (13.3%, CI 9.2-17.4%), in sheep (12.7%, CI 7.8-17.6%) and in donkeys (10.7%, CI 4.1-17.4%).
With the ITL (Table 2), T. evansi-specific antibodies were detected only in 34 animals (30 camels, 2 goats, 1 sheep and 1 donkey). Thus, the seroprevalence in ITL was 1.9% (34/1811, CI 1.3-2.5%).
Kappa statistics indicated a poor but significant agreement between CATT/T.evansi and ITL (p < 0.001, Table 3).
Molecular diagnosis
The overall molecular prevalence of T. evansi type A assessed with RoTat 1.2 PCR was 145/1811 or 8.0% (CI 6.8-9.3%) with 8.0% (CI 6.3-9.8%) in Tigray and 8.0% (CI 6.2-9.8%) in Afar (Table 2). The molecular prevalence of T. evansi type A in camels (11.7%, CI 9.4-14.0%) was significantly higher (p < 0.001) than in cattle (6.1%, CI 4.0-8.2%), donkeys (6.0%, CI 0.9-11.0%), goats (3.8%, CI 1.5-6.1%), and sheep (2.2%, 0.1-4.4%). The molecular prevalence of T. evansi type A was 28.0% (CI 10.4-45.6%) in horse and 10.0% (CI 7.6-27.6%) in mule. Kappa statistics indicated a poor but significant agreement between RoTat 1.2 PCR and the antibody detection tests, ITL and CATT/T. evansi (p < 0.001, Table 3). Among the 145 RoTat 1.2 PCR positives, only 71 were positive in CATT/T.evansi and only 18 were positive in ITL. Four camels, all from Awash Fentale district in Afar, were found positive in EVAB PCR indicating the presence of T. evansi type B. All four were negative in CATT/T.evansi and ITL although one of them was also positive in RoTat 1.2 PCR suggesting a mixed infection.
The overall molecular prevalence of T. vivax assessed with TvPRAC PCR was 54/1811 or 3.0% (CI 2.2-3.8%) with 3.3% (CI 2.2-4.5%) in Tigray and 2.6% (CI 1.5-3.7%) in Afar (Table 2). The molecular prevalences of T. vivax were 3.5% (CI 2.2-4.8%) in camels, 3.0% in goats (CI 1.0-5.1%), 2.6% (CI 1.2-4.1%) in cattle and 2.2% (CI 0.1-4.4%) in sheep and were not significantly different (p = 0.925). All horses and mules were negative in TvPRAC PCR. The molecular prevalence of T. vivax in cattle from Tigray was 3.2% (13/411) but was 0% in Afar. Among the 54 TvPRAC PCR positives, 10 were also positive in CATT/T.evansi but were negative in RoTat 1.2 PCR. Only two camels and one goat were positive in both TvPRAC PCR and RoTat 1.2 PCR.
Among the 68 parasitologically positive animals, 32 cattle, 1 camel and 1 sheep were negative in the RoTat 1.2 PCR, EVAB PCR and TvPRAC PCR. To check for the possibility that mHCT detected T. theileri and T. congolense, ITS1-PCR was run on their specimens. Four cattle were positive for T. vivax and two cattle specimens were positive for T. theileri. Ten were negative. No single one was positive for T. congolense. Eighteen cattle specimens showed a profile with amplicons of different lengths that could not be interpreted unequivocally.
Among the CATT/T.evansi positive animals, 269 (77%) were negative in all PCR tests (165 cattle, 42 camels, 33 goats, 22 sheep and 7 donkeys).
Packed cell volume (PCV)
In Table 4, the average PCV values and standard deviations (SD) are given according to the status of the animals in the mHCT, CATT/T. evansi, RoTat 1.2 PCR and TvPRAC PCR. Camels that were found positive in those tests had a significantly lower average PCV than the animals that were negative in the different tests. The average PCV in ITL positive camels (24.2% ± 3.4%) was not significantly different from ITL negatives (25.7% ± 3.59%) (p = 0.05). In cattle and equines, the average PCV value was significantly lower only in CATT/T.evansi positive animals. In sheep and goats, no significant differences in average PCV were observed.
Discussion
In this cross sectional study, the mHCT, CATT/T.evansi, RoTat 1.2 ITL and RoTat 1.2 PCR, EVAB PCR and TvPRAC PCR were used to assess the non-tsetse transmitted AAT prevalence in domestic animals in two regions of northern Ethiopia, Tigray and Afar. The overall prevalence of AAT as assessed with mHCT was 3.75% which was similar to AAT prevalence reported in cattle from other tsetse-free areas in Ethiopia (3.2% in Gondar and Bale Lowlands) using the same technique [44]. This is probably underestimating the real prevalence since mHCT is acknowledged to detect <50% of infections with low parasitaemia [49,76]. Although only one goat and one sheep were positive in mHCT, this finding confirms the presence of trypanosomosis in small ruminants [38,77-79]. The fact that no single equine was positive in mHCT while some of them were positive in the T. evansi specific RoTat 1.2 PCR and the T. vivax specific TvPRAC PCR, indicates that in these animals the parasitaemia level remained under the lower detection limit of mHCT (about 60 trypanosomes/mL, [80].
With RoTat 1.2 PCR, it was confirmed that all domestic animals are susceptible to infection with T. evansi type A but that camels and horses are particularly at risk [21,22]. With EVAB PCR, we report for the first time the presence of T. evansi type B in camel in Ethiopia. Till today, T. evansi type B has only been isolated from camel in Kenya although indirect evidence exists that it also circulates in Sudan [31,32,81,82]. Furthermore, Hagos et al. suggested the existence of non-RoTat 1.2 T. evansi in camels from Bale zone in Ethiopia based on their finding that about one third of parasitologically positive camels were negative in CATT/T.evansi [45]. Also in our study, all four camels with T. evansi type B were negative in CATT/T. evansi. These data suggest that T. evansi type B is not confined to Kenya but may occur in more East African countries and even beyond, thus calling for the adaptation of serological and molecular diagnostic tests, like CATT/T.evansi and RoTat 1.2 PCR, to ensure detection of both types of T. evansi without compromising specificity.
Our study also confirms that T. vivax can infect diverse domestic animal species, including donkeys [4]. The overall molecular prevalence of T. vivax as assessed with TvPRAC PCR was lower than reported in other studies [44,56]. The present study shows that camels in Ethiopia can be infected with T. vivax and that infection is associated with morbidity reflected by a significant reduction in PCV. Co-infections with T. vivax and T. evansi were rare (2 camels, 1 goat) but characterised by low PCV (20–22.5%). Mixed infection by both parasites was also reported in [83].
As expected, ITS1 PCR confirmed the absence of T. congolense in the mHCT positive animals that were negative in RoTat 1.2 PCR and TvPRAC PCR but revealed four T. vivax infections that were not picked up by TvPRAC PCR. Interestingly, ten mHCT positive animals remained negative in all PCRs. In the single sheep, the presence of the non-pathogenic T. melophagium cannot be ruled out but the other nine negatives remain unexplained [84,85]. Also unexplained remain the 18 cattle specimens showing a complex amplicon profile in ITS1 PCR, including a putative T. vivax specific 150 bp amplicon. In a previous study, which led to the development of the TvPRAC PCR, we observed that the ITS1 PCR can generate non-specific amplicons in the presence of cattle DNA rendering unequivocal interpretation of the results impossible [44]. Although the analytical sensitivity of TvPRAC is lower than of ITS1 PCR, it is still much higher than of mHCT [56]. Therefore, mHCT positive and TvPRAC negative but ITS1 T. vivax positive specimens may be due to particular T. vivax strains not detectable in TvPRAC. To further investigate these unexplained results, it will be necessary to isolate the trypanosomes detected in the mHCT, which will be particularly challenging in case of T. vivax. Indeed, T. vivax is notoriously difficult to grow in laboratory rodents and/or in culture [86,87].
Seroprevalence, as assessed with CATT/T.evansi was much higher than molecular prevalence which is not unexpected for several reasons. First, CATT/T.evansi cannot distinguish current from cured infection as detectable level of antibodies can persist for 2.3–22.6 month after trypanocidal treatment [88,89]. Secondly, in particular in chronic infections, parasitaemia can be well below the detection limit of parasitological and even molecular diagnostic tests, a phenomenon well known in human African trypanosomosis but less studied in AAT [90,91]. Finally, as CATT/T.evansi is not 100% specific, false positive cases do occur [92].
Still, the poor agreement between CATT/T.evansi and ITL is puzzling. Both serological tests detect antibodies against the VSG RoTat 1.2 that is considered specific for T. evansi type A. Although a limited loss in sensitivity of ITL when performed on filter paper eluates may be expected other factors may cause this discrepancy [70,93]. While ITL detects exclusively variant specific antibodies, CATT/T.evansi detects also antibodies directed against non-variant specific epitopes of VSG RoTAt 1.2 and other surface exposed antigens. Thus, infection with other trypanosomes, e.g. T. vivax, may lead to a positive result in CATT/T.evansi as was suggested in a study on bovine trypanosomosis in Suriname [47,60,94]. This cross-reactivity caused by T. vivax infection may explain why all CATT/T.evansi positive cattle specimens remained negative in ITL. However, it provides no explanation why the 30 cattle specimens that were positive in RoTat 1.2 PCR remained negative in ITL and why from the 145 RoTat 1.2 PCR positives, only 71 were also positive in CATT/T.evansi. Is it possible that the target sequence of RoTat 1.2 PCR is also present in some particular T. vivax strains circulating in Afar and Tigray but that the gene containing that sequence is a pseudogene or a gene that is not expressed during an infection? As we were not able to isolate T. vivax strains during this study, a conclusive answer to this question cannot be given.
If one considers a low PCV as a morbidity marker, it is striking that mainly camels are susceptible to AAT as disease. Indeed, camels that were positive in mHCT, CATT/T.evansi, RoTat 1.2 PCR and TvPRAC PCR had a significantly lower PCV than camels that were negative in all these tests. Among the other host species, only cattle and equines that were positive in CATT/T.evansi had a significantly lower PCV than CATT/T.evansi negative animals again suggesting that most CATT/T.evansi positive animals were actually infected, whether with T. evansi or T. vivax.
Among the parasitologically positive animals, three quarter presented without or with only mild symptoms. As in the study region, it is common to treat only sick camels and bovines with trypanocidal drugs such as diminazine and isometamidum, asymptomatic infections remain without treatment and constitute an uncontrolled reservoir for the disease.
Our study has some limitations. Although intended, it was not possible to compare the AAT prevalence between Tigray and Afar since the number of examined individuals per animal species was considerably different between two study regions. Also, no stained blood preparations were prepared that would have allowed morphological characterisation of those parasites that were detected in the mHCT but that remained negative in the species-specific PCRs.
Conclusions
This study shows that non-tsetse transmitted AAT is an important threat to the health of camels, equine and ruminants in Afar and Tigray regions in Ethiopia. In these regions, AAT is caused by T. vivax and T. evansi type A and type B, the latter only in camels. Hence, improving serological and molecular diagnostic tests to detect both types of T. evansi as well as T. vivax is necessary. Unexplained results obtained with the current diagnostic tests in bovine specimens warrant particular efforts to isolate and characterise trypanosome strains that circulate in Northern Ethiopia.
Abbreviations
- CATT:
-
Card agglutination test for trypanosomiasis
- NTTAT:
-
Non-tsetse transmitted animal trypanosomosis
- RoTat:
-
Rhode Trypanozoon antigenic type
- TvPRAC:
-
Trypanosoma vivax proline racemase
- VSG:
-
Variant surface glycoprotein
References
CSA. Central Statistical Authority (CSA) of Federal Democratic Republic of Ethiopia: Agricultural sample report on livestock and livestock characterstics. Addis Ababa, Ethiopia: 2012:9–22. [http://www.csa.gov.et/images/documents/pdf_files/nationalstatisticsabstract/2012/agriculture%202012.pdf].
MOA. Ministry of Agriculture (MOA) Ethiopia Animal Health Year Book (2009/10). Addis Ababa: Federal Ministry of Agriculture Animal and Plant Health Regulatory Directorate (APHRD). Addis Ababa, Ethiopia: 2010:1–63. [http://www.disasterriskreduction.net/fileadmin/user_upload/drought/docs/Ethiopian%20Animal%20Health%20Year%20Book%202009-%2020101.pdf].
Abebe G. Trypanosomosis in Ethiopia. Ethiop J Biol Sci. 2005;4:75–121.
Hoare CA. The Trypanosomes of Mamals. A Zoological Monograph. Oxford, UK: Blackwell Scientific Publications; 1972.
Da Silva AS, Garcia Perez HA, Costa MM, Franca RT, De Gasperi D, Zanette RA, et al. Horses naturally infected by Trypanosoma vivax in southern Brazil. Parasitol Res. 2011;108:23–30.
Van den Bossche P. The Development of A New Strategy For The Sustainable Control of Bovine Trypanosomosis In South Africa, PhD thesis. Pretoria, South Africa: University of Pretoria; 2000.
Da Silva AS, Pierezan F, Wolkmer P, Costa MM, Oliveiro CB, Tonin AA, et al. Pathological findings associated with experimental infection by Trypanosoma evansi in cats. J Comp Pathol. 2010;142:170–6.
Losos GJ. Infectious Diseases of Tropical Diseases of Domestic Animals. New York: Churchill Livingstone; 1986.
Gutierrez C, Corbera JA, Juste MC, Doreste F, Morales I. An outbreak of abortions and high neonatal mortality associated with Trypanosoma evansi infection in dromedary camels in the Canary Islands. Vet Parasitol. 2005;130:163–8.
Lohr KF, Pholpark S, Siriwan P, Leesirikul N, Srikitjakarn L, Staak C. Trypanosoma evansi infection in buffaloes in North-East Thailand. II. Abortions. Trop Anim Health Prod. 1986;18:103–8.
Dagnatchew Z. Trypanosomiasis in Ethiopia. In: Third International Symposium on Veterinary Epidemiology and Economics. Arlington, Virginia, USA: Veterinary Medicine Publishing Company; 1982. p. 467–73.
Abebe G, Yilma J. Trypanosomosis: a threat to cattle production in Ethiopia. Rev Med Vet. 1996;147:897–902.
Moloo SK, Gettinby G, Olubayo RO, Kabata JM, Okumu IO. A comparison of African buffalo, N’Dama and Boran cattle as reservoirs of Trypanosoma vivax for different Glossina species. Parasitology. 1993;106:277–82.
Osório AL, Madruga CR, Desquesnes M, Soares CO, Ribeiro LR, Costa SC. Trypanosoma (Duttonella) vivax: its biology, epidemiology, pathogenesis, and introduction in the New World-a review. Mem Inst Oswaldo Cruz. 2008;103:1–13.
Mihok S, Maramba O, Munyoki E, Kagoiya J. Mechanical transmission of Trypanosoma spp. by African Stomoxyinae (Diptera: Muscidae). Trop Med Parasitol. 1995;46:103–5.
Oluwafemi A, Ilemobade A, Laseined E. The impact of African animal trypanosomosis and tsetse on the livelihood and wellbeing of cattle and their owners in the BICOT study area of Nigeria. Sc Res Essays. 2007;2:380–3.
Cortez AP, Ventura RM, Rodrigues AC, Batista JS, Paiva F, Añez N, et al. The taxonomic and phylogenetic relationships of Trypanosoma vivax from South America and Africa. Parasitology. 2006;133:159–69.
Raina AK, Kumar R, Rajora VS, Sridhar, Singh RP. Oral transmission of Trypanosoma evansi infection in dogs and mice. Vet Parasitol. 1985;18:67–9.
Sinha PK, Mukherjee GS, Das MS, Lahiri RK. Outbreak of trypanosomiasis evansi amongst tigers and jaguars in the zoological garden, Calcutta. Indian Vet J. 1971;48:306–10.
Langridge WP. A tsetse and Trypanosomiasis Survey of Ethiopia. London: Ministry of Overseas Development; 1976.
Desquesnes M, Holzmüller P, Lai DH, Dargantes A, Lun ZR, Jittaplapong S. Trypanosoma evansi and surra: a review and perspectives on origin, history, distribution, taxonomy, morphology, hosts, and pathogenic effects. Biomed Res Int. 2013;2013:ID 194176.
Desquesnes M, Dargantes A, Lai DH, Lun ZR, Holzmüller P, Jittapalapong S. Trypanosoma evansi and surra: a review and perspectives on transmission, epidemiology and control, impact, and zoonotic aspects. Biomed Res Int. 2013;2013:ID 321237.
Gutierrez C, Corbera JA, Morales M, Büscher P. Trypanosomosis in goats: current status. Ann N Y Acad Sci. 2006;1081:300–10.
Holland WG, Do TT, Huong NT, Dung NT, Thanh NG, Vercruysse J, et al. The effect of Trypanosoma evansi infection on pig performance and vaccination against classical swine fever. Vet Parasitol. 2003;111:115–23.
Singla LD, Juyal PD, Sharma NS. Immune responses to haemorrhagic septicaemia (HS) vaccination in Trypanosoma evansi infected buffalo-calves. Trop Anim Health Prod. 2010;42:589–95.
Holland WG, My LN, Dung TV, Thanh NG, Tam PT, Vercruysse J, et al. The influence of T. evansi infection on the immuno-responsiveness of experimentally infected water buffaloes. Vet Parasitol. 2001;102:225–34.
Songa Bajyana E, Paindavoine P, Wittouck E, Viseshakul N, Muldermans S, Steinert M, et al. Evidence for kinetoplast and nuclear DNA homogeneity in Trypanosoma evansi isolates. Mol Biochem Parasitol. 1990;43:167–79.
Ou YC, Giroud C, Baltz T. Kinetoplast DNA analysis of four Trypanosoma evansi strains. Mol Biochem Parasitol. 1991;46:97–102.
Lun ZR, Brun R, Gibson W. Kinetoplast DNA and molecular karyotypes of Trypanosoma evansi and Trypanosoma equiperdum from China. Mol Biochem Parasitol. 1992;50:189–96.
Verloo D, Magnus E, Büscher P. General expression of RoTat 1.2 variable antigen type in Trypanosoma evansi isolates from different origin. Vet Parasitol. 2001;97:183–9.
Borst P, Fase-Fowler F, Gibson WC. Kinetoplast DNA of Trypanosoma evansi. Mol Biochem Parasitol. 1987;23:31–8.
Ngaira JM, Olembo NK, Njagi EN, Ngeranwa JJ. The detection of non-RoTat 1.2 Trypanosoma evansi. Exp Parasitol. 2005;110:30–8.
Njiru ZK, Constantine CC, Ndung’u JM, Robertson I, Okaye S, Thompson RC, et al. Detection of Trypanosoma evansi in camels using PCR and CATT/T. evansi tests in Kenya. Vet Parasitol. 2004;124:187–99.
Njiru ZK, Constantine CC, Masiga DK, Reid SA, Thompson RC, Gibson WC. Characterization of Trypanosoma evansi type B. Infect Genet Evol. 2006;6:292–300.
Gibson WC, Wilson AJ, Moloo SK. Characterisation of Trypanosoma (Trypanozoon) evansi from camels in Kenya using isoenzyme electrophoresis. Res Vet Sci. 1983;34:114–8.
Bajyana Songa E, Hamers-Casterman C, Hamers R, Pholpark M, Pholpark S, Leidl K, et al. The use of the card agglutination test (Testryp CATT) for the detection of T. evansi infection: a comparison with other trypanosomiasis diagnostic tests under field conditions in Thailand. Ann Soc Belg Med Trop. 1987;67:137–48.
Claes F, Radwanska M, Urakawa T, Majiwa PA, Goddeeris B, Büscher P. Variable Surface Glycoprotein RoTat 1.2 PCR as a specific diagnostic tool for the detection of Trypanosoma evansi infections. Kinetoplastid Biol Dis. 2004;3:3.
Sinshaw A, Abebe G, Desquesnes M, Yoni W. Biting flies and Trypanosoma vivax infection in three highland districts bordering lake Tana, Ethiopia. Vet Parasitol. 2006;142:35–46.
Codjia V, Mulatu W, Majiwa PA, Leak SG, Rowlands GJ, Authié E, et al. Epidemiology of bovine trypanosomiasis in the Ghibe valley, southwest Ethiopia. 3. Occurrence of populations of Trypanosoma congolense resistant to diminazene, isometamidium and homidium. Acta Trop. 1993;53:151–63.
Rowlands GJ, Mulatu W, Authié E, d’Ieteren GD, Leak SG, Nagda SM, et al. Epidemiology of bovine trypanosomiasis in the Ghibe valley, southwest Ethiopia. 2. Factors associated with variations in trypanosome prevalence, incidence of new infections and prevalence of recurrent infections. Acta Trop. 1993;53:135–50.
Hagos A, Degefa G, Yacob H, Fikru R, Alemu T, Feseha G, et al. Seroepidemiological survey of trypanozoon infection in horses in the suspected dourine-infected Bale highlands of the Oromia region, Ethiopia. Rev Sci Tech. 2010;29:649–54.
Miruk A, Hagos A, Yacob HT, Asnake F, Basu AK. Prevalence of bovine trypanosomosis and trypanocidal drug sensitivity studies on Trypanosoma congolense in Wolyta and Dawero zones of southern Ethiopia. Vet Parasitol. 2008;152:141–7.
Cherenet T, Sani RA, Speybroeck N, Panandam JM, Nadzr S, Van den Bossche P. A comparative longitudinal study of bovine trypanosomiasis in tsetse-free and tsetse-infested zones of the Amhara Region, northwest Ethiopia. Vet Parasitol. 2006;140:251–8.
Fikru R, Goddeeris BM, Delespaux V, Moti Y, Tadesse A, Bekana M, et al. Widespread occurrence of Trypanosoma vivax in bovines of tsetse- as well as non-tsetse-infested regions of Ethiopia: a reason for concern? Vet Parasitol. 2012;190:355–61.
Hagos A, Yilkal K, Esayass T, Alemu T, Fikru R, Fesseha G, et al. Parasitological and serological survey on trypanosomosis (surra) in camels in dry and wet areas of Bale Zone, Oromyia Region, Ethiopia. Rev Med Vet. 2009;160:569–73.
Aradaib Imadeldin E, Majid Ali A. A simple and rapid method for detection of Trypanosoma evansi in the dromedary camel using a nested polymerase chain reaction. Kinetoplastid Biol Dis. 2006;5:1–6.
Büscher P. Diagnosis of human and animal African trypanosomiasis. In: Black SJ, Seed JR, editors. The African Trypanosomes. Boston: Kluwer Academic Publishers; 2001. p. 51–63.
Woo PTK. The haematocrit centrifuge for the detection of trypanosomes in blood. Can J Zool. 1969;47:921–3.
Murray M, Murray PK, McIntyre WI. An improved parasitological technique for the diagnosis of African trypanosomiasis. Trans R Soc Trop Med Hyg. 1977;71:325–6.
Lanham SM, Godfrey DG. Isolation of salivarian trypanosomes from man and other mammals using DEAE-cellulose. Exp Parasitol. 1970;28:521–34.
Lumsden WH, Kimber CD, Evans DA, Doig SJ. Trypanosoma brucei: miniature anion-exchange centrifugation technique for detection of low parasitaemias: Adaptation for field use. Trans R Soc Trop Med Hyg. 1979;73:312–7.
Zillmann U, Konstantinov SM, Berger MR, Braun R. Improved performance of the anion-exchange centrifugation technique for studies with human infective African trypanosomes. Acta Trop. 1996;62:183–7.
Büscher P, Mumba ND, Kabore J, Lejon V, Robays J, Jamonneau V, et al. Improved models of mini Anion Exchange Centrifugation Technique (mAECT) and Modified Single Centrifugation (MSC) for sleeping sickness diagnosis and staging. PLoS Negl Trop Dis. 2009;3:e471.
Konnai S, Mekata H, Mingala CN, Abes NS, Gutierrez CA, Herrera JR, et al. Development and application of a quantitative real-time PCR for the diagnosis of surra in water buffaloes. Infect Genet Evol. 2009;9:449–52.
Desquesnes M, McLaughlin G, Zoungrana A, Davila AM. Detection and identification of Trypanosoma of African livestock through a single PCR based on internal transcribed spacer 1 of rDNA. Int J Parasitol. 2001;31:610–4.
Fikru R, Hagos A, Rogé S, Reyna-Bello A, Gonzatti MI, Merga B, et al. A proline racemase based PCR for identification of Trypanosoma vivax in cattle blood. PLoS One. 2014;9:e84819.
World Animal Health (OIE). Trypanosoma evansi Infection (surra). 2012 [http://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/2.01.17_TRYPANO_SURRA.pdf].
Bajyana Songa E, Hamers R. A card agglutination test (CATT) for veterinary use based on an early VAT RoTat 1/2 of Trypanosoma evansi. Ann Soc Belg Med Trop. 1988;68:233–40.
Camargo RE, Uzcanga GL, Bubis J. Isolation of two antigens from Trypanosoma evansi that are partially responsible for its cross-reactivity with Trypanosoma vivax. Vet Parasitol. 2004;123:67–81.
Uzcanga GL, Perrone T, Noda JA, Perez-Pazos J, Medina R, Hoebeke J, et al. Variant surface glycoprotein from Trypanosoma evansi is partially responsible for the cross-reaction between Trypanosoma evansi and Trypanosoma vivax. Biochemistry. 2004;43:595–606.
Uzcanga G, Mendoza M, Aso PM, Bubis J. Purification of a 64 kDa antigen from Trypanosoma evansi that exhibits cross-reactivity with Trypanosoma vivax. Parasitology. 2002;124:287–99.
Pillay D, Izotte J, Fikru R, Büscher P, Mucache H, Neves L, et al. Trypanosoma vivax GM6 antigen: a candidate antigen for diagnosis of African animal trypanosomosis in cattle. PLoS One. 2013;8:e78565.
Tassew W. Economic Analysis and Policy Implications of Farm And Off-Farm Employment: A Case Study In The Tigray Region Of Northern Ethiopia, PhD thesis. Wageningen University: Agricultural Economics and Rural Policy Group; 2000.
CSA. Central Statistical Authority (CSA) of Federal Democratic Republic of Ethiopia. Agricultural sample survey. Volume I: Report on area and production of major crops. Addis Ababa, Ethiopia. 2011:1–123. [http://harvestchoice.org/sites/default/files/downloads/publications/Ethiopia_2010-1_Vol_1.pdf].
David HS, Thomas PO. Historical Dictionary of Ethiopia. 2nd ed. Maryland, USA: Scarecrow Press; 2013.
Thrusfield M. Veterinary Epidemiology. 3rd ed. Edinburgh, UK: Blackwell Publishing; 2007.
Woo PTK. Evaluation of the haematocrit centrifuge and other techniques for the field diagnosis of human trypanosomiasis and filariasis. Acta Trop. 1971;28:298–303.
De Winne K, Büscher P, Luquetti AO, Tavares SB, Oliveira RA, Solari A, et al. The Trypanosoma cruzi satellite DNA OligoC-TesT and Trypanosoma cruzi kinetoplast DNA OligoC-TesT for diagnosis of Chagas disease: a multi-cohort comparative evaluation study. PLoS Negl Trop Dis. 2014;8:e2633.
Pyana Pati P, Ngay Lukusa I, Mumba Ngoyi D, Van Reet N, Kaiser M, Karhemere Bin Shamamba S, et al. Isolation of Trypanosoma brucei gambiense from cured and relapsed sleeping sickness patients and adaptation to laboratory mice. PLoS Negl Trop Dis. 2011;5:e1025.
Camara O, Camara M, Lejon V, Ilboudo H, Sakande H, Leno M, et al. Immune trypanolysis test with blood spotted on filter paper for epidemiological surveillance of sleeping sickness. Trop Med Int Health. 2014;19:828–31.
Herbert WJ, Lumsden WHR. Trypanosoma brucei: A rapid “matching” method for estimating the host’s parasitemia. Exp Parasitol. 1976;40:427–31.
Aref M, Yasin SM, Bahear W, Ghulam Z, Hastie L, Dennison T, et al. Canine Trypanosoma evansi infection in Afghanistan. Vet Parasitol. 2013;197:638–41.
Kocher TD, Thomas WK, Meyer A, Edwards SV, Paabo S, Villablanca FX, et al. Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci. 1989;86:6196–200.
StataCorp. Stata: Release 13. Statistical software. College Station. Texas, USA: StataCorp LP; 2013.
Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33:159–74.
Monzón CM, Mancebo OA, Roux JP. Comparison between six parasitological methods for diagnosis of Trypanosoma evansi in the subtropical area of Argentina. Vet Parasitol. 1990;36:141–6.
Samson L, Frehiwot M. Prevalence of Small Ruminant Trypanosomosis and Tsetse Fly Challenge In Upper Didessa Valley, Ethiopia, G V, vol. 5. 2010. p. 215–9.
Tadesse A, Tsegaye B. Bovine trypanosomosis and its vectors in two districts of Bench Maji zone, South Western Ethiopia. Trop Anim Health Prod. 2010;42:1757–62.
Kebede N, Fetene T, Animut A. Prevalence of Trypanosomosis of small ruminants in Guangua district of Awi Zone, northwestern Ethiopia. J Infect Dev Ctries. 2009;3:245–6.
World Animal Health (OIE). Trypanosomosis (tsetse-transmitted). 2013 [http://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/2.04.18_TRYPANOSOMOSIS.pdf].
Salim B, Bakheit MA, Kamau J, Nakamura I, Sugimoto C. Molecular epidemiology of camel trypanosomiasis based on ITS1 rDNA and RoTat 1.2 VSG gene in the Sudan. Parasit Vectors. 2011;4:31.
Boid R. Isoenzyme characterisation of 15 stocks of Trypanosoma evansi isolated from camels in the Sudan. Trop Med Parasitol. 1988;39:45–50.
Takeet MI, Fagbemi BO, De Donato M, Yakubu A, Rodulfo HE, Peters SO, et al. Molecular survey of pathogenic trypanosomes in naturally infected Nigerian cattle. Res Vet Sci. 2013;94:555–61.
Gibson W, Pilkington JG, Pemberton JM. Trypanosoma melophagium from the sheep ked Melophagus ovinus on the island of St Kilda. Parasitology. 2010;137:1799–804.
Büscher G, Friedhoff KT. The morphology of ovine Trypanosoma melophagium (Zoomastigophorea: Kinetoplastida). J Protozool. 1984;31:98–101.
Gathuo HK, Nantulya VM, Gardiner PR. Trypanosoma vivax: adaptation of two East African stocks to laboratory rodents. J Protozool. 1987;34:48–53.
D’Archivio S, Medina M, Cosson A, Chamond N, Rotureau B, Minoprio P, et al. Genetic engineering of Trypanosoma (Dutonella) vivax and in vitro differentiation under axenic conditions. PLoS Negl Trop Dis. 2011;5:e1461.
Hilali M, Abdel-Gawad A, Nassar A, Abdel-Wahab A, Magnus E, Büscher P. Evaluation of the card agglutination test (CATT/T. evansi) for detection of Trypanosoma evansi infection in water buffaloes (Bubalus bubalis) in Egypt. Vet Parasitol. 2004;121:45–51.
Monzón CM, Mancebo OA, Russo AM. Antibody levels by indirect ELISA test in Trypanosoma evansi infected horses following treatment with quinapyramine sulphate. Vet Parasitol. 2003;111:59–63.
Büscher P. Diagnosis of African trypanosomiasis. In: Magez S, Radwanska M, editors. Trypanosomes and Trypanosomiasis. Wien: Springer; 2014. p. 189–216.
Deborggraeve S, Büscher P. Recent progress in molecular diagnosis of sleeping sickness. Expert Rev Mol Diagn. 2012;12:719–30.
Verloo D, Tibayrenc R, Magnus E, Büscher P, Van Meirvenne N. Performance of serological tests for Trypanosoma evansi infections in camels from Niger. J Protozool Res. 1998;8:190–3.
Holland WG, Thanh NG, My LN, Magnus E, Verloo D, Büscher P, et al. Evaluation of whole fresh blood and dried blood on filter paper discs in serological tests for Trypanosoma evansi in experimentally infected water buffaloes. Acta Trop. 2002;81:159–65.
Van Vlaenderen G. In Search of Cattle Trypanosomiasis in Suriname, M.Sc. thesis. Antwerp: Prince Leopold Institute of Tropical Medicine; 1996.
Acknowledgments
The PhD fellowships of Hadush Birhanu and Regassa Fikru were financed by the Belgian Directorate General for Development Cooperation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We acknowledge the Mekelle University in Tigray, Ethiopia and the Katholieke Unversiteit Leuven, Belgium for supporting laboratory and field work. We are grateful to Kumulachew Belay, Habtu Siyoum, Semere Kiros, Tsehaye, Gidena Desta, Berihu Gebrekidan and Abreha Tesfay, Stijn Deborggraeve, Stijn Rogé, Nicolas Bebronne, Fatima Balharbi and Tessa De Block for technical assistance. The animal owners, contact persons and local administrators are acknowledged for their collaboration during the field work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HB and PB conceived this study, generated, analysed and interpreted the data and prepared the manuscript. BG and GT contributed for the conception of the study and provided technical and infrastructural support to the field and laboratory work in Ethiopia. HA, RF, KW and AT participated in the field and part of the laboratory work. DB and SM performed the statistical analysis on the data. DT designed the map of the study areas. All authors revised and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Birhanu, H., Fikru, R., Said, M. et al. Epidemiology of Trypanosoma evansi and Trypanosoma vivax in domestic animals from selected districts of Tigray and Afar regions, Northern Ethiopia. Parasites Vectors 8, 212 (2015). https://doi.org/10.1186/s13071-015-0818-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-015-0818-1