
Abstract
Background
The primary objective of rapeseed breeding is to enhance oil content, which is predominantly influenced by environmental factors. However, the molecular mechanisms underlying the impact of these environmental factors on oil accumulation remain inadequately elucidated. In this study, we used transcriptome data from two higher (HOC) and two lower oil content (LOC) inbred lines at 35 days after pollination (DAP) to investigate genes exhibiting stable expression across three different environments. Meanwhile, a genome-wide association study (GWAS) was utilized to detect candidate genes exhibiting significant associations with seed oil content across three distinct environments.
Results
The study found a total of 405 stable differentially expressed genes (DEGs), including 25 involved in lipid/fatty acid metabolism and 14 classified as transcription factors. Among these genes, BnBZIP10-A09, BnMYB61-A06, BnAPA1-A08, BnPAS2-A10, BnLCAT3-C05 and BnKASIII-C09 were also found to exhibit significant associations with oil content across multiple different environments based on GWAS of 50 re-sequenced semi-winter rapeseed inbred lines and previously reported intervals. Otherwise, we revealed the presence of additive effects among BnBZIP10-A09, BnKASIII-C09, BnPAS2-A10 and BnAPA1-A08, resulting in a significant increase in seed oil content. Meanwhile, the majority of these stable DEGs are interconnected either directly or indirectly through co-expression network analysis, thereby giving rise to an elaborate molecular network implicated in the potential regulation of seed oil accumulation and stability.
Conclusions
The combination of transcription and GWAS revealed that natural variation in six environment-insensitive gene regions exhibited significant correlations with seed oil content phenotypes. These results provide important molecular marker information for us to further improve oil content accumulation and stability in rapeseed.
Similar content being viewed by others


Introduction
With the development of society, the consumption demands for edible oil are increasing rapidly. The majority of vegetable oils are derived from four major crops, namely soybean, rapeseed, oil palm, and sunflower [1]. The global supply of edible oil is boosted by rapeseed, accounting for over 15% [2]. The cultivation of rapeseed plays a pivotal role in providing essential edible oil in the European Union, Canada, and China. The increasing demand for edible oil among consumers necessitates the imperative to enhance both seed oil content and oil production per unit area of land in rapeseed breeding.
The oil content of seeds is highly susceptible to environmental factors and exhibits variations ranging from 35 to 55% across diverse ecological zones and climatic conditions [3]. Low temperature increases the polyunsaturated fatty acid content in plants, thereby contributing to the maintenance of biological membrane fluidity [4]. Bellaloui et al. demonstrated that elevated temperatures had an impact on both the production and composition of oil, potentially due in part to the restricted availability and transport of carbohydrates from leaves to seeds [5]. Zhou et al. suggested that temperature causes changes in the expression of lipid metabolism genes, thereby regulating lipid accumulation in rapeseed [3]. The expression stability of the FAD2 and FAD3 genes during lipid accumulation may be directly influenced by temperature, as suggested by some research [6,7,8]. Meanwhile, light intensity is an essential factor in determining photosynthetic efficiency. Light intensity affects gene expression in lipid metabolism, which regulates the seed oil content in developing seeds [9, 10]. The expression of WRINKLED1, a crucial gene involved in regulating lipid biosynthesis, is closely linked to the photosynthetic activity of the silique wall during seed development [11]. Although the molecular mechanisms underlying the regulation of lipid/fatty acid accumulation by temperature and light remain poorly understood, identifying genes involved in lipid/fatty acid metabolism that are insensitive to environmental factors may offer a more effective approach to improve oil content stability and optimize fatty acid composition.
Environmental and genotypic interactions lead to gene expression pattern differences that result in phenotypic diversity. With the next-generation sequencing development, gene expression variation can be measured quantitatively, and DEGs related to phenotypes and/or environments can be explored. The application of transcriptome analysis has been instrumental in unraveling the DEGs implicated in modulating the oil content of rapeseed [12, 13]. Numerous DEGs could be identified in short times by this application. However, the transcriptome was only interpreted as phenotypic variation in terms of gene expression and failed to fully interpret genetic variations [14]. Meanwhile, numerous differentially expressed genes make explaining transcriptome results and enacting breeding strategies more difficult. GWAS is an application that studies complex phenotypes by investigating genetic variations in the whole genome and has been extensively applied in rapeseed [15,16,17]. Therefore, combining transcriptome analysis and GWAS to identify differentially expressed genes and explore genetic variations is a novel strategy. For example, Zhang et al. used GWAS combined with transcriptome analysis to reveal that HCTs and WRKYs interact to regulate the defence response of poplar [18]. Xiao et al. identified a few key genes of the lipid biosynthesis pathway controlling oil content by combining GWAS and transcriptome analysis in rapeseed [19]. The combination of GWAS and transcriptome analysis was employed to detect ten candidate genes implicated in the regulation of flax seed fatty acid metabolism in rapeseed [20].
In this study, we carried out transcriptome sequencing of two HOC and LOC accessions at 35 DAP across three different environments. Meanwhile, a GWAS was conducted to identify candidate genes that demonstrate significant associations with seed oil content across three distinct environmental conditions. Our aim was to identify environment-insensitive genes in the process of oil accumulation. The research results will provide theoretical basis for breeding varieties with high and stable oil content in rapeseed.
Results
Changes in oil content in developmental stages and different environments
To investigate the dynamics of oil accumulation during seed development, we measured the seed oil content of two HOC and LOC accessions at 20, 25, 30, 35, 40, and 45 DAP under three distinct environmental conditions. CS and HZ are characterized by low temperatures and high levels of precipitation, in contrast to KM (Fig. 1). The seed oil content accumulation in CS and HZ occurred between 20 and 40 DAP, with the period from 30 to 35 DAP exhibiting the most rapid increase, followed by a decline observed at 40 to 45 DAP. While the seed oil content at 20–35 DAP showed a faster increase, the period at 35–45 DAP exhibited a downward increase in KM (Additional file 1: Fig. S1a). HOC and LOC reached significant differences at 35 DAP in the three different environments (Additional file 1: Fig. S1a). All genes expression patterns of HOC and LOC at 35 DAP across the three environments were characterized through a principal component analysis (PCA). The PC1 explained 14.0% of the variation and exhibited distinct separation between the HOC and LOC groups (Additional file 1: Fig. S1b). These results indicate that gene expression is more stable in different environments at 35 DAP. Therefore, we particularly aimed at transcriptome data from 35 DAP to explore the stably expression genes involved in the process of lipid accumulation in different environments.

Climate data for three environments tested. The graphs show sunlight (monthly mean values), temperature (monthly mean values), and precipitation (monthly sums) for each environment. The environments are abbreviated as follows: Changsha (CS), Hangzhou (HZ) and Kunming (KM) in China
Identification of environment-insensitive genes during the accumulation of oil
To investigate environment-insensitive genes associated with seed oil content, we analyzed the stable differentially expressed genes (DEGs) between two HOC and LOC accessions at 35 DAP in three different environments: CS, HZ, and KM. The DEGs were defined as those with a fold change of FPKM expression values that were at least 2 in either direction, when the q value or FDR < 0.001 and |Log2 fold change| ≥ 1.
We identified 1738, 1535, and 3359 up-regulated genes in two HOCs compared with two LOCs under CS, HZ, and KM, respectively, and 2495, 1589, and 3291 down-regulated genes were discovered, respectively (Additional file 2: Fig. S2b). As illustrated in Fig. 2a, 220 and 185 genes were stably up- and down-regulated in the three environments, respectively, indicating that these genes were environment-insensitive genes. The KEGG enrichment analysis revealed a significant enrichment of these 405 stable DEGs in various pathways, including flavonoid biosynthesis, lipid metabolism, energy metabolism, and more (Additional file 2: Fig. S2c). Among them, we found 25 stable DEGs involved in fatty acid/lipid metabolism, and 14 stable DEGs were transcription factors (Fig. 2c, Additional file 7: Table S1). Next, these 39 stable DEGs were selected to evaluate the accuracy of RNA‒Seq by qRT‒PCR analysis. The expression patterns of these stable DEGs by RNA-Seq and qRT‒PCR that exhibited a high degree of correlation (R2 = 0.70–0.77), thereby providing further validation and consistency with the RNA sequencing data (Fig. 2d).

Summary of DEGs between the extremely HOC and LOC inbred lines. Number of stable up-regulated (a) and down-regulated (b) DEGs in CS, HZ and KM. c The heatmap of 39 selected stable DEGs from the comparison between HOC and LOC inbred lines in three different environments. Three biological replicates were used to calculate expression values with three technical replicates, and normalized by log10(mean expression values). d Transcriptome sequencing verified by qRT-PCR. Normalized expression (2−∆∆Ct) in HOC inbred lines was divided by normalized expression in LOC inbred lines and log2 transformed. The correlation coefficient (R2) was calculated for comparison. The orange, green and turquoise lines represent the CS, HZ and KM environment fitted curves, respectively. p values show the significance of pairwise comparisons
GWAS was employed to detect associations between SNPs and oil content in 50 semi-winter rapeseed inbred lines. The Manhattan plots in Fig. 3d depict numerous significant SNP associations with seed oil content observed in glasshouse and field experiments. The study identified 35 haplotype (Hap) regions that exhibited a significant association with seed oil content, including 23 regions that overlapped with previously reported intervals (Fig. 3c and d; Additional file 8: Table S2). The combination of the aforementioned 39 stable DEGs revealed that 31 of them were situated within these overlapping regions (Fig. 3b; Additional file 8: Table S2).

The circos plot of 39 selected stable DEGs, previously reported QTLs regions and GWAS for oil content. a Chromosomes; b position of 39 stable DEGs, the red font represents genes that are located in the overlapping interval between reported interval and the significantly associated haplotype regions; c QTLs and association region genetic intervals from published studies; d GWAS of seed oil content, the blue line represents −log10p−value ≥ 4.0. Red fill represents significant QTLs
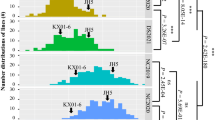
Of these 31 DEGs, two transcription factors BnMYB61-A06 and BnBZIP10-A09 were located in QTL-A06 (2,847,280–3,181,818 bp) and QTL-A09.1 (142,667–1,638,715 bp), respectively (Fig. 4). Two and three haplotype alleles were identified within these two gene regions, and BnMYB61-A06-Hap1 and BnBZIP10-A09-Hap1 corresponded to inbred lines exhibiting relatively high oil content (Fig. 4c and f; Additional file 9: Table S3). Four lipid/fatty acid synthesis genes, namely, BnAPA1-A08, BnPAS2-A10, BnLCAT3-C05 and BnKASIII-C09 located within QTL-A08 (16,454,214–17,628,998 bp), QTL-A10 (14,388,914–15,045,397 bp), QTL-C05 (41,242,900–42,812,067 bp), and QTL-C09.2 (6,631,263–7,574,558 bp), respectively (Additional file 3: Fig. S3 and Additional file 4: Fig. S4). Two, four, two, and three haplotype alleles were found in these four gene regions, and BnAPA1-A08-Hap1, BnPAS2-A10-Hap1, BnLCAT3-C05-Hap1, and BnKASIII-C09-Hap1 had higher oil contents than the other haplotype alleles (Additional file 3: Fig. S3c and f; Additional file 4: Fig. S4c and f; Additional file 9: Table S3).

Analysis of candidate genes in the significantly associated QTL-A06 and QTL-A09 regions. Regional Manhattan plot surrounding the peak signals on QTL-A06 (a) and QTL-A09 (d). Green dots indicate SNPs located in the BnMYB61-A06 (a) and BnBZIP10-A09 (d) gene regions which significantly associated with oil content. Genetic structure variations of BnMYB61-A06 (b) and BnBZIP10-A09 (e). c, f Boxplots showing comparative analysis between haplotypes related to the oil content phenotype. p values show the significance of pairwise comparisons
Additive effects analysis of environment-insensitive genes
To perform a single-variant-additive-effect analysis in GWAS, we investigated the interaction effects on oil content among both significant and non-significant SNP markers within these six candidate gene regions. Our findings revealed that the combination of BnKASIII-C09 and BnAPA1-A08, BnAPA1-A08 and BnPAS2-A10, as well as BnKASIII-C09 and BnBZIP10-A09, exhibited an additive effect (Fig. 5a, Additional file 12: Table S6). Additionally, we identified three combinations of haplotype alleles (BnKASIII-C09-Hap1 + BnAPA1-A08-Hap1, BnKASIII-C09-Hap1 + BnBZIP10-A09-Hap1, BnAPA1-A08-Hap1 + BnPAS2-A10-Hap1) that correspond to inbred lines exhibiting relatively high oil content compared to single haplotype allele (BnKASIII-C09-Hap1, BnAPA1-A08-Hap1, BnBZIP10-A09-Hap1, and BnPAS2-A10-Hap1) (Fig. 5b). These results suggest potential additive effects of these four genes on oil content.

Additive effect analysis between candidate genes. a Circos plot showing candidate gene interaction analysis by the R package “SIPI”. The dots in the outer arcs represent −log10(p−value) of GWAS, and the secondary arcs represent the candidate gene structure. The links positioned at the center of the circle symbolize the additive-by-additive relationship between two SNPs. b Boxplots showing the oil content of additive-by-additive haplotypes which is higher than other of a single haplotype. p values show the significance of pairwise comparisons
Co-expression network analysis of environment-insensitive genes
To provide additional context for the proposed functions of BnKASIII-C09, BnBZIP10-A09, BnAPA1-A08, BnLCAT3-C05, BnPAS2-A10, and BnMYB61-A06, we utilized transcriptome data from four accessions across three different environments at 35 DAP to construct intricate co-expression networks. This analysis resulted in the identification of 12 gene modules. The BnKASIII-C09, BnBZIP10-A09, BnAPA1-A08, and BnLCAT3-C05 genes fell in the blue module, which showed a significant positive correlation with oil content (r 0.84; Additional file 5: Fig. S5b). The turquoise module including the BnPAS2-A10 and BnMYB61-A06 genes showed a significant positive correlation with oil content (r 0.53; Additional file 5: Fig. S5b, Additional file 6: Fig. S6a). KEGG enrichment analysis of module genes was performed. The module genes were subjected to KEGG enrichment analysis. The blue module exhibited significant enrichment in unsaturated fatty acid biosynthesis, fatty acid degradation, and flavonoid biosynthesis, whereas the turquoise module showed significant enrichment in fatty acid biosynthesis, flavonoid biosynthesis, and photosynthesis (Additional file 6: Fig. S6b).
The subnetwork contained a total of 286 genes, including 125 stable DEGs (Fig. 6, Additional file 10: Table S4). The set of stable DEGs consisted of 30, 34, 15, and 46 genes involved in fatty acid/lipid metabolic processes, carbohydrate metabolic processes, flavonoid metabolism pathways, and plant hormone metabolic pathways, respectively. Further analysis of candidate gene subnetworks revealed that BnBZIP10-A09 is directly linked to BnKASIII-C09, BnAPA1-A08, BnLCAT3-C05, and BnPAS2-A10, while BnMYB61-A06 is directly linked to BnPAS2-A10 (Fig. 6). These results suggest that these genes establish a potential molecular network that impacts the accumulation and stability of oil content in rapeseed.

Co-expression network analysis. Hexagon nodes represent eight candidate genes, and rhombus nodes represent common DEGs in three environments. Co-expression network into the following groups based on functional annotation: red nodes representing lipid/fatty acid biosynthetic process, turquoise nodes representing transcription factors, wathet blue nodes representing flavonoid metabolic pathway, brown nodes representing carbohydrate metabolic pathway, green nodes representing photosynthesis, and purple nodes representing plant hormones
Discussion
The augmentation of seed oil content stands as a paramount objective in the realm of rapeseed breeding. The regulation of oil accumulation in seeds is governed by a complex network of genes and is highly susceptible to environmental influences [21,22,23]. To date, extensive investigations have been conducted on numerous QTLs associated with seed oil content in rapeseed, including some environment-insensitive QTLs identified across different environments [24,25,26,27]. Building on previous research findings, environment-insensitive genes might exist and regulate oil content accumulation in rapeseed.
In this study, we initially investigated the dynamics of oil accumulation at six different developmental stages (20, 25, 30, 35, 40, and 45 DAP) under three distinct environmental conditions. Our findings revealed key stages of oil synthesis occurring between 30 and 35 DAP, which aligns with the findings reported in previous studies [12]. To identify genes that show insensitivity to environmental changes, we conducted a transcriptome analysis of HOC and LOC inbred lines at 35 DAP across three distinct environments. As a result, we discovered 25 stable DEGs involved in lipid/fatty acid metabolism, while 14 stable DEGs were identified as transcription factors (Fig. 2c), indicating that these genes are environment-insensitive and associated with the oil content accumulation of rapeseed.
Meanwhile, BnAPA1-A08, BnPAS2-A10, BnLCAT3-C05, BnKASIII-C09, BnMYB61-A06, and BnBZIP10-A09 were identified as environment-insensitive genes affecting the oil content accumulation in rapeseed by transcriptome analysis and GWAS. APA1 and LCAT3 are involved in lipid metabolic processes [28, 29]. The interaction between PAS2 and CER10 suggests their participation in very long-chain fatty acid biosynthesis [30]. Dehesh et al. suggested that BnKASIII affects oil content accumulation in rapeseed [31]. BZIP10 forms a ternary complex with BZIP53 and ABI3 to promote the expression of seed maturation genes and affect seed oil accumulation [32,33,34]. The positive regulation of MYB61 on GL2 exerts a partial inhibition the accumulation of oil content, partly by modulating the formation of mucilage in the seed coat [35, 36]. Moreover, the co-expression network revealed that BnBZIP10-A09 is directly linked to BnKASIII-C09, BnAPA1-A08, BnLCAT3-C05, and BnPAS2-A10. BnMYB61-A06 is directly linked to BnPAS2-A10 and BnGL2-A07. The results suggest that fatty acid/lipid metabolism and transcription factors potentially interact and play a role in regulating the accumulation and stability of seed oil content in rapeseed.
The constant updates in sequencing technology and the continuous decrease in sequencing costs have stimulated large-scale germplasm sequencing projects in crops, thereby creating exciting opportunities for utilizing haplotypes in breeding applications. The observed haplotype alleles had a significantly higher level of diversity compared to the typical intra-species variation, indicating that successful reconstruction of haplotypes in polyploid species will have a substantial impact on crop breeding in the future [37]. Recently, researchers have utilized large-scale population sequencing data to detect favorable haplotype alleles associated with enhanced cold tolerance in rice [38], drought tolerance in maize [39], and head blight resistance in wheat [40]. In our study, we identified six favorable haplotype alleles corresponding to inbred lines exhibiting relatively high oil content, and three of these alleles had additive effects with each other. Voss-Fels et al. revealed the presence of an additive × additive epistasis effect between the two haplotypes, resulting in a significant increase in root biomass in wheat [41]. An additive effect was found between the two haplotypes, which significantly increased the chlorophyll content [42]. Our results will provide favorable haplotype alleles for further enhancement of seed oil content accumulation and stability in rapeseed.
Conclusions
The combination of transcription and GWAS revealed that natural variation in six environment-insensitive gene regions (BnBZIP10-A09, BnMYB61-A06, BnAPA1-A08, BnPAS2-A10, BnLCAT3-C05, and BnKASIII-C09) correlated with the seed oil content phenotypic variation. Additionally, we also found the presence of additive effects among BnBZIP10-A09, BnKASIII-C09, and BnAPA1-A08, resulting in a significant increase in seed oil content. Meanwhile, co-expression network analysis revealed that most of these stable DEGs are interconnected either directly or indirectly, thereby forming a molecular network implicated in the potential regulation of the concentration accumulation and stability of seed oil in rapeseed. The findings will offer valuable molecular markers for enhancing the high and stable oil content varieties in B.napus.
Materials and methods
Plant materials
Four rapeseed inbred lines, XY777 and XY015 (LOC) and CS136 and CS511 (HOC), came from Hunan Agricultural University, China, and were planted in Changsha (CS; E112.938888, N28.228272), Hangzhou (HZ; E120.15358, N30.287458) and Kunming (KM; E102.71225, N25.040609) in China. These four accessions were sown on October 10, 2015, in CS and HZ and in KM on May 15, 2016. Seed tissues were sampled with three biological replicates at 20, 25, 30, 35, 40, and 45 DAP. To measure the seed oil content using the Soxhlet extraction method to determine fat in foods (GB 5009.6-2016, standardization administration, China). The origin and oil content phenotypes of 50 Chinese semi-winter rapeseed inbred lines have been described in detail by Yao et al. [43].
Gene expression analysis
A detailed description of transcriptome sequencing in four inbred lines has been provided by Jia et al. [44]. The R package DESeq2 v1.24.0 [45] was employed to determine the differential expression genes when comparing each HOC with each LOC under CS, HZ, and KM, respectively, q value < 0.001 and |Log2 fold change| ≥ 1 were defined as differential expression genes. DEGs that were up- or down-regulated in all three environments were defined as environment-insensitive genes. Gene sequences from the B. napus reference genome (http://www.genoscope.cns.fr/brassicanapus/) blast to the Arabidopsis genome database (http://www.arabidopsis.org/) were used to assign putative gene functions.
KEGG enrichment analysis was conducted by TBtools [46]. The results were visualized using “ggplot2” [47].
Real-Time Quantitative PCR verification
cDNAs were synthesized using the same RNAs as those used for RNA-seq. The analysis of the results was conducted using LightCycler 480 SYBR Green I Mastermix and a LightCycler 480II real-time PCR system (Roche, Switzerland). The expression pattern of all selected stable DEGs was verified by qRT-PCR using gene copy-specific primers, and data were normalized by BnEF [48] independently (primer showen in Additional file 11: Table S5). The 2−ΔΔCT method [49] was used to access the fold change. The relative expression quantified by qRT‒PCR was converted to log2-fold change for direct comparison with RNA-Seq data.
Genome-wide association analysis
The resequencing of 50 rapeseed inbred lines and the acquisition of 532,005 high-quality SNPs were comprehensively described by Dong et al. [50]. TASSEL 5.0 software was used to perform the relative kinship and PCA [51]. Based on PCA and relative kinship (P and K matrix), the calculation was performed with a mixed linear model (MLM) incorporated into TASSEL 5.0 software. The critical p value for assessing the significance of SNP-trait associations was calculated for seed oil content based on the false discovery rate (FDR) [52]. An FDR < 0.05 was used to identify significant marker‒trait associations for oil content at P value threshold of 1 × 10−4.
Additive effect analysis
The R package “SIPI” [53] was used to evaluate pairwise interactions between the candidate gene SNP markers. In this study, only additive–additive interactions were considered. Wald p values < 0.01 were defined as significant SNP pairs.
Weighted gene co-expression network analysis (WGCNA) for candidate genes
A total of 14,019 genes had FPKM max and a mean value greater than 10 and 2, respectively. The module identification was implemented by merging modules with similar expression profiles using a merge Cut Height of 0.25. The co-expression network was constructed by the R package WGCNA v1.69 [54] and visualized through Cytoscape 3.5.1 [55].
Availability of data and materials
The entirety of the data generated or analyzed throughout this study has been incorporated within this published article and its additional files.
Abbreviations
- GWAS:
-
Genome-wide association study
- DEGs:
-
Differentially expressed genes
- CS:
-
Changsha
- HZ:
-
Hangzhou
- KM:
-
Kunming
- SNP:
-
Single nucleotide polymorphism
- DAP:
-
Days after pollination
- QTL:
-
Quantitative trait locus
- HOC:
-
High-oil content accessions
- LOC:
-
Low-oil content accessions
- Hap:
-
Haplotype
- qRT‒PCR:
-
Quantitative real-time PCR
- WRINKLED1 :
-
WRINKLED 1
- FAD2 :
-
Fatty acid desaturase 2
- FAD3 :
-
Fatty acid desaturase 3
- FPKM:
-
The fragments per kilobase of transcript per million reads
- PCR:
-
Polymerase chain reaction
- KASIII :
-
3-Ketoacyl-ACP synthase III
- BZIP10/53 :
-
Basic leucine zipper 10/53
- APA1 :
-
Aspartic proteinase A1
- LCAT3 :
-
Lecithin: cholesterol acyltransferase 3
- PAS2 :
-
PASTICCINO 2
- MYB61 :
-
MYB domain protein 61
- CER10 :
-
Eceriferum 10
- ABI3 :
-
Abscisic acid insensitive 3
- GL2 :
-
Glabra 2
References
Dyer JM, Mullen RT. Engineering plant oils as high-value industrial feedstocks for biorefining: the need for underpinning cell biology research. Physiol Plant. 2008;132:11–22.
USDA ERS—oil crops yearbook. 2019. https://www.ers.usda.gov/data-products/oil-crops-yearbook.aspx. Accessed 21 Sept 2019.
Zhou L, Yan T, Chen X, Li Z, Wu D, Hua S, Jiang L. Effect of high night temperature on storage lipids and transcriptome changes in developing seeds of oilseed rape. J Exp Bot. 2018;69:1721–33.
Los DA, Murata N. Structure and expression of fatty acid desaturases. Biochim Biophys Acta. 1998;1394:3–15.
Bellaloui N, Mengistu A, Kassem MA. Effects of genetics and environment on fatty acid stability in soybean seed. Food Nutr Sci. 2013;4:165–75.
Román Á, Andreu V, Hernández ML, Lagunas B, Picorel R, Martínez-Rivas JM, Alfonso M. Contribution of the different omega-3 fatty acid desaturase genes to the cold response in soybean. J Exp Bot. 2012;63:4973–82.
Zhu Y, Cao Z, Xu F, Huang Y, Chen M, Guo W, Zhou W, Zhu J, Meng J, Zou J. Analysis of gene expression profiles of two near-isogenic lines differing at a QTL region affecting oil content at high temperatures during seed maturation in oilseed rape (Brassica napus L.). Theor Appl Genet. 2012;124:515–31.
Li Q, Zheng Q, Shen W, Cram D, Fowler DB, Wei Y, Zou J. Understanding the biochemical basis of temperature-induced lipid pathway adjustments in plants. Plant Cell. 2015;27:86–103.
Vuorinen AL, Kalpio M, Linderborg KM, Kortesniemi M, Lehto K, Niemi J, Yang B, Kallio HP. Coordinate changes in gene expression and triacylglycerol composition in the developing seeds of oilseed rape (Brassica napus) and turnip rape (Brassica rapa). Food Chem. 2014;145:664–73.
Zhu L, Zhao X, Xu Y, Wang Q, Wang H, Wu D, Jiang L. Effect of germination potential on storage lipids and transcriptome changes in premature developing seeds of oilseed rape (Brassica napus L.). Theor Appl Genet. 2020;133:2839–52.
Hua W, Li R-J, Zhan G-M, Liu J, Li J, Wang X-F, Liu G-H, Wang H-Z. Maternal control of seed oil content in Brassica napus: the role of silique wall photosynthesis. Plant J. 2012;69:432–44.
Yu L, Liu D, Yin F, Yu P, Lu S, Zhang Y, Zhao H, Lu C, Yao X, Dai C, Yang Q-Y, Guo L. Interaction between phenylpropane metabolism and oil accumulation in the developing seed of Brassica napus revealed by high temporal-resolution transcriptomes. BMC Biol. 2023;21:202.
Tan H, Zhang J, Qi X, Shi X, Zhou J, Wang X, Xiang X. Correlation analysis of the transcriptome and metabolome reveals the regulatory network for lipid synthesis in developing Brassica napus embryos. Plant Mol Biol. 2019;99:31–44.
Van Dam S, Craig T, De Magalhães JP. GeneFriends: a human RNA-seq-based gene and transcript co-expression database. Nucleic Acids Res. 2015;43:D1124–32.
Chen K, Yin Y, Liu S, Guo Z, Zhang K, Liang Y, Zhang L, Zhao W, Chao H, Li M. Genome-wide identification and functional analysis of oleosin genes in Brassica napus L. BMC Plant Biol. 2019;19:294.
He Y, Wu D, Wei D, Fu Y, Cui Y, Dong H, Tan C, Qian W. GWAS, QTL mapping and gene expression analyses in Brassica napus reveal genetic control of branching morphogenesis. Sci Rep. 2017;7:15971.
Zhou Q, Han D, Mason AS, Zhou C, Zheng W, Li Y, Wu C, Fu D, Huang Y. Earliness traits in rapeseed (Brassica napus): SNP loci and candidate genes identified by genome-wide association analysis. DNA Res. 2018;25:229–44.
Zhang J, Yang Y, Zheng K, Xie M, Feng K, Jawdy SS, Gunter LE, Ranjan P, Singan VR, Engle N. Genome-wide association studies and expression-based quantitative trait loci analyses reveal roles of HCT2 in caffeoylquinic acid biosynthesis and its regulation by defense-responsive transcription factors in Populus. New Phytol. 2018;220:502–16.
Xiao Z, Zhang C, Tang F, Yang B, Zhang L, Liu J, Huo Q, Wang S, Li S, Wei L. Identification of candidate genes controlling oil content by combination of genome-wide association and transcriptome analysis in the oilseed crop Brassica napus. Biotechnol Biofuels. 2019;12:216.
Xie D, Dai Z, Yang Z, Tang Q, Deng C, Xu Y, Wang J, Chen J, Zhao D, Zhang S, Zhang S, Su J. Combined genome-wide association analysis and transcriptome sequencing to identify candidate genes for flax seed fatty acid metabolism. Plant Sci. 2019;286:98–107.
Boem FHG, Lavado RS, Porcelli CA. Note on the effects of winter and spring waterlogging on growth, chemical composition and yield of rapeseed. Field Crops Res. 1996;47:175–9.
Jensen CR, Mogensen VO, Mortensen G, Fieldsend JK, Milford GFJ, Andersen MN, Thage JH. Seed glucosinolate, oil and protein contents of field-grown rape (Brassica napus L.) affected by soil drying and evaporative demand. Field Crops Res. 1996;47:93–105.
Si P, Mailer RJ, Galwey N, Turner DW. Influence of genotype and environment on oil and protein concentrations of canola (Brassica napus L.) grown across southern Australia. Aust J Agric Res. 2003;54:397–407.
Zhao J, Becker HC, Zhang D, Zhang Y, Ecke W. Oil content in a European\times Chinese rapeseed population: QTL with additive and epistatic effects and their genotype–environment interactions. Crop Sci. 2005;45:51–9.
Li C, Li B, Qu CM, Yan XY, Fu FY, Liu LZ, Chen L, Li JN. Analysis of difference QTLs for oil content between two environments in Brassica napus L. Acta Agron Sin. 2011;37:249–54.
Teh L, Möllers C. Genetic variation and inheritance of phytosterol and oil content in a doubled haploid population derived from the winter oilseed rape Sansibar× Oase cross. Theor Appl Genet. 2016;129:181–99.
Sun F, Liu J, Hua W, Sun X, Wang X, Wang H. Identification of stable QTLs for seed oil content by combined linkage and association mapping in Brassica napus. Plant Sci. 2016;252:388–99.
Gaudet P, Livstone MS, Lewis SE, Thomas PD. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief Bioinform. 2011;12:449–62.
Noiriel A, Benveniste P, Banas A, Stymne S, Bouvier-Navé P. Expression in yeast of a novel phospholipase A1 cDNA from Arabidopsis thaliana. Eur J Biochem. 2004;271:3752–64.
Bach L, Michaelson LV, Haslam R, Bellec Y, Gissot L, Marion J, Da Costa M, Boutin J-P, Miquel M, Tellier F. The very-long-chain hydroxy fatty acyl-CoA dehydratase PASTICCINO2 is essential and limiting for plant development. Proc Natl Acad Sci USA. 2008;105:14727–31.
Dehesh K, Tai H, Edwards P, Byrne J, Jaworski JG. Overexpression of 3-ketoacyl-acyl-carrier protein synthase IIIs in plants reduces the rate of lipid synthesis. Plant Physiol. 2001;125:1103–14.
Alonso R, Onate-Sanchez L, Weltmeier F, Ehlert A, Diaz I, Dietrich K, Vicente-Carbajosa J, Dröge-Laser W. A pivotal role of the basic leucine zipper transcription factor bZIP53 in the regulation of Arabidopsis seed maturation gene expression based on heterodimerization and protein complex formation. Plant Cell. 2009;21:1747–61.
Fatihi A, Boulard C, Bouyer D, Baud S, Dubreucq B, Lepiniec L. Deciphering and modifying LAFL transcriptional regulatory network in seed for improving yield and quality of storage compounds. Plant Sci. 2016;250:198–204.
Kumar N, Chaudhary A, Singh D, Teotia S. Transcriptional regulation of seed oil accumulation in Arabidopsis thaliana: role of transcription factors and chromatin remodelers. J Plant Biochem Biotechnol. 2020;29:754–68.
Shi L, Katavic V, Yu Y, Kunst L, Haughn G. Arabidopsis glabra2 mutant seeds deficient in mucilage biosynthesis produce more oil. Plant J. 2012;69:37–46.
Matías-Hernández L, Jiang W, Yang K, Tang K, Brodelius PE, Pelaz S. AaMYB1 and its orthologue AtMYB61 affect terpene metabolism and trichome development in Artemisia annua and Arabidopsis thaliana. Plant J. 2017;90:520–34.
Sun H, Jiao WB, Krause K, Campoy JA, Goel M, Folz-Donahue K, Kukat C, Huettel B, Schneeberger K. Chromosome-scale and haplotype-resolved genome assembly of a tetraploid potato cultivar. Nat Genet. 2022;54:342–8.
Zhang Z, Li J, Pan Y, Li J, Zhou L, Shi H, Zeng Y, Guo H, Yang S, Zheng W. Natural variation in CTB4a enhances rice adaptation to cold habitats. Nat Commun. 2017;8:14788.
Wang X, Wang H, Liu S, Ferjani A, Li J, Yan J, Yang X, Qin F. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat Genet. 2016;48:1233–41.
Hao C, Wang Y, Hou J, Feuillet C, Balfourier F, Zhang X. Association mapping and haplotype analysis of a 3.1-Mb genomic region involved in Fusarium head blight resistance on wheat chromosome 3BS. Plos ONE. 2012;7: e46444.
Voss-Fels KP, Qian L, Parra-Londono S, Uptmoor R, Frisch M, Keeble-Gagnère G, Appels R, Snowdon RJ. Linkage drag constrains the roots of modern wheat. Plant Cell Environ. 2017;40:717–25.
Qian L, Voss-Fels K, Cui Y, Jan HU, Samans B, Obermeier C, Qian W, Snowdon RJ. Deletion of a stay-green gene associates with adaptive selection in Brassica napus. Mol Plant. 2016;9:1559–69.
Yao M, Guan M, Yang Q, Huang L, Xiong X, Jan HU, Voss-Fels KP, Werner CR, He X, Qian W. Regional association analysis coupled with transcriptome analyses reveal candidate genes affecting seed oil accumulation in Brassica napus. Theor Appl Genet. 2021;134:1545–55.
Jia Y, Yao M, He X, Xiong X, Guan M, Liu Z, Guan C, Qian L. Transcriptome and regional association analyses reveal the effects of oleosin genes on the accumulation of oil content in Brassica napus. Plants. 2022;11:3140.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia R. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13:1194–202.
Villanueva RAM, Chen ZJ. ggplot2: elegant graphics for data analysis. Meas-Interdiscip Res. 2019;17:160–7.
Nesi N, Lucas M-O, Auger B, Baron C, Lécureuil A, Guerche P, Kronenberger J, Lepiniec L, Debeaujon I, Renard M. The promoter of the Arabidopsis thaliana BAN gene is active in proanthocyanidin-accumulating cells of the Brassica napus seed coat. Plant Cell Rep. 2009;28:601–17.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–8.
Dong H, Tan C, Li Y, He Y, Wei S, Cui Y, Chen Y, Wei D, Fu Y, He Y, Wan H, Liu Z, Xiong Q, Lu K, Li J, Qian W. Genome-wide association study reveals both overlapping and independent genetic loci to control seed weight and silique length in Brassica napus. Front Plant Sci. 2018;9:921.
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–5.
Storey JD. A direct approach to false discovery rates. J R Stat Soc B. 2002;64:479–98.
Lin H-Y, Chen D-T, Huang P-Y, Liu Y-H, Ochoa A, Zabaleta J, Mercante DE, Fang Z, Sellers TA, Pow-Sang JM. SNP interaction pattern identifier (SIPI): an intensive search for SNP–SNP interaction patterns. Bioinformatics. 2017;33:822–33.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Funding
This study was funded by the National Key Research and Development Program of China (Grant no. 2022YFD1200404), the Science Foundation for Distinguished Youth Scholars of Hunan Province, China (Grant number: 2022JJ10027) and the Research Foundation of Education Bureau of Hunan Province, China (Grant number: 21A0135).
Author information
Authors and Affiliations
Contributions
All authors planned and supervised the research. MY performed data curation, investigation, and visualization and wrote the original draft. DH, WL, XHX, and XH assisted with the data for the investigation. ZSL and CYG provided the resources. LWQ provided funding and performed supervision and led the conceptualization, project administration, resources, writing, review, and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Oil content phenotype of the development period and PCA distribution for LOC and HOC inbred lines using 35 DAPs FPKM. a The seed oil content of LOC and HOC inbred lines at different environments in different growth stage, the blue and gray, red and orange lines represent LOC and HOC inbred lines, respectively. b PCA distribution for LOC and HOC inbred lines using 35 DAPs FPKM. CS, HZ and KM was signed by cycle, tangle and rhombus, respectively.
Additional file 2: Figure S2.
Overview of DEGs in 35 DAP seeds of the HOC compared to LOC inbred lines. a DEG number of different comparisons at 35 DAP seed. b DEGs overlapped in the same environment under study. c Top 20 KEGG enhancement of common DEGs in three environments.
Additional file 3: Figure S3.
The analysis of candidate genes in the significant associated QTL-A08 and QTL-A10 regions. Regional Manhattan plot surrounding the peak signals on QTL-A08 (a) and QTL-A10 (d). Green dot indicates the SNPs located in the BnAPA1-A08 (a) and BnPAS2-A10 (d) gene region which are associated with oil content. Genetic structure variations of BnAPA1-A08 (b) and BnPAS2-A10 (e). c and f Boxplots showing comparative analysis between haplotypes related to oil content phenotype. p values show the significance of pairwise comparisons.
Additional file 4: Figure S4.
The analysis of candidate genes in the significant associated QTL-C05 and QTL- QTL-C09.2 regions. Regional Manhattan plot surrounding the peak signals on QTL-C05 (a) and QTL-C09.2 (d). Green dot indicates the SNPs located in BnLCAT3-C05 (a) and BnKASIII-C09 (d) which associated with oil content. Genetic structure variations of BnLCAT3-C05 (b) and BnKASIII-C09 (e), numbers indicate the SNP positions from gene start site. c and f Boxplots showed comparative analysis between haplotypes related to oil content phenotype. p values show the significance of pairwise comparisons.
Additional file 5: Figure S5.
The result of co-expression network analysis. a Cluster dendrogram of WGCNA gene modules. b The information of module-trait coefficient and module gene numbers.
Additional file 6: Figure S6.
Co-expression network analysis. a Whole co-expression network exhibit, hexagon nodes represent eight candidate genes, triangle nodes represent genes directly linked to the candidate gene. The blue and turquoise nodes represent blue and turquoise module genes. b Top 20 KEGG enhancement of blue and turquoise module genes.
Additional file 7: Table S1.
The detailed information of 39 stable DEGs.
Additional file 8: Table S2.
The information of chromosome positions of 50 stable DEGS, GWAS of oil content and previous reported QTL intervals.
Additional file 9: Table S3.
Haplotype analysis of candidate genes region in 50 resequenced accessions.
Additional file 10: Table S4.
Gene information in the co-expression network.
Additional file 11: Table S5.
Primers information of qRT‒PCR.
Additional file 12: Table S6.
The detailed information of additive affect between haplotypes in 50 resequenced accessions.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yao, M., He, D., Li, W. et al. Identification of environment-insensitive genes for oil content by combination of transcriptome and genome-wide association analysis in rapeseed. Biotechnol Biofuels 17, 29 (2024). https://doi.org/10.1186/s13068-024-02480-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-024-02480-x