
Abstract
The simultaneous assay of duloxetine hydrochloride (DLX) and avanafil (AVN) in their pure forms, synthetic mixtures, and spiked human plasma was achieved using a novel, eco-friendly, sensitive, and specific HPTLC methodology that have been established and validated. Measuring the levels of co-administered antidepressants and sexual stimulants in biological fluids is an important step for individuals with depression and sexual problems. Separation was performed successfully using pre-coated silica gel 60-F254 as a stationary phase and a mobile phase composed of methanol, acetone, and 33% ammonia (8:2:0.05, v/v/v). Compact bands were produced by the optimized mobile phase that was chosen for development (Rf values were 0.23 and 0.75 for DLX and AVN, individually) after dual-wavelength detection for DLX and AVN at 232 and 253 nm, respectively. The results of polynomial regression analysis were exceptional (r = 0.9999 for both medicines) over concentration ranges of 5-800 and 10-800ng/spot for DLX and AVN, respectively. The quantitation limits were 4.69 and 9.53 ng/spot (0.31 and 0.94 µg/mL), whereas the detection limits were 1.55 and 3.15 ng/spot (0.63 and 1.91 µg/mL), for DLX and AVN, respectively. The International Council for Harmonization (ICH) criteria served as the basis for validating the established approach. Moreover, the proposed technique was evaluated in terms of greenness using four contemporary ecological metrics: The Analytical Greenness software (AGREE), the Green Analytical Procedure Index (GAPI), Eco-Scale, and the National Environmental Method Index (NEMI). Additionally, the Blue Applicability Grade Index (BAGI), a newly developed tool for evaluating the practicality (blueness) of procedures, was taken into consideration when evaluating the sustainability levels of the established approach.
Similar content being viewed by others


Introduction
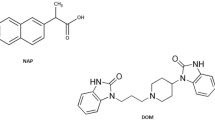
Duloxetine hydrochloride is an antidepressant drug with the chemical name (+) - (S) - N-methyl -(gamma)- (1- naphthyloxy) -2-thiophenepropylamine hydrochloride1 (Fig. 1) [1]. Duloxetine hydrochloride received FDA approval in 2004 for the therapeutic management of major depressive disorder (MDD), pain triggered as a result of Diabetes-related neuropathy in the peripheral nerves, and urinary incontinence due to stress (SUI) [2]. It is a potent norepinephrine and serotonin reuptake inhibitor. No significant affinity exists between DLX and the cholinergic, dopaminergic, adrenergic, histaminergic, glutamate, opioid, or GABA receptors [3]. It helps to elevate mood, alleviate anxiety, promote sleep, and increase energy and appetite [4]. The drug is efficiently absorbed after oral administration, binds to protein 96%, reaches its peak plasma levels (C max) at a median time of 6 h, and has a mean elimination half-life of 12 h. DLX can be regarded as a selective reuptake inhibitor at the 5HT and NE transporters because it does not have any noticeable affinity for dopaminergic, cholinergic, histaminergic, opioid, glutamate, or GABA reuptake transporters. DLX is extensively metabolized, however the main circulating metabolites have little or no effect on the drug’s pharmacologic efficacy [5]. For the assay of DLX in pharmaceutical products, many approaches were employed, these methods include spectrophotometry [1, 2, 5, 6], spectrofluorimetry [3, 7,8,9], high-performance liquid chromatography (HPLC) [10,11,12], Thin-layer chromatography (TLC) [4, 13,14,15], potentiometry [16,17,18] and voltammetry [19,20,21,22].

The chemical structures of the studied drugs
Avanafil directly inhibits the cGMP-specific type 5 phosphodiesterase. It is employed for the treatment of erectile dysfunction [23]. Avanafil has the advantage of having a much faster beginning of effect than other PDE5 inhibitors. It is readily absorbed and reaches its peak concentration in 30–45 min [24]. The antiproliferative and vasodilatory effects of endogenous nitric oxide, which is excreted by endothelial cells that lining the arteries, are mediated by AVN by inhibiting phosphodiesterase type-5, which increases cGMP in the penile vasculature and helps guarantee a strong erection. AVN has been developed because it is more highly selective for the PDE5 isoenzyme than other inhibitors that target PDE5 [25]. Avanafil overdose symptoms include blurred vision, sudden hearing or visual loss, dyspnea, rapid pulsation. Its use is still preferred above that of the other PDE-5 medications, despite all of these adverse reactions. Avanafil has the following chemical name: (S)-4-[(3-chloro-4-methoxybenzyl)amino].-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide (Fig. 1) [26]. AVN was given approval by the US FDA in 2012 and the European Medicines Agency did the same in 2013 [27]. Literature survey illustrates that a variety of techniques have been documented for the quantitative measurement of avanafil including spectrophotometry [28, 29], spectrofluorimetry [24, 26, 30], HPLC [23, 25, 31], LC/MS [32,33,34], TLC [35], voltammetry [36,37,38] and capillary electrophoresis [39]. Additionally, a recent review article was published which summarized most of the analytical methods for determination of sex stimulants including avanafil [40].
Medications for depression particularly selective serotonin reuptake inhibitors (SSRIs) and serotonin norepinephrine reuptake inhibitors (SNRIs) can cause sexual dysfunction as a side effect [41]. The unpleasant sexual side effects including erectile dysfunction can be treated with AVN. For the remedying of male reproductive problems induced by duloxetine, a number of physicians suggested a private prescription regarding phosphodiesterase-5 (PDE5) inhibitors like Avanafil [42, 43]. Therefore, the focus of our investigation was the simultaneous determination of DLX and AVN. The current study presents a novel, specific, and environmentally friendly HPTLC approach for the separation and quantification of both drugs in bulk, laboratory-prepared combinations, and human plasma with good precision, sensitivity, and selectivity results in accordance with ICH recommendations. This study is novel because there is right now no published HPTLC protocol for quantitative separation of DLX and AVN together.
Experimental
Instrumentation
The chromatographic analysis was performed using a CAMAG (Muttenz, Switzerland) TLC apparatus. To customize the data, Scanner 3 was operated by Vision CATS version software TLC. High-pressure mercury vapor lamps were employed as the scanner’s radiation source. The slit’s dimensions were 5 × 0.2 mm, and the scanning speed was 20 mm/s. The samples were applied to the plate using a Hamilton syringe (100 µL; Bonaduz, Switzerland) with a Linomat 5 auto-sampler while being lightly streamed with nitrogen. The development of the plate was carried out in a twin-trough chamber (27.0 × 26.5 × 7.0 cm) in the ascending mode.
Chemicals are used and materials
DLX powder had been generously provided by Mash Premiere pharmaceutical company (Badr City, Cairo, Egypt), and AVN powder received via Andalous Pharma (6th of October city, Cairo, Egypt). HPLC grade solvents (acetone and methanol) were purchased from Merck (Darmstadt, Germany) and 33% ammonia solution was obtained from El Nasr Pharmaceutical Chemical Co. (Cairo, Egypt).
Preparation of standard solution
25 mg were weighed precisely of each medication, was dissolved in 10 mL of methanol via a sonicator before being diluted to a final volume of 25 mL with methanol to produce the stock solutions (1.0 mg/mL) for DLX and AVN separately. Following that, standard working solutions were produced by diluting the stock solutions using the same diluent to produce concentrations of 1, 10, 20, 40, 80, and 160 µg/mL for DLX, and 2, 4, 20, 40, 80, and 160 µg/ mL for AVN. Lastly, to acquire final concentrations, a small portion of Lastly, a small portion of 5 µl of each working solution was spotted onto the plate to reach final concentrations of DLX of 5, 50, 100, 200, 400, and 800 ng/spot and AVN of 10, 20, 100, 200, 400, and 800 ng/spot.
Procedures
Chromatographic conditions
On HPTLC plates (Merck, Darmstadt, Germany) pre-coated with silica gel 60 F254, all chromatographic investigations were performed. The plates were divided into 20 × 5-centimeter segments. The mobile phase was allowed to developed until reaching a migration distance of 3.5 cm from the origin [44,45,46]. For the purpose of preventing the drug spot from dissolving when the TLC plate was inserted inside the chamber of development, the band was positioned 1 cm farther away the lowest edge of the plate, with a width of 4 mm and a distance of 6 mm between each two subsequent bands displacing a 10 mm distance from the plate’s beginning. The bands were steamed with nitrogen to dry. Nitrogen drying prevents the band from widening, what benefits HPTLC by enabling the simultaneous assessment of numerous samples in a single run. Before starting chromatographic development, the plates that were used had previously been methanol-washed.
Methanol: acetone: 33% ammonia (8:2:0.05, v/v/v) utilized to be the mobile phase for chromatographic development. the chamber had been completely saturated with the mobile phase solvents for 20 min at the ambient temperature., and the mobile phase volume was actually 10 mL. Hair dryer was used to dry the plate and then it was canned at slit dimension of 5 × 0.2 mm with CAMAG TLC scanner at dual wavelength of 232 and 253 nm for DLX and AVN, respectively, in the absorbance reflectance mode.
Procedure for pharmaceutical preparations
Ten tablets of Erovanafile ® 200 mg were finely crushed. From this, a carefully weighed amount of tablet powder equivalent to 50 mg of AVN was transferred into a 50 mL volumetric flask. The contents of 10 capsules of Cymbatex® 30 mg were carefully mixed, and an exactly weighed quantity of powder equal to 50 mg DLX was added to the same flask. Subsequently, the drugs were extracted using methanol through sonication for 30 min. Then, the volume was adjusted to 50 ml using the same solvent after which the initial part of the filtrate was cast-off. The standard assay procedure was followed, involving five measurements for each concentration.
Procedure for spiked human plasma
Samples of blood from healthy participants were collected at Misr Hospital (Sohag, Egypt) via the forearm vein and then immediately put into heparinized tubes. The work’s purpose was explained to the volunteer before he signed his assent. The method for using human plasma samples was accepted by the Committee on Ethics of Scientific Research, Faculty of Pharmacy, Sohag University, and complied with the Declaration of Helsinki Recommendations [47]. In all cases, informed written consent was obtained from each participant before donating the blood samples. Centrifugation was used to separate the plasma for 30 min. at 4000 rpm. 1.0 ml of drug-free plasma, 1.0 ml of drug mixture solution with concentrations of 10, 40, and 80 µg mL− 1 for both drugs, and 2.0 ml of acetonitrile as a protein precipitator were added to a centrifugation tube. The sample was then thoroughly mixed. The tube was centrifuged at 4000 rpm for 30 min. The collected supernatant was clear and was analyzed in accordance with the steps outlined under “Chromatographic conditions.”
Results and discussion
TLC is one of the most familiar and adaptable techniques used in detection and simultaneous quantification of several pharmaceuticals. It has several advantages including; simplicity, cost-effectiveness, rapidness as well as batch analysis with accurate quantification of multicomponent mixture [48, 49]. TLC method is an effective approach for simultaneously determining mixtures because it has great selectivity and accuracy, requires little sample preparation, and uses little solvent.
Successful treatment of sexual dysfunction caused by DLX could be attained through simultaneous administration of AVN. Consequently, there is a critical need to establish an innovative method for the simultaneous determination of the examined medications. In the present TLC approach, a dual wavelength procedure was used that is highly sensitive, quick, simple, selective, and affordable. The absorption spectra for both medications are illustrated in (Fig. 2). It was possible to quantify several samples containing both DLX and AVN in a single run, which preserves time, chemical substances, and manpower. The suggested approach was able to measure both medications in human plasma that has been spiked without any interference from the plasma components. Thus, the method can be applied for the measurement of blood levels of the investigated medications following co-administration.

2D HPTLC densitogram of the mixture containing (400 ng/band) of DLX and (400 ng/band) of AVN measured at dual wavelength (232 nm for DLX and 253 nm for AVN)
Method development and optimization
Numerous variables were examined to determine their effects on the approach so as to achieve the best resolution and separation with appropriate values of retardation factor (Rf) and sharp symmetrical peaks. The mobile phase’s composition, saturation period of time, and scanning wavelengths have been adjusted as parameters affecting the suggested TLC-densitometric methodology.
Mobile phase composition
In order to separate the binary mixture of DLX and AVN, various mobile phase compositions investigated. Numerous trials for optimizing the mobile phase composition had been performed by adjusting the ratios of several organic solvents as shown in (Table 1). For example, methanol-acetone (8:2, v/v) was tried where DLX was very near to the base line with tailing. Toluene-methanol (7:3, v/v) could achieve good separation but with tailing. Upon using n-hexane-methanol (8:2, v/v), both DLX and AVN remained at the baseline. Additionally, when, acetone-ethanol-ammonia (8:1:1, v/v/v) were used, the two peaks were near the solvent front line. In addition, ethyl acetate-acetone-ammonia (8:1:1, v/v/v) could not well separate the two peaks. Finally, the optimum mobile phase which could efficiently separate DLX and AVN combination was methanol-acetone-ammonia (8:2:0.05, v/v/v) as shown in Fig. 2. The Rf values of the separated peaks were 0.23 for DLX and 0.75 for AVN, it was necessary to use 33% ammonia solution for the purpose of preventing bands from tailing. The AVN band appears close to the solvent front with higher ammonia volumes. Therefore, a mixture of methanol-acetone-33% ammonia solution (8:2:0.05, v/v/v) was used as the mobile phase that could attain the most effective separation between the two components.
Time of saturation
It is crucial to fully saturate the chamber where development is happening by adding the mobile phase. because it significantly affects chromatographic separation. Prior to chromatographic development, the development jar was permitted to get saturated with the mobile phase for varying periods of time (10–30 min). It was observed that 20 min was adequate to obtain good separation.
Scanning wavelength
In order to determine the best wavelength for detecting both medications, a plate that had been spotted with both DLX and AVN was developed and then scanned in the UV region between 200 and 400 nm. The UV spectra of the suggested substances are shown in Fig. 3. The optimal scanning wavelengths that exhibit the best sensitivity and selectivity were 232 nm for DLX and 253 nm for AVN. The peaks that were produced are shown in (Fig. 2) and were sharp, symmetrical, well-separated, and noise-free.

Absorption spectra of DLX and AVN (6 µg/mL) in methanol solvent
Method validation
The suggested TLC methodology has been validated to be compliant with the ICH guidelines [50]. The evaluated parameters were; the linearity range, detection and quantitation limits, accuracy, precision, and robustness. All results are presented as percentages.
Linearity
Several medication solutions at varying concentrations were examined using the analysis procedure described above. The peak area values were recorded and displayed vs. drug concentrations in ng/spot for constructing the calibration curves after plate development and scanning at 232 and 253 nm. Three measurements were made for each concentration. It readily became apparent that a residual graph of a second order polynomial fit the data considerably much better than the linear regression model with higher values for the determination and correlation coefficient. The following polynomial equation was employed to fit the data; y = ax2 + bx + c. This relationship was proportionate within concentration ranges of 5-800 ng/spot (1–160 µg/mL) for DLX and 10–800 ng/spot (2–160 µg/mL) for AVN (Fig. 4). For the quadratic polynomial model fit, the estimated correlation and determination coefficients were found to be r = 0.9999 and r2 = 0.9999. It showed that the examined concentrations and the determined peak areas had a strong correlation. Table 2 displays the additional statistical variables for the second order polynomial regression equation and other validation parameters for both medications.

Three-dimensional HPTLC densitogram showing recorded intensities against Rf values (5–800 ng/band) for DLX and (10–800 ng/band) for AVN, measured at dual wavelength (232 nm for DLX and 253 nm for AVN)
Limits of detection and quantification
The limits of quantitation (LOQ) and detection (LOD) have been estimated to assess the sensitivity of the approach. The recommended approach was discovered to be sensitive due to the low values of LOD and LOQ for DLX and AVN, as shown in (Table 2). The coefficient of the X variable from the polynomial regression equation and the standard deviation of the intercept were used to determine LOD and LOQ. The formulas used were LOD = 3.3σ/S and LOQ = 10σ/S, where (S) is the coefficient of the x variable and (σ) is the standard deviation of the intercept. The calculated LOD values were 1.55 and 3.15 ng/spot (0.31 and 0.63 µg/mL) for DLX and AVN, respectively, and the calculated LOQ values were 4.69 and 9.53 ng/spot (0.94 and 1.91 µg/mL) for DLX and AVN, respectively (Table 2).
Accuracy
The recovery percentage of each of the drugs under study was calculated at 3 different concentrations, covering the linear range (low, medium, and high), after being determined by the recommended procedure. Three replicate assessments for each concentration were carried out in order to judge the accuracy of the suggested method. The obtained findings demonstrated the excellent accuracy of the suggested procedure for the reason that the standard deviation values were low and the estimated recovery value percentages were close to 100% (Table 3).
Precision
Two types of precision, intra-day (repeatability) and inter-day (intermediate), were assessed for the proposed approach. The recommended approach was used to analyze a total of three concentrations covering the range of linearity (200, 400, and 600 ng/spot for both drugs) Three separate times on one day to evaluate the repeatability, while the suggested method was used to analyze three concentrations over three consecutive days to estimate the intermediate precision. As shown in Table 4, relative standard deviation (%RSD) values were fairly small.
Robustness
Minor but intentional variations in the chromatographic procedure variables were made, and their influence on resulting data were examined, in order to evaluate the robustness of the proposed approach. Minor changes were made to the detection wavelength, saturation period of time, and constitution of the mobile phase system. According to the findings, none of the analyses of the two medications were significantly impacted by the modification made to the parameters under investigation. (Table 5). This demonstrated that the suggested approach was robust.
Specificity
As seen in the HPTLC chromatogram (Fig. 3) that shows entirely separation of DLX and AVN serves as a demonstration of the proposed method’s specificity. Additionally, the lack of anypeaks at the Rf of the investigated medication and the successful application of the technique to the synthetic mixture, as shown in (Table 6), demonstrate the absence of excipient impact.
Application of the suggested approach
Application to pharmaceutical preparations
The suggested HPTLC approach was successfully applied to analyze laboratory-made solutions of the Erovanafile® tablets and Cymbalta (the commercial dosage forms in the local market). As indicated in (Table 6), the final results were compared to those attained using the previously published approaches [2] for DLX and [51] for AVN, as shown in Table 6. According to t and F tests, it was found that there was no statistically significant difference between the findings produced by the established method and those obtained by the methods that were published for the mixture ingredients. The findings obtained showed that there is no interference from either the co-administered medication or those frequently experienced additives and that the proposed approach is beneficial for the examined medications to achieve satisfactory recovery. The suggested approach is sensitive, specific, precise, and accurate. It is appropriate for determining the dosage forms of the investigated medications and for use in quality-control tests in laboratories.
Application to spiked human plasma
The extremely sensitive HPTLC approach used in the current work allowed for an analysis of the DLX-AVN mixture in human plasma. The samples of plasma have been spiked with three different concentrations of the drug combination. The drugs concentrations were determined in accordance with the established protocol after a simple precipitation of proteins using acetonitrile and centrifugation. The chromatogram of the analyzed samples under investigation effectively provided acceptable identification and acceptable separation of the two drugs on the TLC plate, as demonstrated in (Fig. 5b). A sample of plasma that was not previously spiked with any drugs was also subjected to the procedure as a blank analysis, as seen in (Fig. 5a). The peaks of the plasma components are weak, well-resolved and completely separated from the peaks of the drugs. Consequently, the plasma components did not significantly interfere with the findings, which are shown in (Table 7). Because the found % recoveries were high enough it was suggested that the present technique is suitable for determining the considered drugs in plasma from humans after concurrent administration. Because collecting samples of blood from individuals receiving experimental medications was problematic, we preferred to conduct the research study using human plasma samples that had been prepared in vitro in combination with the medications under investigation. The results presented here showed that there is a minor variability that resulted from the influence of matrix n the estimation of DLX and AVN in human plasma.

2D HPTLC densitogram of plasma sample (a) before and (b) after spiking with (400 ng/band) of DLX and (200 ng/band) of AVN measured at dual wavelength (232 nm for DLX and 253 nm for AVN)
It should be noted that almost all the previously published methods were utilized to determined avanafil or duloxetine in their single form or in combination with other drugs. Thus, the merit of the present works comes from its ability to simultaneously determine both avanafil and duloxetine in presence of each other. However, recently a new spectrofluorimetric method was published for the simultaneous determination. Although the published method was more sensitive, the proposed method consumes less chemical and faster owing to its ability to analyze several samples in the same run. A summary comparing the proposed method with the reported methods is presented in the supplementary data (S1 and S2).
Evaluation of method greenness
The three aspects that make up a “green” analysis are the absence of waste, the limited or absence use of hazardous chemicals, and the reduction in energy use. There are many tools available to assess how ecologically friendly the suggested approach is [52, 53]. The greenness profile, Eco-Scale methodology, and the Green Analytical Procedure Index (GAPI) were investigated to assess the greenness of the proposed TLC densitometric approach.
The National Environmental Method Index (NEMI) was used to assess the established method’s greenness profile [54]. It depends on the use of PBTs, which are non-permanent, bio accumulative, and toxic solvents. The proposed methodology used acetonitrile and ethyl acetate as solvents, none of which are PBT. The pH of the developing system was also in the middle range of the pH scale therefore it was determined that it was not corrosive. The substances are safe to use, so not hazardous. Additionally, the waste volume was under 50 ml. Therefore, the procedure meets all the criteria for being a green procedure. Accordingly, the suggested TLC-densitometric methodology was deemed an environmentally green method as it passed all four greenness profile quadrants (Fig. 6).

Evaluation of the proposed HPTLC method using NEMI pictograms (A), GAPI (B), AGREE (C), and BAGI (D) tools for greenness and blueness
Eco-scale is a simple method that is easily applied in the work of quality control laboratories. The analytical Eco-Scale score is calculated by utilizing the following equation (analytical Eco-Scale score = 100 − total penalty). Penalty points were assigned for each variable of the studied technique, including the amount of chemicals, workplace risks, waste, and energy consumption [55]. When the final score is greater than 75, the analytical strategy is regarded as an excellent green analysis. The solvents used in the current research were non-toxic. There was extremely little waste and very little energy utilized in the present study. These features allow the developed TLC-densitometric technique achieve an eco-Scale score of 86, which is an excellent level of environmental friendliness (Table 8).
The Green Analytical Procedure Index (GAPI) is an additional tool that could be used to assess how environmentally friendly the suggested approach is [56]. Further characteristics of the analytical method are taken into account while evaluating the method’s greenness in this tool. To evaluate each step of the procedure of analysis that might have an effect on the environment, five pentagrams were generated. The three unique color codes of green, yellow, and red were used to represent minimum, medium, and major environmental implications, respectively. The GAPI pentagrams showed that the currently employed technique has an acceptable degree of greenness because it includes 7 green, 5 yellow, and 3 red shaded fields, as shown in Fig. 6.
For a deeper understanding of the unique green features of the suggested approach, a further green metric was included. This measure is referred to as the AGREE analytical tool [57]. The twelve sections that make up the AGREE framework are arranged in accordance with the twelve major GAC principles. Indeed, every component in these sections is given a distinct color that corresponds to its degree of greening, ranging from 0.0 (shown by the color red) to 1.0 (shown by the color green). This score is represented graphically as a clock-shaped pictogram in the middle of the framework, which functions as a color and score indicator for the whole procedure. The evaluation can be carried out with software, which automatically produced a report and a graph. The present methodology received an ultimate score of (0.84) upon evaluating all of these principles.
Blueness evaluation
A new metric tool called the Blue Applicability Grade Index (BAGI) is utilized to assess the analytical method’s practicality [58]. Two different sets of results are produced by the BAGI metric tool: a graphical representation in the form of an asteroid pictogram and a numerical score at the center. The asteroid-shaped pictogram, which is made up of several hues of blue to represent varying degrees of compliance (dark blue for high, blue for moderate, light blue for low, and white for non-compliance), serves as a visual representation of the result of the evaluation. In order to develop a pictogram and a score that illustrate the practicality and functionality of an analytical approach, BAGI takes into account 10 factors (S3, Table). It is advised that the final score be greater than 60, so that the analytical method can be considered “practical”. As seen in the pictogram’s center, the suggested approach receives a high score of 80.0.
Conclusion
The determination of DLX-AVN combination was performed for the first time using a precise, simple, ecologically friendly, and highly sensitive TLC approach. The evaluation of the proposed method was carried out using the Eco-scale, NEMI, GAPI, AGREE, and BAGI tools. The findings clearly demonstrate the sustainability and environmental friendliness of the presented approach. As a result, it can potentially be used in quality control laboratories. The technique uses a low-cost procedure with great precision and accuracy and is sensitive, straightforward, quick, selective, and doesn’t involve any complicated extraction steps. Without any noticeable interference from the plasma ingredients, the approach appeared sensitive enough to estimate the examined medications in plasma. For depressive patients, determining the concentrations of co-administered antidepressants and sexual stimulant medications in biological fluids is a crucial and important step. The recommended method offered a number of desirable benefits, including direct sample application for simplicity of use, quick analysis, a high number of samples per run, and minimal solvent use.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Yunoos M, Sankar DG, Kumar BP, Hameed S, Hussain A. Simple UV spectrophotometric determination of duloxetine hydrochloride in bulk and in pharmaceutical formulations. E-J Chem. 2010;7:785–8.
Kamila MM, Mondal N, Ghosh LK. A validated UV spectrophotometric method for determination of duloxetine hydrochloride. Die Pharm Int J Pharm Sci. 2007;62:414–5.
Abu-hassan AA, Derayea SM, Ali R, Abu-hassan AA. Two facile approaches based on association complex with erythrosine-B for nano-level analysis of duloxetine: application to content uniformity. R Soc Open Sci. 2021;8:210922.
Abdelhamid NS, Naguib IA, Anwar BH, Magdy MA. A validated HPTLC method for the quantitative determination of duloxetine hydrochloride and 1-naphthol in bulk and pharmaceutical formulation. JPC–Journal Planar Chromatogr TLC. 2020;33:391–6.
Samal L, Prusty A. Development and validation of Uv-Visible Spectrophotometric Method for determination of Duloxetine. Int J Pharm Pharm Sci. 2019;11:27–31.
Vijay Amirtha Raj R, Ramesh T, Phani Kumar A. A validated UV spectrophotometric determination of an antidepressant drug - duloxetine hydrochloride from capsule formulations. Int J Pharma Bio Sci. 2011;2:716–20.
Alarfaj NA, Ammar RA, El-Tohamy MF. Cationic-enhanced spectrofluorimetric method for determination of selective serotonin reuptake inhibitor duloxetine hydrochloride in its dosage forms. Asian J Chem. 2013;25:6416–20.
Lakshmana Prabu S, Shahnawaz S, Dinesh Kumar C, Vasantharaju SG, Abraham J. Spectrofluorimetric method for determination of tramadol hydrochloride in bulk and pharmaceutical dosage forms. Indian Drugs. 2009;46:32–4.
Sagirli O, Toker SE, Önal A. Development of sensitive spectrofluorimetric and spectrophotometric methods for the determination of duloxetine in capsule and spiked human plasma. Luminescence. 2014;29:1014–8.
prabu SL, Shahnawaz S, Karthik A, Kumar CD, Vasantharaju SG. High-performance liquid chromatography method for the quantification of duloxetine in rat plasma. 2008;49:283–92.
Boopathy D, Jawarkar RD, Prakash M, Mathew B, Perumal P. New RP-HPLC method development and validation determination for estimation of duloxetine HCl in enteric coated capsules. Int J Chem Tech Res. 2010;2:239–41.
Srinivasulu P, Srinivas KSV, Reddy RS, Mukkanti K, Buchireddy R. A validated stability indicating rapid LC method for duloxetine HCl. Die Pharm Int J Pharm Sci. 2009;64:10–3.
Pushpalatha P, Sarin RK, Idris M, Rao MA, Baggi TRR, Varma MS. Determination of free duloxetine in human serum by high-performance thin-layer chromatography. JPC–Journal Planar Chromatogr TLC. 2013;26:354–7.
Dhaneshwar SS, Deshpande P, Patil M, Vadnerkar G, Dhaneshwar SR. Development and validation of a HPTLC method for estimation of duloxetine hydrochloride in bulk drug and in tablet dosage form. Indian J Pharm Sci. 2008;70:233–6.
Patel SK, Patel NJ, Patel PU, Patel DB, Prajapati AM, Patel SA. Validation of a stability-indicating HPTLC method for analysis of duloxetine hydrochloride in capsule dosage form. Separation and analysis of duloxetine hydrochloride and olanzapine in a synthetic mixture. JPC–Journal Planar Chromatogr TLC. 2009;22:121–6.
Al-Qahtani SD. Potentiometric determination of Duloxetine Hydrochloride using coated Wire Electrode in pure and Pharmaceutical preparations. Asian J Pharm. 2017;11:S854–858.
Ammar RA, Otaif H, Al-Warthan A. A novel ion-selective membrane electrode for the determination of duloxetine. Anal Methods. 2012;4:1427–31.
Alarfaj NA, Ammar RA, El-Tohamy MF. Disposable screen-printed sensors for determination of duloxetine hydrochloride. Chem Cent J. 2012;6:1–8.
Manrique GRP, Salamanca-Neto CAR, Tobias Moraes J, Sartori ER. Fast surface water quality analysis based on an ultra-sensitive determination of the antidepressant drug duloxetine hydrochloride on a diamond electrode by voltammetry. Int J Environ Anal Chem. 2022;102:5680–94.
Hassanein AM, Moharram YI, Oraiby NF, Ebied SE. Trace determination of duloxetine HCl in formulation and spiked human serum at a carbon paste electrode. Am J Anal Chem. 2017;8:708–25.
Lović J, Bogdanović A, Tadić V, Mijin D, Vuković D, Petrović S, Ivić MA. Electrochemical Behavior of Duloxetine hydrochloride at au and GC solid electrodes: its quantitative determination and degradation. J Electrochem Soc. 2022;169:76507.
Jyoti, Żołek T, Maciejewska D, Gilant E, Gniazdowska E, Kutner A, Noworyta KR, Kutner W. Polytyramine film-coated single-walled carbon nanotube electrochemical chemosensor with molecularly imprinted polymer nanoparticles for duloxetine-selective determination in human plasma. ACS Sens. 2022;7:1829–36.
Kumar N, Sangeetha D, Kalyanraman L, Sainath K. Stability-indicating HPLC method for simultaneous determination of degradation products and process-related impurities of avanafil in avanafil tablets. Acta Chromatogr. 2018;30:158–63.
Mohamed TA, Atty SA. Native and synchronous fluorescence spectroscopy for determination of avanafil in presence of its co-formulated drug (dapoxetine hydrochloride): application to pharmaceutical produc. Spectrochim Acta Part Mol Biomol Spectrosc. 2020;229:117898.
Fahmy UA, Aljaeid BM. Stability indicating HPLC method for analysis of Avanafil using diode array detector. Int J Adv Pharm Biol Chem. 2016;5:59–64.
Hasan MA, Madkour W, El-. EA. Second derivative synchronous fluorescence determination of avanafil in the presence of its acid-induced degradation product aided by powerful Lean Six Sigma tools augmented with D-optimal design. RSC Adv. 2021;11:3834–3842.
Can NÖ. Development of validated and stability-indicating LC-DAD and LC-MS/MS methods for determination of avanafil in pharmaceutical preparations and identification of a novel degradation product by LCMS-IT-TOF. Molecules. 2018;23:1771.
Darwish MK, Soliman MM, Abdel-Razeq SA. Validated stability indicating HPTLC and UV-spectrophotometric techniques for the determination of avanafil. Asian J Chem Sci. 2019;6:1–16.
Patel MN, Kothari CS. Multivariate approaches for simultaneous determination of avanafil and dapoxetine by UV chemometrics and HPLC-QbD in binary mixtures and pharmaceutical product. J AOAC Int. 2016;99:649–63.
Abd-AlGhafar WN, Abo Shabana R, El‐Shaheny R, Tolba MM. Environmentally benign first derivative synchronous spectrofluorimetry for the analysis of two binary mixtures containing duloxetine with avanafil or tadalafil in spiked plasma samples. Luminescence. 2024;39:e4696.
Hegazy M, Kessiba A, Abdelkawy M. RP-HPLC with Time Programmed Fluorescence Detection for Quantitation of Avanafil and Dapoxetine Hydrochloride: application to Pharmaceutical Dosage Form and Biological Fluid RP-HPLC with Time Programmed Fluorescence Detection for Quantitation of Avanafil. J Liq Chromatogr Relat Technol. 2015;38:1660–5.
Er EÖ, Akkaya E, Özbek B, Bakırdere S. Development of an analytical method based on citric acid coated magnetite nanoparticles assisted dispersive magnetic solid-phase extraction for the enrichment and extraction of sildenafil, tadalafil, vardenafil and avanafil in human plasma and urine prior to determination by LC-MS/MS. Microchem J. 2019;147:269–76.
Abou-Omar MN, Annadi AM, El Zahar NM, Youssef AO, Amin MA, Attia MS, Mohamed EH. Simultaneous determination of Avanafil and Dapoxetine in human plasma using liquid chromatography/tandem mass spectrometry (LC-MS/MS) based on a protein precipitation technique. RSC Adv. 2021;11:29797–806.
CAN NÖ. Development of validated and stability-indicating LC-DAD and LC-MS / MS methods for determination of Avanafil in Pharmaceutical preparations and. Molecules. 2018;23:1771.
Shah DA, Vegad KL, Patel ED, Prajapati HK, Patel RN, Patel YK. Analytical method validation for estimation of avanafil and dapoxetine hydrochloride tablet dosage form by HPTLC method. Pharm Biol Eval. 2017;4:171–9.
Ali A-MBH, Rageh AH, Abdel-aal FAM, Mohamed A-MI. Anatase titanium oxide nanoparticles and multi-walled carbon nanotubes-modified carbon paste electrode for simultaneous determination of avanafil and doxorubicin in plasma samples. Microchem J. 2023;185:108261.
Ali A-MBH, Abdel-aal FAM, Rageh AH, Mohamed A-MI. Hybrid NiO nanostructured/sulfanilamide polymeric film for studying possible pharmacokinetic interaction between avanafil and nimodipine in real human serum by their simultaneous determination using square-wave voltammetry. Microchem J. 2022;172:106895.
Ali MB, Abdel-Raoof AM, Hendawy HAM, Talaat W, Omran GA, Morshedy S. An eco-friendly solid-state electrode modified with ZnO nanoparticles decorated with MWCNT as an Electrochemical Sensor for the determination of Avanafil in pure form, dosage form and human plasma. J Electrochem Soc. 2021;168:87510.
Ali MB, Talaat W, Omran GA, Hendawy HAM, Morshedy S. Validated Capillary Zone Electrophoretic determination of Avanafil and Dapoxetine Hydrochloride in their pure form and Pharmaceutical Preparation. J Pharm Res Int. 2021;33:446–59.
Ali A-MBH, Abdel-Aal FAM, Rageh AH, Mohamed A-MI. A Comprehensive Review on Analytical techniques for determination of sex stimulants, PDE5 inhibitors in different matrices with Special Focus on the Electroanalytical methods. Crit Rev Anal Chem. 2022;:1–30.
Higgins A. Antidepressant-associated sexual dysfunction: impact, effects, and treatment. Drug Healthc Patient Saf. 2010;2:141–50.
Mazzilli R, Angeletti G, Olana S, Delfino M, Zamponi V, Rapinesi C, Del Casale A, Kotzalidis GD, Elia J, Callovini G, Girardi P, Mazzilli F. Erectile dysfunction in patients taking psychotropic drugs and treated with phosphodiesterase-5 inhibitors. Arch Ital Urol Androl. 2018;90:44–8.
Maruca-Sullivan PE, Kleinfeld SA, Wilkins KM. Treatment approaches to sexual dysfunction in late life. Curr Treat Options Psychiatry. 2018;5:255–74.
Wadie M, Abdel-Moety EM, Rezk MR, Marzouk HM. A novel smartphone HPTLC assaying platform versus traditional densitometric method for simultaneous quantification of alfuzosin and solifenacin in their dosage forms as well as monitoring content uniformity and drug residues on the manufacturing equipment. RSC Adv. 2023;13:11642–51.
Wadie M, Abdel-Moety EM, Rezk MR, Marzouk HM. Smartphone-based high-performance thin layer chromatographic method along with benchtop densitometry for simultaneous quantification of co-formulated dutasteride with silodosin and their residuals on manufacturing equipment’s surfaces. Bull Fac Pharm Cairo Univ. 2023;61:29–45.
Wadie M, Abdel-Moety EM, Rezk MR, Marzouk HM. Sustainable and smart HPTLC determination of silodosin and solifenacin using a constructed two illumination source chamber with a smartphone camera as a detector: comparative study with conventional densitometric scanner. Sustain Chem Pharm. 2023;35:101095.
Rits IA. Declaration of Helsinki. Recommendations guidings doctors in clinical research. World Med J. 1964;11:281.
Kelani KM, Hegazy MA, Hassan AM, Tantawy MA. A green TLC densitometric method for the simultaneous detection and quantification of naphazoline HCl, pheniramine maleate along with three official impurities. BMC Chem. 2022;16:1–11.
Zhou B, Tan M, Lu J, Zhao J, Xie A, Li S. Simultaneous determination of five active compounds in chimonanthus nitens by double-development HPTLC and scanning densitometry. Chem Cent J. 2012;6:1–5.
Conference I, Harmonisation ON, Technical OF, For R, Of R, For P. Requirements for registration of pharmaceuticals for human ich harmonised tripartite guideline validation of a nalytical p rocedures: Parent Guideline: Text on Validation of Analytical Procedures. 2005;1994 November.
Savaliya SD, Chaudhary AB, Rathod BG, Dobariya TD, Mohan S. Development and validation of UV spectrophotometric method for estimation of Avanafil in tablet dosage form. Inven Rapid Pharm Anal Qual Assur. 2013.
Wadie M, Abdel-Moety EM, Rezk MR, Marzouk HM. A novel eco-friendly HPLC method with dual detection modes for versatile quantification of dutasteride and silodosin in pharmaceutical formulation, dissolution testing and spiked human plasma. Microchem J. 2024;197:109753.
Rostom Y, Rezk MR, Wadie M, Abdel-Moety EM, Marzouk HM. State-of-the-art mathematically induced filtration approaches for smart spectrophotometric assessment of silodosin and solifenacin mixture in their new challenging formulation: multi-tool greenness and whiteness evaluation. Spectrochim Acta Part Mol Biomol Spectrosc. 2024;307:123650.
Keith LH, Gron LU, Young JL. Green analytical methodologies. Chem Rev. 2007;107:2695–708.
Gałuszka A, Migaszewski ZM, Konieczka P, Namieśnik J. Analytical Eco-scale for assessing the greenness of analytical procedures. TrAC Trends Anal Chem. 2012;37:61–72.
Płotka-Wasylka J. A new tool for the evaluation of the analytical procedure: Green Analytical Procedure Index. Talanta. 2018;181:204–9.
Pena-Pereira F, Wojnowski W, Tobiszewski M. AGREE - Analytical GREEnness Metric Approach and Software. Anal Chem. 2020;92:10076–82.
Manousi N, Wojnowski W, Płotka-Wasylka J, Samanidou V. Blue applicability grade index (BAGI) and software: a new tool for the evaluation of method practicality. Green Chem. 2023;25:7598–604.
Acknowledgements
Not applicable to this study.
Funding
Open access funding provided by The Science, Technology & Innovation. Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
Hadeer A. Elhamdy: Methodology, Formal analysis, Investigation, Writing – original draft. Sayed M. Derayea: Supervision, Conceptualization, Resources, Project administration. Mohamed Oraby: Validation, Writing – review & editing. Khalid M. Badr El-Din: Data curation, Visualization.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental protocols in the current study were approved by the Committee of Research Ethics in the Faculty of Pharmacy, Sohag University, Sohag, Egypt. All methods were carried out in accordance with relevant regulations and guidelines. In all cases, informed written consent was obtained from each participant before donating the blood samples.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Derayea, S.M., Elhamdy, H.A., Oraby, M. et al. Simultaneous measurement of duloxetine hydrochloride and avanafil at dual-wavelength using novel ecologically friendly TLC-densitometric method: application to synthetic mixture and spiked human plasma with evaluation of greenness and blueness. BMC Chemistry 18, 92 (2024). https://doi.org/10.1186/s13065-024-01195-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-024-01195-2