
Abstract
The QSAR models are employed to predict the anti-proliferative activity of 81 derivatives of flavonol against prostate cancer using the Monte Carlo algorithm based on the index of ideality of correlation (IIC) criterion. CORAL software is employed to design the QSAR models. The molecular structures of flavonols are demonstrated using the simplified molecular input line entry system (SMILES) notation. The models are developed with the hybrid optimal descriptors i.e. using both SMILES and hydrogen-suppressed molecular graph (HSG). The QSAR model developed for split 3 is selected as a prominent model (\({R}_{Validation}^{2}\)= 0.727, \({IIC}_{validation}\)= 0.628, \({Q}_{Validation}^{2}\)= 0.642, and \({\overline{r} }_{m}^{2}\)=0.615). The model is interpreted mechanistically by identifying the characteristics responsible for the promoter of the increase or decrease. The structural attributes as promoters of increase of pIC50 were aliphatic carbon atom connected to double-bound (C…=…, aliphatic oxygen atom connected to aliphatic carbon (O…C…), branching on aromatic ring (c…(…), and aliphatic nitrogen (N…). The pIC50 of eight natural flavonols with pIC50 more than 4.0, were predicted by the best model. The molecular docking is also performed for natural flavonols on the PC-3 cell line using the protein (PDB: 3RUK).
Similar content being viewed by others

Introduction
Flavonoids are a class of polyphenolic compounds which possess a phenyl benzopyrone structure (C6–C3–C6) and are present in all vascular plants. These are produced as secondary plant metabolites, which are known to demonstrate broad-spectrum pharmacological activities, but the human body is unable to produce them [1,2,3]. These compounds according to saturation level subdivided into flavanols, flavonols, flavones, flavanones, isoflavones, flavanonols, and chalcones [4, 5].
The CYP17A1 has an important role in the biosynthesis of dehydroepiandrosterone (DHEA) as the precursor of androgens and overexpression of this enzyme can cause prostate cancer. Abiraterone as an approved anti-prostate cancer drug is a CYP17A1 inhibitor [6, 7]. Flavonols are characterized by a hydroxyl group present at C-3 of the flavone skeleton and there are some reports about the CYP17A1 inhibitory activity of flavonoids like rutin, morusflavone, quercetin, kaempferol and isorhamnetin [8,9,10].
These have also been attracted by medicinal chemists because of their effective anti-prostate cancer properties. Prostate cancer is the most common type of diagnosed cancer among males worldwide with the incidence of 28 cases per 100,000 and mortality being 7 per 100,000 [11,12,13]. Normal growth and maintenance of the prostate is dependent on androgen hormones that act through the androgen receptor. Activation of the androgen receptor drives the development of prostate cancer. It has been reported that the agents such as flavonols that down-regulate androgen receptors can inhibit the development of prostate cancer cells [14,15,16].
The influence of chemical structures of flavonols over their anticancer activities has been investigated experimentally and shown that structural modification can further increase its anti-cancer activity and ability to activate PC-3 cell apoptosis. However, the structure–activity relationship for flavonols as anti-prostate cancer agents has captured attention by quantitatively correlating the molecular structures or properties with variation in pharmacological activity [17, 18].
The anti-prostate cancer activity is expressed typically with IC50 (half maximal inhibitory concentration) values. Quantitative structure–activity relationships (QSARs) are a powerful tool to predict IC50 of flavonoids in general. Already, no study has been reported on QSAR modeling for predicting the IC50 of flavonols against prostate cancer.
QSAR model is a mathematical equation which is widely employed to estimate and predict pharmacological activity or physical, chemical properties/activities of chemicals using descriptors derived from chemical structure [19,20,21,22]. The CORAL (Correlation and Logic) freeware software is employed for designing the Quantitative structure–activity/activity relationships (QSPRs/QSARs) models in compliance with OECD principles [23,24,25,26]. In CORAL software, the SMILES notations of the molecular structure are used as an input file and produce the best model based on Monte Carlo optimization [27,28,29,30]. It can be applied to compute the optimal descriptor by using solely SMILES or molecular graph-based descriptor or a combination of both descriptors (so-called hybrid descriptor). A literature survey reveals that the index of ideality of correlation (IIC) parameter of CORAL software can be employed to build robust QSAR models [31,32,33,34].
Molecular docking simulation is a computational methodology that purveys automatic tools to measure the conformation of a protein–ligand complex. The aim of molecular docking is to regulate the position of the ligand in the protein. An energy-based scoring function is commonly used in docking procedures to find the energetically most advantageous ligand conformation when attached to the target. Intermittently, the Monte Carlo computational methodologies are also applied in molecular docking simulation [35, 36].
Since ancient times various natural products have been used as traditional medicine against various human diseases. Moreover, natural products are easily applicable, cheap, accessible and acceptable treatment method with minimum cytotoxicity [37]. As a results of QSAR modeling, the pIC50 activity of some natural flavonols as anti-proliferative agents were predicted and reported.
The goal of this report is to devise reliable first QSAR models utilizing CORAL software to predict pIC50 of 81 flavonols against prostate cancer. In the development of QSAR models, a hybrid optimal descriptor, a combination of SMILES and hydrogen suppressed graph (HSG), is employed. The index of ideality of correlation (IIC) is used to improve the predictive potential of QSAR models. Further, the pIC50 is also calculated for a series of eight natural flavonols using the QSAR models of all splits. As mentioned above flavonols show their anti-prostate cancer activity through different mechanism of actions. However, molecular docking is also performed for eight natural flavonol derivatives in order to evaluate their potential affinity to CYP17A1 (PDB: 3RUK).
Methods
Data
Experimental data on anti-prostate cancer (PC-3) activities of 86 flavonols were taken from the four literature reports (Additional file 1: Table S1) [11, 38,39,40]. The numerical values of activity were converted to a negative logarithmic scale, pIC50 (− logIC50) (Molar) for QSAR modelling. The range of pIC50 for PC-3 cell line was from 3.39 to 6.28. The current dataset was not previously used for QSAR modeling. The molecular structures of the flavonol derivatives were sketched by BIOVIADraw 2019 and transferred to the SMILES code for modeling with the CORAL software. Three splits were made from the dataset and each split was further randomly divided into four sets i.e., training (≈ 35%), invisible training (≈ 25%), calibration (≈ 15%), and validation (≈ 25%) sets. In CORAL-based QSAR modeling, each set was assigned its specific accountability. The task of the training set (TRN) was to compute correlation weights and the task of the invisible training set (iTRN) was to control the adaptability of the data which were not employed in the training set. The assignment of the calibration set (CAL) was to detect the overtraining whereas the final estimation of the predictive potential of the designed QSAR model was assigned to the validation set (VAL) [34, 41].
Hybrid optimal descriptor
Herrin, the optimal hybrid optimal descriptor based on SMILES and HSG was employed to create QSAR models for pIC50 of flavonol compounds. The literature reports showed that the QSPR models produced through the ‘hybrid’ optimal descriptor had better statistical parameters than the model designed by individually SMILES or HSG descriptors [42, 43].
The QSAR model employed to predict pIC50 of flavonol derivates is demonstrated in the following equation:
Here, C0 is the regression coefficient and C1 is the slope computed by the least-squares method; DCW (descriptor of correlation weights) is computed with correlation weights of molecular features extracted from HSG and SMILES notations. The following equation is employed to compute DCW:
where AK is an attribute of SMILES or HSG, the T* and N* define the threshold value and number of epochs of the Monte Carlo optimization, respectively.
The DCW of HSG and SMILES employed here are illustrated as Eqs. (4) and (5):
The SMILES attributes and HSG invariant applied in Eqs. (4) and (5) are depicted in Table 1.
A flowchart of a Monte Carlo optimization cycle is presented by Sokolovic et al. [44]. At first cycle, the CW(x) of features is randomly generated and then optimized based on the proposed objective function. Herein, two kinds of target functions consisting of the balance of correlation without IIC (TF1) and the balance of correlation with IIC (TF2) are studied.
The following mathematical equation is employed to compute the TF1 and TF2:
The Rtraining and RinvTraining are the correlation coefficients for the training and invisible training sets, respectively. The empirical constant (Const) is usually fixed [45, 46].
The IICCAL is calculated with data on the calibration (CAL) set as the following:
RCAL is the correlation coefficient for the calibration set. The negative and positive mean absolute errors are shown with −MAE and +MAE, which are computed using the following equations:
The ‘k’ is the index (1, 2,…N). The observedk and calculatedk are related to numerical values of the endpoint.
This IIC is obtained by using the correlation coefficient between the observed and predicted values of the endpoint for the calibration set, taking into account the positive and negative dispersions between the observed and calculated values [47].
Applicability domain
The applicability domain (AD) is another key guideline that should be included in a built QSPR/QSAR model. It was defined by the OECD as "the response and chemical structure space in which the model produces predictions with a specified reliability" [48, 49]. The CORAL-based QSAR model computes AD based on the dispersion of SMILES features in the training and calibration sets [50]. The AD is defined as ‘DefectAK’, which was computed with the following equation:
\({P}_{TRN}{(A}_{K})\) and \({P}_{CAL}{(A}_{K})\) are the probability of an attribute 'Ak' in the training and the calibration sets; \({N}_{TRN}{(A}_{K})\) and \({N}_{CAL}{(A}_{K})\) are the number of times of Ak in the training and calibration sets, respectively.
The statistical defect is computed using the following equation:
NA is the number of active SMILES attributes for the given compounds.
In CORAL, a substance is an outlier if inequality 14 is fulfilled:
\({\overline{\mathrm{Defect}} }_{\mathrm{TRN}}\) is an average of statistical defect for the dataset of the training set.
Validation of the model
It is most important to validate the predictive potential of a constructed QSAR model. In the present manuscript, the reliability and robustness of the QSAR models were verified using the following three methodologies: i) internal validation or cross-validation by considering the training dataset, ii) external validation by considering the prediction set and iii) data randomization or Y-scrambling.
The various standard statistical metrics such as correlation coefficient (R2), cross-validated correlation coefficient (Q2), concordance correlation coefficient (CCC), the IIC, \({Q}_{F1}^{2}\), \({Q}_{F2}^{2}\), and \({Q}_{F3}^{2}\), standard error of estimation (s), mean absolute error (MAE), Fischer ratio (F), novel metrics (\({r}_{m}^{2}\)) and Y-scrambling (\({\mathrm{c}}_{{R}_{p}^{2}})\) were employed to validate the developed QSAR models. The mathematical equations of various validation metrics are shown in Table 2.
R2 statistic is a metric to evaluate the goodness of fit of a regression analysis. It measures the variation of experimental data with the predicted ones. The range of R2 is between 0 (no correlation) and 1 (perfect fit). R2 cross‐validated (Q2) is used for internal validation. The concordance correlation coefficient (CCC) is calculated to measure both precision and accuracy detecting how far each observation deviate from the best-fit. The CCC is calculated to detect both precision and accuracy distance of the observations from the fitting line and the degree of deviation of the regression line from that passing through the origin, respectively [51]. A lower value of MAE and s is desirable for good internal/external predictivity. Roy et al. [54] introduced a new metric \({\mathrm{r}}_{\mathrm{m}}^{2}\) that penalizes the r2 value of a model when there is large deviation between r2 and \({\mathrm{r}}_{0}^{2}\) values (Table 2). For a reliable QSAR model, the \(\overline{{r }_{m}^{2}}\) and \(\Delta {r}_{m}^{2}\) should be greater than 0.5 and smaller than 0.2, respectively. Y-scrambling or Y-randomization is an assessment to ensure the developed QSAR model is not due to chance, thereby giving an idea of model robustness [52]. For a robust QSAR model, Todeschini \({\mathrm{c}}_{{R}_{p}^{2}}\) parameter [55] is also calculated which should be more than 0.5. One of the important statistical parameters to judge different QSAR models is \(\overline{{r }_{m}^{2}}\) for test set. Here, this parameter is used to select best model between six proposed models.
Model interpretation
A straightforward process for the structural interpretation of QSPR/QSAR models is provided by the CORAL application. Three types of attributes may be identified by computing the correlation weights across several iterations of the Monte Carlo optimization algorithm. The positive numerical value of CWs in every iteration is considered for endpoint increase, the attributes with a negative value of CWs in every iteration is a notation for endpoint decrease. The unstable numerical value in the different runs is not considered for predicting the promoter of the increase/decrease endpoint [19, 56].
Molecular docking
Molecular docking is a method commonly employed in drug discovery and development to identify protein–ligand binding configurations This approach involves the docking of a molecule with a specific macromolecule and then computing the binding free energy between the ligand and receptor[35]. The structure was sketched in ChemDraw 16.0, and the energy was minimised in Chem3D using the MM2 technique [57]. The crystallographic structure of Human cytochrome P450 CYP17A1 in complex with abiraterone was obtained from the Protein Data Bank (PDB: 3RUK) and used for molecular docking [58]. AutoDock Vina was employed for docking studies (Molecular Graphics Lab, CA, USA) [59]. The value of exhaustiveness was 8 and the dimensions of the grid box were 20.0, 20.0, and 20.0 Å in size. The findings and illustration were examined visually using Discovery Studio visualizer 2021.
Results and discussion
QSPR modelling for pIC50
Three types of outliers affect the model quality in QSPR/QSAR study. The first is the outliers in the dependent variable y, the second is the outliers in the direction of the independent variable X, and the third type of outliers indicates a different relationship between X and y. [60]. Here, based on several preliminary QSAR models, six compounds (compounds No. 31, 32, 36, 37, 67, and 80) identified as outliers, these molecules showed a large absolute error (> 3 s). These compounds fall in first type of outlier. The structure of these compounds is similar to the main body of the samples. So, they were removed from the data set before further data processing.
In this study, the balance of correlation approach was employed to generate QSAR models. A total of six QSAR models was generated utilizing two kinds of target functions i.e. TF1 (WIIC = 0.0) and TF2 (WIIC = 0.2). To obtain the preferable threshold value (T*) and the number of epochs (N*), the range of 1–10 for threshold and 1 to 50 for epoch were employed. In the case of TF1, the value of T* and N* were 1 and 10 for split 1; 1 and 3 for split 2; 1 and 7 for split 3, respectively. However, in the case of TF2, the value of optimum (T*, N*) for splits 1, 2, and 3 were (1, 10), (1, 10), and (1, 7), respectively.
The mathematical relationship for the developed QSAR model of pIC50 using TF1 and TF2 for three splits are displayed below:
The Monte Carlo optimization with target function TF1
The Monte Carlo optimization with target function TF2
The statistical results of designed QSAR models for three splits utilizing TF1 and TF2 are presented in Table 3. As can be seen, all developed QSAR models were acceptable statistically and agreed with the requirements of various validation criteria.
According to the results presented in Table 3, it was found that the models constructed using TF2 (with IIC) had better statistical results than the models constructed using TF1 (without IIC). The results of calibration and validation sets were better for the models constructed by using TF2, but the inferior quality of the model for the training sets was obtained. Hence, it can be expressed that the models designed with the IIC are more statistically considerable and robust for the present dataset. Based on validation metric study of QSPR/QSAR models by Ojha et al., the \({\overline{r} }_{m}^{2}\) value of models is used to judge the quality of the predictions by different models. The QSAR model developed by TF2 for split 3 was selected as a prominent model with highest \({\overline{r} }_{m}^{2}\) (\({\overline{r} }_{m}^{2}\)=0.615).
The plot of observed pIC50 versus predicted pIC50 for three models designed with TF2 is displayed in Fig. 1. In the QSAR model generated by utilizing the Monte Carlo method, the outliers were introduced by the statistical defects. So, in the present QSAR model created by TF2, the number of outliers was found six for all splits. Table 4 displays flavonols IDs, SMILES codes, and descriptor of correlation weights (DCWs) with their experimental and predicted pIC50.

Observed pIC50 versus predicted pIC50 values for three CORAL models constructed based on TF2
Interpretation of the QSAR model
The mechanistic interpretation of a QSAR model is the fifth principle of OECD. The mechanistic interpretation of the QSAR model provides a correlation and a relationship between the chemical structure of the compounds and their property/activity. It also enunciates the molecular features which are responsible for the increase/decrease of endpoints that can be computed from QSAR models. Information on the mechanistic interpretation of flavonols as a promoter of pIC50 increase/decrease may aid in the design and development of new flavonol derivatives.
In CORAL, correlation weights (CWs) of structural attributes (SAk) are calculated in three or more runs and the mechanistic interpretation is achieved by analysis of CWs. If in all probes of the optimization, the numerical value of CW of structural attributes is found greater than zero, then these attributes are considered as a promoter of increase. Whereas, if the numerical value of CW of structural attributes is found smaller than zero, then these attributes are defined as the promoter of decrease [61, 62].
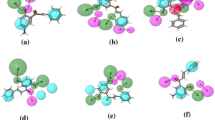
The list of attributes and their correlation weights for three runs of all splits computed with TF2 is presented in Table 5. The most significant structural attributes as the promoter of pIC50 increase were distinguished and extracted. The structural attributes as promoters of increase of pIC50 were aliphatic carbon atom connected to double-bound (C…=…, aliphatic oxygen atom connected to aliphatic carbon (O…C…), branching on aromatic ring (c…(…), and aliphatic nitrogen (N…). The good fingerprints obtained from Monte Carlo optimization method are indicated in Fig. 2. These attributes for two compounds with the highest pIC50 are shown in Fig. 2 (compound no. 60 and 64).

Good fingerprints obtained from Monte Carlo optimization method
A series of natural flavonols with unknown pIC50 was selected and their pIC50 was calculated from the QSAR models of best split (split 3). Names, chemical structure and corresponding predicted pIC50 of selected natural flavonol derivatives with pIC50 more than 4, are depicted in Table 6. These compounds were also considered for molecular docking studies.
Molecular docking studies
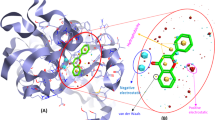
The docking for abiraterone was performed into the active site of Human Cytochrome P450 CYP17A1 (PDB: 3RUK) to validate the binding energy of ligand–protein interactions. The validation results showed a binding energy of − 10.3 kcal/mol for abiraterone and a root-mean-square deviation (RMSD) value 1.172 Å (Fig. 3). The active pocket consisted of amino acid residues such as Val366, Val483, Val482, Ala367, Glu305, Gly301, Leu209, Asn220, Tyr201, Ile206, Ile205, Arg239, Phe114, ala302, Ile371, Ala113, Thr306, and Cys442, which play fundamental roles by hydrophobic interactions and forming H-bond (Fig. 4).

Superposition of the abiraterone output docked ligand (blue) and the co-crystallized ligand (green) of 3RUKA

3D docking mode and 2D schematic interaction diagram for the best pose of abiraterone redocked into 3RUK crystal structure
In addition, the docking studies for eight natural flavonols with predicted pIC50 more than 4.0 based on the best model (split 3), were conducted along with compound number 60, which has high experimental activity. Natural flavonols azaleatin, gossypetin, isorhamnetin, myricetin, pachypodol, quercetin, rhamnazin, and rhamnetin exhibited binding energies of − 8.1, − 8.5, − 8.0, − 8.2, − 7.9, − 8.4, − 8.3, and − 8.2 kcal/mol, respectively (Table 6). The docking outcomes matched the calculated pIC50 of flavonols. The superimposition image of the optimum binding pose for each suggested flavonol is displayed in Fig. 5. Figure 6 shows the 3D docking mode and 2D schematic depiction of interactions for some natural flavonols and the active ligand. The oxygen atom was involved in hydrogen bond interactions with the active site amino acid residues, and so the oxygen of flavonols was particularly significant for the anti-prostate cancer effect of flavonols. The positive contribution of oxygen atom on pIC50 of flavonol derivatives was seen in the mechanistic interpretation of the above-mentioned QSAR models. So, the present QSAR models are acceptable for a wide range of flavonols derivatives.

Superimposed poses of docked molecules and the co-crystallized abiraterone (violet) into the active site of 3RUKA

3D docking mode and 2D schematic interaction diagram for the best pose of some natural flavonols against 3RUK crystal structure (for interpretation of the references to color in this figure legend, the reader is referred to the web version of this article)
Conclusion
In the present study, a reliable QSAR model was described to predict the anti-prostate cancer activities of 81 flavonol derivatives using the Monte Carlo optimization technique of CORAL software. To date, the QSAR models to predict the pIC50 of this dataset were not previously reported. Six QSAR models were constructed utilizing the balance of correlation method with two target functions TF1 (WIIC = 0.0) and TF2 (WIIC = 0.2). The IIC was employed to improve the reliability and robustness of the models. The QSAR models developed by using TF2 were found better than the models developed by TF1. The predictability and robustness of designed models were evaluated by the various statistical parameters such as R2, Q2, IIC, CCC, MAE, s, \(\overline{{r }_{m}^{2}}\), Δ\({r}_{m}^{2}\), \({C}_{{R}_{p}^{2}}\), F and Y-test. Based on ‘statistical defect’, d(A) for a SMILES attribute, the AD was also analysed and the outliers were extracted. The structural attributes as promoters of increase/decrease of pIC50 were identified and used to predict the pIC50 of natural flavonols. The mechanistic interpretation was also confirmed by molecular docking of natural flavonols into the active site of Human Cytochrome P450 CYP17A1 (PDB: 3RUK).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Middleton E, Kandaswami C, Theoharides TC. The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease, and cancer. Pharmacol Rev. 2000;52(4):673–751.
Panche AN, Diwan AD, Chandra SR. Flavonoids: an overview. J Nutr Sci. 2016; 5.
Yan W, et al. Flavonoids potentiated anticancer activity of cisplatin in non-small cell lung cancer cells in vitro by inhibiting histone deacetylases. Life Sci. 2020;258: 118211.
Liu HL, Jiang WB, Xie MX. Flavonoids: recent advances as anticancer drugs. Recent Pat Anti-Cancer Drug Discov. 2010;5(2):152–64.
Ravishankar D, et al. Flavonoids as prospective compounds for anti-cancer therapy. Int J Biochem Cell Biol. 2013;45(12):2821–31.
Yu T, et al. Exploring the chemical space of CYP17A1 inhibitors using cheminformatics and machine learning. Molecules. 2023;28(4):1679.
Wróbel TM, et al. Non-steroidal CYP17A1 inhibitors: discovery and assessment. J Med Chem. 2023;66:6542.
Fei Q, et al. Rutin inhibits androgen synthesis and metabolism in rat immature Leydig cells in vitro. Andrologia. 2021;53(11): e14221.
Xin-Guang S, et al. New prenylated flavonoid glycosides derived from Epimedium wushanense by β-glucosidase hydrolysis and their testosterone production-promoting effects. Chin J Nat Med. 2022;20(9):712–20.
Abdi SAH, et al. Morusflavone, a new therapeutic candidate for prostate cancer by CYP17A1 inhibition: exhibited by molecular docking and dynamics simulation. Plants. 2021;10(9):1912.
Britton RG, et al. Synthesis and biological evaluation of novel flavonols as potential anti-prostate cancer agents. Eur J Med Chem. 2012;54:952–8.
Khan I, et al. Biodegradable nanoparticulate co-delivery of flavonoid and doxorubicin: mechanistic exploration and evaluation of anticancer effect in vitro and in vivo. Biomater Biosyst. 2021;3: 100022.
Le Marchand L. Cancer preventive effects of flavonoids—a review. Biomed Pharmacother. 2002;56(6):296–301.
Rajamahanty S, et al. Growth inhibition of androgen-responsive prostate cancer cells with Brefeldin A targeting cell cycle and androgen receptor. J Biomed Sci. 2010;17(1):1–8.
Tavsan Z, Kayali HA. Flavonoids showed anticancer effects on the ovarian cancer cells: involvement of reactive oxygen species, apoptosis, cell cycle and invasion. Biomed Pharmacother. 2019;116: 109004.
Isaacs JT, Isaacs WB. Androgen receptor outwits prostate cancer drugs. Nat Med. 2004;10(1):26–7.
Menezes JC, et al. Natural and synthetic flavonoids: structure–activity relationship and chemotherapeutic potential for the treatment of leukemia. Crit Rev Food Sci Nutr. 2016;56(sup1):S4–28.
Chen I-L, et al. Synthesis and antiproliferative evaluation of amide-containing flavone and isoflavone derivatives. Bioorg Med Chem. 2008;16(16):7639–45.
Ahmadi S, Habibpour E. Application of GA-MLR for QSAR modeling of the arylthioindole class of tubulin polymerization inhibitors as anticancer agents. Anti-Cancer Agents Med Chem. 2017;17(4):552–65.
Ahmadi S, et al. Predictive QSAR modeling for the antioxidant activity of natural compounds derivatives based on Monte Carlo method. Mol Divers. 2021;25(1):87–97.
Toropova AP, Toropov AA. CORAL software: prediction of carcinogenicity of drugs by means of the Monte Carlo method. Eur J Pharm Sci. 2014;52:21–5.
Ghasedi N, et al. DFT based QSAR study on quinolone-triazole derivatives as antibacterial agents. J Recept Signal Transduction. 2022;42(4):418–28.
Kumar P, Kumar A. Monte Carlo method based QSAR studies of Mer kinase inhibitors in compliance with OECD principles. Drug Research. 2018;68(04):189–95.
Lotfi S, Ahmadi S, Kumar P. The Monte Carlo approach to model and predict the melting point of imidazolium ionic liquids using hybrid optimal descriptors. RSC Adv. 2021;11(54):33849–57.
Toropova AP, et al. The system of self-consistent models for vapour pressure. Chem Phys Lett. 2022;790: 139354.
Jafari K, et al. Correlation Intensity Index (CII) as a criterion of predictive potential: applying to model thermal conductivity of metal oxide-based ethylene glycol nanofluids. Chem Phys Lett. 2020;754: 137614.
Duhan M, et al. Quantitative structure activity relationship studies of novel hydrazone derivatives as α-amylase inhibitors with index of ideality of correlation. J Biomol Struct Dyn. 2022;40(11):4933–53.
Ahmadi S, et al. CORAL: Monte Carlo based global QSAR modelling of Bruton tyrosine kinase inhibitors using hybrid descriptors. SAR QSAR Environ Res. 2021;32(12):1013–31.
Azimi A, et al. SMILES-based QSAR and molecular docking study of oseltamivir derivatives as influenza inhibitors. Polycyclic Aromat Compd. 2022;43:3257.
Hamzehali H, et al. Quantitative structure–activity relationship modeling for predication of inhibition potencies of imatinib derivatives using SMILES attributes. Sci Rep. 2022;12(1):1–9.
Kumar A, Kumar P, Singh D. QSRR modelling for the investigation of gas chromatography retention indices of flavour and fragrance compounds on Carbowax 20 M glass capillary column with the index of ideality of correlation and the consensus modelling. Chemom Intell Lab Syst. 2022;224: 104552.
Kumar P, Kumar A. Nucleobase sequence based building up of reliable QSAR models with the index of ideality correlation using Monte Carlo method. J Biomol Struct Dyn. 2020;38(11):3296–306.
Kumar A, Kumar P. Prediction of power conversion efficiency of phenothiazine-based dye-sensitized solar cells using Monte Carlo method with index of ideality of correlation. SAR QSAR Environ Res. 2021;32(10):817–34.
Toropov AA, et al. “Ideal correlations” for biological activity of peptides. Biosystems. 2019;181:51–7.
Javidfar M, Ahmadi S. QSAR modelling of larvicidal phytocompounds against Aedes aegypti using index of ideality of correlation. SAR QSAR Environ Res. 2020;31(10):717–39.
Ahmadi S, et al. SMILES-based QSAR and molecular docking study of xanthone derivatives as α-glucosidase inhibitors. J Recept Signal Transduct. 2021;42:361.
Dutta S, et al. Natural products: an upcoming therapeutic approach to cancer. Food Chem Toxicol. 2019;128:240–55.
Li X, et al. A new class of flavonol-based anti-prostate cancer agents: design, synthesis, and evaluation in cell models. Bioorg Med Chem Lett. 2016;26(17):4241–5.
Li X, et al. 3-O-Substituted-3′, 4′, 5′-trimethoxyflavonols: synthesis and cell-based evaluation as anti-prostate cancer agents. Bioorg Med Chem. 2017;25(17):4768–77.
Li X, et al. Structure–activity relationship and pharmacokinetic studies of 3-O-substitutedflavonols as anti-prostate cancer agents. Eur J Med Chem. 2018;157:978–93.
Lotfi S, Ahmadi S, Kumar P. A hybrid descriptor based QSPR model to predict the thermal decomposition temperature of imidazolium ionic liquids using Monte Carlo approach. J Mol Liq. 2021;338: 116465.
Kumar A, Sindhu J, Kumar P. In-silico identification of fingerprint of pyrazolyl sulfonamide responsible for inhibition of N-myristoyltransferase using Monte Carlo method with index of ideality of correlation. J Biomol Struct Dyn. 2021;39(14):5014–25.
Toropova AP, et al. QSAR models for HEPT derivates as NNRTI inhibitors based on Monte Carlo method. Eur J Med Chem. 2014;77:298–305.
Sokolović D, et al. Monte Carlo-based QSAR modeling of dimeric pyridinium compounds and drug design of new potent acetylcholine esterase inhibitors for potential therapy of myasthenia gravis. Struct Chem. 2016;27:1511–9.
Ahmadi S. Mathematical modeling of cytotoxicity of metal oxide nanoparticles using the index of ideality correlation criteria. Chemosphere. 2020;242: 125192.
Ghiasi T, et al. The index of ideality of correlation: QSAR studies of hepatitis C virus NS3/4A protease inhibitors using SMILES descriptors. SAR QSAR Environ Res. 2021;32(6):495–520.
Toropov AA, Toropova AP. The index of ideality of correlation: a criterion of predictive potential of QSPR/QSAR models? Mutat Res/Genetic Toxicol Environ Mutagen. 2017;819:31–7.
Ahmadi S, Khazaei MR, Abdolmaleki A. Quantitative structure–property relationship study on the intercalation of anticancer drugs with ct-DNA. Med Chem Res. 2014;23(3):1148–61.
Ahmadi S. A QSPR study of association constants of macrocycles toward sodium cation. Macroheterocycles. 2012;5(1):23–31.
Ahmadi S, Akbari A. Prediction of the adsorption coefficients of some aromatic compounds on multi-wall carbon nanotubes by the Monte Carlo method. SAR QSAR Environ Res. 2018;29(11):895–909.
Lawrence I, Lin K. Assay validation using the concordance correlation coefficient. Biometrics. 1992;58:599–604.
Rücker C, Rücker G, Meringer M. y-Randomization and its variants in QSPR/QSAR. J Chem Inf Model. 2007;47(6):2345–57.
Ojha PK, et al. Further exploring rm2 metrics for validation of QSPR models. Chemom Intell Lab Syst. 2011;107(1):194–205.
Roy K, et al. Comparative studies on some metrics for external validation of QSPR models. J Chem Inf Model. 2012;52(2):396–408.
Todeschini R. Milano Chemometrics. 2010: University of MilanoBicocca, Milano, Italy.
da Silva Costa J, et al. Virtual screening and statistical analysis in the design of new caffeine analogues molecules with potential epithelial anticancer activity. Curr Pharm Des. 2018;24(5):576–94.
Morris GM, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19(14):1639–62.
DeVore NM, Scott EE. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature. 2012;482(7383):116–9.
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–61.
Cao DS, et al. A new strategy of outlier detection for QSAR/QSPR. J Comput Chem. 2010;31(3):592–602.
Ghosh K, et al. Identification of structural fingerprints for ABCG2 inhibition by using Monte Carlo optimization, Bayesian classification, and structural and physicochemical interpretation (SPCI) analysis. SAR QSAR Environ Res. 2020;31(6):439–55.
Jain S, et al. Exploration of good and bad structural fingerprints for inhibition of indoleamine-2, 3-dioxygenase enzyme in cancer immunotherapy using Monte Carlo optimization and Bayesian classification QSAR modeling. J Biomol Struct Dyn. 2020;38(6):1683–96.
Acknowledgements
The authors would like to express their deepest gratitude to Dr. Alla P. Toropova and Dr. Andrey A. Toropov for providing the CORAL software.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
FT performed drawing of structures and building the QSAR models. SA did visualization, supervision, performed interpretation of models and molecular docking. SL wrote original draft, and PK did editing of the manuscript. AA conducted molecular docking.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Chemical structures of flavonol derivatives and IC50 values against PC-3 prostate cancer cells.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tajiani, F., Ahmadi, S., Lotfi, S. et al. In-silico activity prediction and docking studies of some flavonol derivatives as anti-prostate cancer agents based on Monte Carlo optimization. BMC Chemistry 17, 87 (2023). https://doi.org/10.1186/s13065-023-00999-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-023-00999-y