
Abstract
A series of quinazolinone derivatives (7a–7h) were synthesized as antiproliferative agents. All compounds, were synthesized through three steps method and structurally evaluated by FTIR, 1H-NMR, 13CNMR and Mass spectroscopy. Their cytotoxic activities were assessed using MTT protocol against three humans cancerous (MCF-7, A549 and 5637) and normal (MRC-5) cell lines. In addition, molecular docking and simulation studies of the synthesized compounds were performed to assessment their orientation, interaction mode against EGFR as plausible mechanism of quinazoline compounds as anticancer agents. The synthesized compounds mostly showed moderate activity against the three studied cell lines. They also indicated an appropriate selectivity against tumorigenic and non-tumorigenic cell line. The molecular docking results also confirmed biological activity. Most of the compounds fulfilled Lipinski rule. Collectively, these compounds with further modification can be considered as potent antiproliferative agents.
Similar content being viewed by others

Introduction
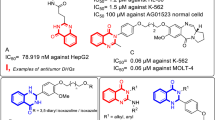
Cancer is a complicated disease due to uncontrolled growth of cells without differentiation, and an increase in abnormal cells leading to tumor formation [1, 2]. In 2020, one out of every 6 deaths in the world was due to cancer and approximately 10 million people died from cancer that year. Breast, lung, colon, rectum and prostate cancers are the most common cancers worldwide [3]. Chemotherapy, surgery, hormone therapy and radiotherapy are the main cancer treatments based on the stage and type of cancer [4]. Nevertheless, multidrug resistance (MDR) and healthy cell damage during cancer treatment are among the main disadvantages of these treatments [5]. Therefore, there is an urgent need to find novel and selective compounds as antiproliferative agents. Quinazoline scaffolds have been shown to have various biological and pharmacological effects including anti-cancer [6,7,8], anti-diabetes [9], antifungal [10], antibacterial [11, 12], antihypertensive [13] and anti-tuberculosis activity [14]. In addition, there are various quinazoline scaffold based compounds in the market such as erlotinib, gefitinib (structures I and II) and structures III–VI, with high cytotoxic activity toward different cancerous cell lines (Fig. 1) [15,16,17,18].

Several marketed and reported anticancer agents with quinazoline scaffold
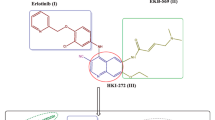
Inhibition of epidermal growth factor receptor (EGFR) enzyme is considered to be one of the main mechanisms of quinazolinone analogs as anticancer agents [19]. EGFR is a receptor with tyrosine kinase (TK) activity with significant designation in the cell proliferation, differentiation, metastasis, and survival cycle [20, 21]. Over activation of EGFR has been reported in some cancerous tissues such as lung, brain, ovarian, colon, breast and prostate tumors [22]. Thus, targeting EGFR can be considered as a rational and worthy approach in cancer therapy. Erlotinib and gefitinib are the most potent and selective EGFR inhibitors with quinazoline scaffold in their structures. Considering the importance of quinazoline and quinazolinone base structures in cancer treatment, in this study, we synthesized some novel quinazolinone-benzyl piperidine derivatives (7a–7h) to obtain more effective anticancer agents. All the synthesized derivatives were elucidated with 1HNMR, 13CNMR, FT-IR and Mass spectroscopy. All compounds were then evaluated against three humans cancerous (MCF-7, A549 and 5367) as well as one normal (MRC-5) cell lines. In addition, these quinazolinone-benzyl piperidine derivatives were applied to a molecular docking simulation to acquire their binding conformations and structural specificities toward EGFR kinase as plausible targets in cancer treatment. Finally, In silico physico-chemical properties were also performed to represent drug-likeness of synthesized compounds. The summery schematic (graphical abstract) of our study was shown in Fig. 2.

The summery schematic of the study
Results and discussion
Chemistry
The synthetic route of the compounds (7a–7h) via three different steps is shown in Fig. 10. The first and second steps were performed as previously described [23]. The final target compounds (7a–7h) were obtained by reacting 4-benzyl piperidine with intermediates (5a–5h) through nucleophilic substitution in the presence of basic catalyst (DIPEA) and acetonitrile as solvent in appropriate yields (50–95%). The chemical structures of all compounds were confirmed by IR, 1H-NMR, 13C-NMR, and mass spectroscopies. The important feature of the 1H NMR spectrum of these compounds is a double peak at 2.54–3.31 ppm belonging to two protons of CH2 placed between quinazolinone and 4-benzyl piperidine ring, except for compounds 7a, 7b and 7d. The peaks appearing as two peaks of doublet may be due to nonequivalent protons in these compounds. Only in compound 7d, the peaks of CH2 between phenyl and piperidine ring appeared as a singlet, while for other compounds it appeared as a doublet, triplet or multiplet. Regarding the 13C NMR spectrum, the significant singlet peak is related to the carbonyl group in quinazoline ring, which was displayed in the range of 161.1–161.6 ppm.
Cytotoxic activity
The antiproliferative activities of all the synthesized compounds were assessed by MTT method on three cancerous (MCF-7, A549 and 5367) as well as one normal cell line (MRC-5). According to the results, among mono-substituted compounds (7a, 7b, 7d–7g), 7b with bromine substitution at the meta position of phenyl ring had greatest effect compared to other studied compounds with IC50 values of 82.1 µΜ, 67.3 µΜ and 51.4 µΜ against cancerous cell lines MCF-7, A549 and 5367 respectively. This effect could be related to the size and electronegativity of bromine. In addition, compound 7e containing chlorine atom at para position of phenyl ring showed IC50 values of 90.2 and 103.04 µΜ for MCF-7 and 5637 cell lines, respectively. Moving of the chlorine atom from para to meta position led to decreasing activity in 7a analogue. In case of disubstituted compounds (7c and 7h), compound 7c containing dimethoxy substitutions is more effective than 7h with methyl and Cl substitutions in MCF-7 and 5637 cancerous cell lines. Overall, the mono-substituted groups showed better effects than the disubstituted compounds (Table 2). All compounds generally represented lesser anti-proliferative effects on the normal cell line (MRC-5) compared to other carcinoma cell lines, which illustrates appropriate selectivity between non-tumorigenic and tumorigenic cell lines [24].
Docking studies
As mentioned previously, EGFR is the most plausible target for the compounds with quinazoline backbone as anticancer agents. Therefore, to evaluate and understand the pattern of interaction and binding mode of the synthesized compounds in the active site of EGFR, molecular modeling was performed. The docking binding energies and interaction of all synthesized compounds were shown in Table 3. As shown in Table 3, compound 7b and 7e with high anti-proliferative activity, showed stronger energies in binding to the active sites compared to the others. Redocking of [6,7-Bis(2-Methoxy-Ethoxy) Quinazoline-4-Yl]- (3-Ethynylphenyl) Amine (erlotinib) as co-crystal ligand, was performed to evaluate the docking accuracy. The RMSD was achieved 1.07 Å compared to its coordination in the crystal structure (Fig. 3). The docking score of erlotinib was obtained − 10.1 (kcal/mol).

Two conformations of co-crystal ligand (erlotinib) (AQ4) in the EGFR active site: The yellow and red color showed the crystal orientation and redocked conformation, respectively
The interactions of two synthesized quinazolinone-benzyl piperidine (7b and 7e), as the most active compounds, with EGFR were investigated. As shown in Fig. 4, compound 7b showed pi-anion interaction between 4-benzyl piperidine and Asp 831 and also, pi-alkyl interactions were seen between quinazoline ring and 3-Br benzyl and Leu 694, Leu 820, Ala 719, Val 702, Lys 721. There are some hydrophobic interactions with Arg 817, Thr 830, Glu 738, Leu 764, Met 742, Thr 766, Met 769, Leu 768, Gly 772.

2D interactions of 7b (left) and 7e (right) with the residues in the binding site of receptor. (Vander waals: green, dark pink: pi-pi, light pink: pi-alkyl, purple: pi-sigma, orange: pi-cation, blue: halogen bond)
The docking scores and detailed interactions of all designed compounds (7a–7h) are shown in Table 3.
The other potent molecule (7e), interacted through six types of interactions including; hydrogen bonding, halogen bonding, pi-pi interaction, and hydrophobic interaction. The carbonyl group of the quinazoline moiety was involved in hydrogen bond interaction with Thr 766. Furthermore, the quinazolinone and phenyl rings interacted with Leu 820 and Val 702, respectively, via pi-sigma interactions. The other interactions were pi-pi and pi-alkyl interactions between quinazolinone moiety and 4-benzyl piperidine with Ala 719, Leu 694 and Phe 699. Some hydrophobic interactions with Gly 697, Gly 772, Met 769, Leu 768, Gln 767, Leu 764, Met 742, Thr 830 were also observed in Fig. 4.
According to the proposed binding mode of erlotinib (Fig. 5), the quinazoline moiety involved in pi–pi interaction with Leu 694, Leu 820 and Ala 719 and also, the 2-methoxy-ethoxy chain interacted via hydrogen bond with Cys 773. 3-ethynyl phenyl ring showed pi-alkyl interaction with Val 702, Lys 721, Met 742 and Leu 764. Besides, there were some hydrophobic interactions with Phe 771, Pro 770, Leu 768, Met 769, GLN 767, Thr 766, Thr 830and Asp 831.

2D interaction of erlotinib with the residues in the binding site of EGFR target. (Vander waals: green, dark pink: pi-pi, light pink: pi-alkyl, purple: pi-sigma, orange: pi-cation, blue: halogen bond)
The results of docking study indicated that, the most active compounds and erlotinib showed the same interactions with key amino acids in the active site of enzyme including Thr 766, Met 769, Ala 719, Lys 721 and Gly 772. However, the less docking score of these compounds compared to erlotinib, probably, can be related to the absence of polar groups attached to the quinazoline ring which increase the inhibitory effect due to the formation of hydrogen bonds in the active site of EGFR enzyme. Altogether, Val 702, Phe 699, Arg 817, Asp 831, Glu 738, Leu 694, Leu 820 and Lys 721 were the most important amino acids for generating interactions with all ligands in the active site of EGFR target.
Insilico physicochemical parameters (ADME) prediction
ADME properties of the all synthesized compounds are represented in Table 4. It can be observed that all compounds follow Lipinski’s rule of five except for compound 7h. Molecular weight (MW) of all compounds are in the accepted range (427–502). All of the compounds also showed reasonable lipophilicity (log P values) for penetration through biological membranes. Furthermore, the hydrogen bond properties (donors or acceptors), total polar surface area (TPSA), and rotatable bond number of all investigated compounds is within the acceptable limit. As a results, it can be proposed that these compounds could potentially be administered orally.
Molecular dynamic simulation
MD simulation is a powerful method to predict the structural refinements as well as behavior of ligand and receptor in biological systems [25, 26]. To investigate the stability and intramolecular interactions of our compounds (7b and 7h) in the active site of EGFR enzyme with respect to time, molecular dynamics simulation was performed [27]. The RMSD, RMSF, number of hydrogen bonds, and radius of gyration (Rg) graphs of 7b and 7h with EGFR receptor are presented in Figs. 6, 7, 8 and 9, respectively.

The RMSD plots for protein- ligand complexes during the simulation time
In general, RMSD analyses shows the stability and equilibrium of the complex during the simulation time. As depicted in Fig. 6, the EGFR complex with 7b has reached equilibrium after 20 ns and remains stable with low fluctuation during the MD simulation time. The average RMSD of 7b in complex with 1M17 was 1A. The complex reached a plateau form and remained stable after 20 ns. The condition for 7h-complex is different and the structural changes were continued until the end of the simulation time. This indicates that 7h may show different performances to the 7b ligand. This finding also revealed that 7b fits more appropriately in the active site of EGFR enzyme.
RMSF (root means square fluctuation) results revealed the region of the fluctuated protein during the simulation time. In fact, the amount of structural movement and flexibility of amino acids was achieved with RMSF analysis [28]. The RMSF of the backbone residue of EGFR was performed to calculate the fluctuation of amino acid residues. The lowest RMSF values represented the flexibility, compactness of the protein, and stability of the complex. In fact, RMSF was applied to evaluate the structural movement and flexibility of EGFR in binding to 7b and 7h. As shown in Fig. 7, the same distribution of RMSFs for both complex 7b and 7h was observed. It can be concluded that the region around 672–722 and 872–972 which indicates lower fluctuation are active sites of EGFR. Interaction and binding conformation of the ligands 7b and 7h in the IM17 binding site was confirmed before and after MD simulation. The key amino acids Met 769, Leu 768, Leu 694, Gly 772, Asp 776, Gly 695, Phe 699, Asp 831, Cys 773, Val 702, Glu 738, Thr 766, Arg 817, Thr 830, Leu 764, Met 742, Leu 768, Asn 819, Lys 851, Cys 721, Ile 735, Gly 833, Leu 834, Leu 820, Ala 719, Lys 721, Gly 697, Leu 775 in the active site of EGFR enzyme with relatively low RMSF values.

RMSF distribution of residues during the simulation time
The radius of gyration (Rg) of a protein is reflected in the compactness of protein during the simulation time [29]. The plot of the EGFR radius of gyration during the MD simulations time is shown in Fig. 8. The low values of Rg reflect the compactness and stability of protein. As shown in Fig. 8, after 20 ns, complex 7b was compacted and maintained constant interaction with 7b. In the case of 7h complex, both the amount and variation of compactness were increased during the simulation time. It is concluded that 7b is a more suitable inhibitor of EGFR in contrast to 7h.

Compactness changes of ligand-EGFR complexes analyzed by Rg parameters
The number of intermolecular hydrogen bonds in the ligand-EGFR complex contribute to the stability of the complex. The analysis of H-bond interactions was performed and shown in Fig. 9. The 7b-EGFR complex showed more interactions with active site residues over the 100 ns simulation compared to 7h ligand.

Number of hydrogen bonds between the EGFR and the selected ligands during the simulation time
Experimental
All reagents and solvents were purchased from Merck and Sigma chemical companies without any purification. Compound purification was done by flash chromatography (silica gel 60) or recrystallization. The 1H NMR, 13C NMR spectra and FT-IR were recorded on a BRUKER DRX-500 AVANCE 400 MHz and a VERTEX70 spectrometer (Bruker, Germany), respectively. Mass spectra were recorded on Agilent Mass instrument using (M++1) mode. Melting point was achieved on Electrothermal 9200 instrument. The chemical structures of all synthesized compounds were shown in Table 1.
Synthesis and characterization
General procedure for the synthesis of substituted 2-((4-benzylpiperidin-1-yl) methyl)-3-phenylquinazolin-4(3H)-one
The benzo oxazine [3] was prepared by mixing 1 mmol of anthranilic acid with chloroacetyl chloride in dichloromethane (10 mL) and diisopropylethylamine (DIPEA) (1.5 mmol). After that intermediate 3 was reacted with eight different substituted of aniline (4a–4h) to obtain compounds (5a–5h) [23]. Finally, 4-benzyl piperidine (1 mmol) was added to various derivatives of compounds (5a–5h) (1 mmol) in the presence of diisopropylethylamine (DIPEA) (2 mmol) in acetonitrile as solvent. The mixture was refluxed for 24 h and the reaction was follow up with TLC. The reaction was then washed with water and extracted with suitable amount of ethyl acetate. The final products (7a–7h) were purified by flash chromatography (Fig. 10).

Synthesis path of designed quinazoline (7a–7h)
Spectra data
2-((4-Benzylpiperidin-1-yl) methyl)-3-(3-chlorophenyl) quinazolin-4(3H)-one (7a)
IR (KBr) v (cm−1): 3024–3072 (C-H, aromatic), 2795–2920 (C–H, aliphatic), 1683(C=O, Stretch), 1597(C=C, aromatic), 1468 (C=N, Stretch), 1335 (C-N, Stretch). 1H-NMR (400 MHz, DMSO) δ(ppm) = 8.24 (d, 1H, J = 10 Hz, H-5-quinazolinone), 7.98 (dd, 1H, J = 10 Hz, J = 1.5 Hz, H-7-quinazolinone), 7.84 (d, 1H, J = 9.5 Hz, H-8-quinazolinone), 7.72–7.73 (m, 1H, H-6-quinazolinone), 7.69–7.70 (m, 1H, aromatic), 7.66–7.67 (m, 1H, aromatic), 7.62–7.64 (m, 1H, aromatic), 7.55 (d, 1H, J = 15 Hz, aromatic), 7.37 (t, 2H, J = 9.5 Hz, benzyl), 7.27 (t, 1H, J = 9 Hz, benzyl), 7.22 (d, 2H, J = 8.5 Hz, benzyl), 3.35 (d, 1H, J = 17, N–CH2–C=N), 3.25 (d, 1H, J = 16.5, N–CH2–C=N), 2.56–2.62 (m, 3H, N-CH2), 2.26–2.30 (m, 1H, CH2–CH–CH2), 1.91–1.98 (m, 2H, Ph-CH2), 1.44–1.50 (m, 3H, N–CH2, N–CH2–CH2), 1.02–1.17 (m, 2H, N–CH2–CH2). 13C-NMR (100 MHz, DMSO) δ (ppm) = 161.5, 153.4, 146.6, 140.2, 138.4, 138.7, 132.4, 130.1, 129.9, 128.9, 128.4, 128.1, 127.6, 127.2, 127.2, 126.3, 125.7, 120.8, 61.5, 52.9, 52.3, 42.4, 36.9, 31.6, 31.5. MS m/z (%):444.0 [M+] (0.82), 269.0 (55.4), 234.0 (14.96), 174.0 (100), 119.0 (9.2), 91.0 (17.8). Elem. anal. calcd. For C27H26ClN3O (443.1); C, 73.04; H, 5.90; N, 9.46. Found: C, 73.05; H, 5.88; N, 9.37.
2-((4-Benzylpiperidin-1-yl) methyl)-3-(3-bromophenyl) quinazolin-4(3H)-one (7b)
IR (KBr) v (cm−1): 3025–3071 (C–H, aromatic), 2795–2922 (C–H, aliphatic), 1684 (C=O Stretch), 1580–1600 (C=C, aromatic), 1466 (C=N, Stretch), 1335 (C-N, Stretch). 1H-NMR (500 MHz, DMSO) δ(ppm) = 8.23 (d, 1H, J = 10 Hz, H-5-quinazolinone), 7.94–7.98 (m, 1H, H-7-quinazolinone), 7.82 (dd, J = 15 Hz, J = 5 Hz, 2H, aromatic), 7.77–7.80 (m, 1H, H-8-quinazolinone), 7.64–7.707 (m, 1H, H-6-quinazolinone), 7.53–7.59 (m, 2H, aromatic), 7.34 (t, 2H, J = 9 Hz, benzyl), 7.23–7.27 (m, 1H, benzyl), 7.20 (d, 2H, J = 8.5 Hz, benzyl), 3.35 (d, 1H, J = 17, N–CH2–C=N), 3.22 (d, 1H, J = 17, N–CH2–C = N), 2.60–2.61 (m, 1H, N–CH2), 2.51–2.57 (m, 2H, N-CH2), 2.21–2.24 (m, 1H, CH2–CH–CH2), 1.89–1.96 (m, 2H, Ph-CH2), 1.41–1.49 (m, 3H, N–CH2, N-CH2-CH2), 1.03–1.15 (m, 2H, N–CH2–CH2). 13C-NMR (125 MHz, DMSO) δ (ppm) = 161.5, 153.4, 146.6, 140.2, 138.5, 134.7, 132.9, 131.2, 130.2, 128.7, 128.1, 127.8, 127.2, 127.1, 126.3, 125.7, 120.8, 120.6, 61.5, 53.0, 52.1, 42.5, 36.9, 31.6, 31.5. MS m/z (%):0.488.0 [M+] (0.54), 315.0 (29.9), 234.0 (21.0), 174.0 (100), 119.0 (7.6), 91.0 (16.6). Elem. anal. calcd. For C27H26BrN3O (488.4); C, 66.40; H, 5.37; N, 8.60. Found: C, 66.37; H, 5.32; N, 8.52.
2-((4-benzylpiperidin-1-yl) methyl)-3-(2, 4-dimethoxyphenyl) quinazolin-4(3H)-one (7c)
IR (KBr) v (cm−1):3072(C–H, aromatic), 2772–2930(C–H, aliphatic), 1686(C=O, Stretch), 1606(C=C, aromatic), 1462–1508 (C=N, Stretch), 1313 (C–N, Stretch), 1050–1150 (COC, Stretch). 1H-NMR (500 MHz, DMSO) δ(ppm) = 8.23 (d, 1H, J = 8.5 Hz, H-5-quinazolinone), 7.93–7.97 (m, 1H, H-7-quinazolinone), 7.90 (d, 1H, J = 10 Hz, H-8-quinazolinone), 7.65 (t, 1H, J = 9.5 Hz, H-6-quinazolinone), 7.34–7.39 (m, 3H, aromatic), 7.27 (d, 1H, J = 9 Hz, benzyl), 7.22 (d, 2H, J = 9 Hz, benzyl), 6.79–6.80 (m, 1H, benzyl), 6.71–6.74 (m, 1H, benzyl), 3.94 (s, 3H, OCH3-phenyl), 3.78 (s, 3H, OCH3-phenyl), 3.34 (d, 1H, J = 16.5, N–CH2), 3.13 (d, 1H, J = 16.5, CH2–CH–CH2), 2.54 (d, 2H, J = 10 Hz, N–CH2–C=N), 2.44–2.47 (m, 2H, N–CH2), 1.92–1.97 (m, 2H, Ph-CH2), 1.45 (s, 3H, N–CH2, N–CH2–CH2), 1.04–1.15 (m, 2H, N–CH2–CH2). 13C-NMR (125 MHz, DMSO) δ (ppm) = 161.1, 160.7, 155.4, 154.8, 146.8, 140.2, 134.6, 130.2, 128.9, 127.1, 126.9, 126.3, 125.7, 120.7, 118.3, 104.4, 98.4, 64.42, 55.5, 53.1, 52.9, 42.3, 36.8, 31.5, 31.3. MS m/z (%): 470.0 \([M+]\) (2.5), 296.0 (32.4), 281.0 (16.4), 265.0 (100), 250.0 (5.9), 234.0 (6.9), 174.0 (96.3), 91.0 (17.1). Elem. anal. calcd. For C29H31N3O3 (470.0); C, 74.18; H, 6.65; N, 8.95. Found: C, 74.12; H, 6.61; N, 8.91.
2-((4-benzylpiperidin-1-yl) methyl)-3-(3-(trifluoromethyl phenyl) quinazolin-4(3H)-one (7d)
IR (KBr) v (cm−1): 3028–3083 (C–H, aromatic), 2764–2929(C–H, aliphatic), 1677 (C=O, Stretch), 1606 (C=C aromatic), 1447 (C=N, Stretch), 1098(C-F, aliphatic). 1H-NMR (500 MHz, DMSO) δ(ppm) = 8.13(d, 1H, J = 7 Hz, Quinazoline),7.73–7.89(m,6H, Quinazoline and Phenyl),7.57(t, 1H, J = 7 Hz, Phenyl), 7.23 (d, 2H, J = 7 Hz, Benzyl), 7.14–7.15(m, 1H, Benzyl), 7.07(d,2H, J = 7 Hz, Benzyl), 3.29(d, 1H, J = 10.8, N–CH2–C=N), 3.13(d, 1H, J = 10.8, N–CH2–C=N),2.35–2.5(m, 2H, N–CH2), 2.11–2.13 (m, 1H, CH2–CH–CH2), 1.8 (s, 2H, Ph-CH2), 1.22–1.36 (m,4H, N–CH2–CH2, N–CH2), 0.79–0.93 (m, 2H, N–CH2–CH2). 13C–NMR (125 MHz, DMSO) δ (ppm) = 161.1, 152.7, 146.1, 139.7, 137.4, 134.1, 132.2, 129.1,128.7, 127.6, 126.7, 126.6, 126.4, 125.8, 125.1, 124.6, 124.5, 120.3, 61.2, 52.4, 51.6, 41.8, 36.2, 30.8, 30.7. MS m/z (%): 477.2 (0.4), 303.1 (25.1), 174.3 (80.8), 145.1 (22.4), 119.1 (25.4), 91.2 (29.4), 57.2 (100), 55.2 (82.1). Elem. anal. calcd. For C28H26F3N3O (477.5); C, 70.43; H, 5.49; N, 8.80. Found: C, 70.23; H, 5.42; N, 8.71.
2-((4-benzylpiperidin-1-yl) methyl)-3-(4-fluorophenyl) quinazolin-4(3H)-one (7e)
IR (KBr) v (cm−1): 3025 (C–H, aromatic), 2774–2937 (C–H, aliphatic), 1693 (C=O, Stretch), 1605–1506 (C=C aromatic), 1464 (C=N, Stretch). 1H-NMR (400 MHz, DMSO) δ(ppm) = 8.13 (dd, 1H, J = 8 Hz, 1.2 Hz, Quinazoline), 7.86(t, 1H, J = 7.6 Hz, Quinazoline), 7.73 (d, 1H, J = 7.6 Hz, Quinazoline), 7.57 (t, 1H, J = 8 Hz, Quinazoline), 7.47–7.51 (m, 2H, Phenyl), 7.34 (t, 2H, J = 8.8 Hz,Phenyl), 7.26 (t, 2H, J = 7.2 Hz, Benzyl), 7.17 (d, 1H, J = 7.2 Hz, Benzyl), 7.12 (d, 2H, J = 7.2 Hz, Benzyl), 3.16 (s, 2H, N–CH2–C=N), 2.43 (d, 2H, J = 6.4 Hz,N–CH2), 2.34(d, 2H, J = 10.8 Hz, Ph-CH2), 1.84(t, 2H, J = 10.8, N-CH2), 1.37 (d, 3H, J = 10.8,N–CH2–CH2,CH2–CH–CH2), 0.96–1.04 (m, 2H, N–CH2–CH2). 13C-NMR (100 MHz, DMSO) δ (ppm) = 161.6, 153.8, 146.7, 146.3, 140.3, 134.6, 133.1, 131.4, 131.3, 128.9, 128.1, 127.2, 127.0,126.3, 125.7, 120.8, 115.3, 115.0, 61.4, 52.7, 42.3, 37.0, 31.4. MS m/z (%):427.2 (0.2), 253.2 (58.8), 174.2 (100), 150.1 (6.3), 119.1 (7.9), 91.2 (19.1), 42.2 (9.8). Elem. anal. calcd. For C27H26FN3O (427.2); C, 75.85; H, 6.13; N, 9.83. Found: C, 75.20; H, 6.09; N, 9.81.
2-((4-benzylpiperidin-1-yl) methyl)-3-(4-chlorophenyl) quinazolin-4(3H)-one (7f)
IR (KBr) v (cm−1): 3025–3069 (C–H, aromatic), 2776–2934 (C–H, aliphatic), 1687 (C=O, Stretch), 1604 (C=C aromatic), 1492 (C=N, Stretch), 783 (C–CL, Stretch). 1H-NMR (500 MHz, DMSO) δ(ppm) = 8.12 (d, 1H, J = 7.5 Hz, Quinazoline), 7.85 (s, 1H, Quinazoline), 7.72 (d, 1H, J = 7.5 Hz, Quinazoline), 7.5 (s, 3H, Quinazoline and Phenyl), 7.46–7.47 (m, 2H, Phenyl), 7.24–7.25 (m, 2H, Benzyl), 7.1–7.15 (m, 3H, Benzyl), 3.18 (s, 2H, N–CH2–C=N), 2.42 (s, 2H, N–CH2), 2.35 (d, 2H, J = 9 Hz, N-CH2), 1.84 (t, 2H, J = 10.5 Hz, Ph–CH2), 1.35–1.37 (m, 3H, CH2–CH–CH2, N–CH2–CH2), 0.98 (d, 2H, J = 10.5 Hz, N–CH2–CH2). 13C-NMR (125 MHz, DMSO) δ (ppm) = 161.0, 153.0,146.2, 139.7, 135.4, 134.1, 132.5, 130.6, 128.4, 127.8, 127.5, 126.7, 126.5, 125.8, 125.1, 120.3, 60.9, 52.2, 41.7, 36.4, 30.9. MS m/z (%): 443.2 (0.1), 269.1 (47.9), 234.2 (11.8), 174.3 (100), 119.2 (13.3), 91.2 (22.07), 65.2 (3.4), 42.2 (9.4). Elem. anal. calcd. For C27H26ClN3O (443.9); C, 73.04; H, 5.90; N, 9.46. Found: C, 72.98; H, 5.86; N, 9.37.
2-((4-benzylpiperidin-1-yl) methyl)-3-(4-phenoxyphenyl) quinazolin-4(3H)-one (7g)
IR (KBr) v (cm−1):3025–3067 (C-H, aromatic), 2758–2907 (C-H, aliphatic), 1688 (C=O, Stretch), 1590 (C=C aromatic), 1505 (C=N, Stretch), 1163 (C-O, Stretch). 1H-NMR (500 MHz, DMSO) δ(ppm) = 8.23 (dd, 1H, J = 6.4 Hz, 1.2 Hz, Quinazoline), 7.97 (t, 1H, J = 7.6 Hz, Quinazoline), 7.83 (d, 1H, J = 7.6 Hz, Quinazoline), 7.68 (t, 1H, J = 7.6 Hz, Quinazoline), 7.52–7.57 (m, 4H, Phenyl), 7.16–7.37 (m, 10H, Phenyl and Benzyl), 3.31 (s, 2H, N–CH2–C=N), 2.46–2.53 (m, 4H, N–CH2, N–CH2),1.99 (t, 2H, J = 10.8 Hz, Ph-CH2), 1.47–1.50 (d, 3H, J = 10.8, CH2–CH–CH2, N–CH2–CH2), 1.09–1.16 (m, 2H, N–CH2–CH2). 13C-NMR (125 MHz, DMSO) δ(ppm) = 162.2, 157.1, 156.9, 154.4, 147.2, 140.8, 135.1, 132.6, 131.5, 130.6,129.4, 128.6, 127.7, 127.5, 126.8, 124.2, 121.3, 119.1, 118.9, 62.0, 53.3, 42.7, 37.5, 32.0. MS m/z (%):501.2 (0.4), 327.2 (13.3), 174.3 (100), 119.2 (7.4), 91.2 (13.07), 55.2 (21.6). Elem. anal. calcd. For C33H31N3O2 (501.2); C, 79.02; H, 6.23; N, 8.38. Found: C, 79.01; H, 6.19; N, 8.31.
2-((4-benzylpiperidin-1-yl) methyl)-3-(5-chloro-2-methoxyphenyl) quinazolin-4(3H)-one (7h)
IR (KBr) v (cm−1):3024–3082 (C–H, aromatic), 2807–2930 (C–H, aliphatic), 1687 (C=O, Stretch), 1608 (C=C aromatic), 1520 (C=N, Stretch), 1018 (C-O, Stretch), 699 (C–CL, Stretch). 1H-NMR (500 MHz, DMSO) δ(ppm) = 8.12 (s, 1H, Quinazoline),7.55–7.86 (m,5H, Quinazoline and Phenyl), 7.1–7.3 (m, 6H, Phenyl and Benzyl), 3.7–4.04 (m, 6H, aliphatic), 3.04 (s, 1H, aliphatic),1.85–2.5 (m, 5H, aliphatic), 1.35–1.46 (m, 3H, aliphatic), 0.93–0.95 (m,1H, aliphatic). 13C-NMR (125 MHz, DMSO) δ (ppm) = 160.3, 153.3,146.2, 144.8, 139.6, 134.4, 134.1, 130.3, 129.6, 129.2, 129.1, 128.4, 128.2, 127.5, 126.7, 126.6, 125.8, 125.1, 122.7, 120.1, 61.1, 55.7, 55.4, 44.9, 41.9, 36.1, 31.1, 30.9. MS m/z (%):473.2 (0.2), 455.2 (1.8), 269.1 (28.8), 234.2 (13.6), 174.3 (100), 154.1 (2.04), 119.2 (4.5), 91.2 (13.3), 55.2 (19.11). Elem. anal. calcd. For C28H28ClN3O (473.2); C, 73.43; H, 6.16; N, 9.17. Found: C, 73.35; H, 6.09; N, 9.12.
MTT assay
Anticancer activity of all designed compounds (7a–7h) were achieved by standard 3-(4,5dimethylthiazol-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay according to our previous protocols [23]. Three human cancer cell lines such as MCF-7 (breast carcinoma), A549 (non-small cell lung carcinoma), 5637 (bladder carcinoma) and normal lung cell (MRC-5) were purchased from National Cell Bank of Iran (NCBI, Pasteur Institute, Tehran, Iran). All cancer cell lines were cultured in RPMI 1640 culture media. For normal cell line (MRC-5) DMEM/Ham’s F12 (Bio Idea, Iran) were used. All media were supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (Gibco, USA). The cells were kept at 37 °C in a humidified CO2 incubator. The cells were harvested using trypsin/EDTA 0.5% solution (Gibco/USA) and were then seeded in 96-well microplates at a density of 1 × 104 cells per well for MCF-7 cell lines and 8 × 103 cells per well for A549, 5637 cell lines and 15 × 103 for MRC-5 cell line in 100 μl of complete culture medium as previously determined [30]. The five different concentrations of the synthesized compounds and cis-platin as positive control (1 to 500 μM) were used for treatment in triplicate manner. For negative controls, three untreated wells were used. The media were replaced by 100 μL fresh MTT solution after 72 h and incubated for 4 h at 37 °C in the incubator to obtain formazan purple crystals [31]. The media were removed and 150 μL of DMSO were added and incubated at 37 °C in dark for 10 min to dissolve the crystals. The absorbance of individual well was read at 490 nm using a microplate ELISA reader. Excel 2016 and Curve Expert 1.4 were used to analyze the data. The data were presented mean ± SD for each analysis.
In silico physicochemical parameters calculations
The SwissADME online software was used to obtain the drug-likeness and ADME properties of all compounds [32].
Docking procedure
The PDB ID of EGFR (1M17) was obtained from Protein Data Bank (http://www.rcsb.org). All synthesized compounds were generated, optimized, and converted to pdbqt format. The final format of receptor was prepared by remove cognate ligand and water molecules and finally, missing hydrogen atoms were added and non-polar hydrogens were merged by using AutoDock Tools package (1.5.6) [33]. An in-house batch script (DOCKFACE) was applied to obtain the grid box with a size of 40 × 40 × 40 points in x, y, and z directions. The other docking parameters were set as default. Binding interactions of docked compounds and the receptor were analyzed by discovery studio client 2016 [34].
Simulation procedure
Gaussian 09 program package was used to optimize all synthesized compounds [35]. MD simulations were performed with GPU accelerated Gromacs 2020 [36]. The topology and coordinate files for protein were obtained using pdb2gmx program taking parameters from AMBER99SB-LIDN force field. The partial atomic charges of ligands was performed based on AM1-BCC method using Antechamber program of AmberTools [37]. The full Amber topology and coordinate files of ligands were generated using parmchk and tleap programs implemented in AmberTools package [38]. Acpype python script was used to convert the AMBER format to the GROMACS format files. Afterwards, the topology file for each complex were made and also, systems were solvated in a cubic periodic box with a side length of 20 Å3 by addition of TIP3P water molecules, and then, they were neutralized. The prepared systems were energy-minimized with steepest descent algorithm for 50,000 steps. A NVT and NPT equilibration was done according to our previous works [39]. The final MD production run was performed in 100 ns with a 0.2 fs time step. VMD software package was applied to visualize protein–ligand complexes interactions and MD trajectories [40].
Conclusion
In effort to find novel antiproliferative compounds, a series of quinazolinone-benzyl piperidine derivatives was prepared and identified. Their activities against three cancerous cell lines (MCF-7, A549 and 5637) were tested and their selectivity also evaluated against tumorigenic and non-tumorigenic cell lines. Most of the compounds, especially 7b, 7e and 7f, were found to have moderate activity against tested cancer cell lines, while assessment of compounds against normal cell line revealed lower toxicity. Molecular docking results also supported the cytotoxic activities of these novel compounds as EGFR inhibitors. Overall, these compounds can be used for development of new anticancer agents with further modifications.
Availability of data and materials
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request. We have presented all data in the form of Tables and Figure. The PDB code (1M17) was retrieved from protein data bank (www.rcsb.org). https://www.rcsb.org/structure/1m17.
Abbreviations
- EGFR:
-
Epidermal growth factor receptor
- MDR:
-
Multidrug resistance
- ADME:
-
Adsorption, Distribution, Metabolism and Discretion
- Rg:
-
Radius of gyration
- RMSD:
-
Root Mean Square Deviation
- RMSF:
-
Root Mean Square Fluctuation
- TPSA:
-
Total polar surface area
- DIPEA:
-
Diisopropy lethylamine
- TK:
-
Tyrosine kinase
- MTT:
-
3-(4, 5 Dimethylthiazol-yl)-2,5-diphenyl-tetrazolium bromide
References
Shao Z, Jahanbani A, Sheikholeslami SM. Multiplicative topological indices of molecular structure in anticancer drugs. Polycyclic Aromat Compd. 2022;42(2):475–88.
Mahmoud HK, Gomha SM, Farghaly TA, Awad HM. Synthesis of thiazole linked imidazo [2, 1-b] thiazoles as anticancer agents. Polycyclic Aromat Compd. 2021;41(8):1608–22.
Auti PS, George G, Paul AT. Recent advances in the pharmacological diversification of quinazoline/quinazolinone hybrids. RSC Adv. 2020;10(68):41353–92.
El-Metwally SA, Abou-El-Regal MM, Eissa IH, Mehany AB, Mahdy HA, Elkady H, et al. Discovery of thieno [2, 3-d] pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg Chem. 2021;112: 104947.
Vass P, Démuth B, Hirsch E, Nagy B, Andersen SK, Vigh T, et al. Drying technology strategies for colon-targeted oral delivery of biopharmaceuticals. J Control Release. 2019;296:162–78.
Wdowiak P, Matysiak J, Kuszta P, Czarnek K, Niezabitowska E, Baj T. Quinazoline derivatives as potential therapeutic agents in urinary bladder cancer therapy. Front Chem. 2021. https://doi.org/10.3389/fchem.2021.765552.
Dohle W, Jourdan FL, Menchon G, Prota AE, Foster PA, Mannion P, et al. Quinazolinone-based anticancer agents: synthesis, antiproliferative SAR, antitubulin activity, and tubulin co-crystal structure. J Med Chem. 2018;61(3):1031–44.
Syed T, Asiri YI, Shaheen S, Gangarapu K. Design, synthesis and anticancer evaluation of structurally modified substituted aryl-quinazoline derivatives as anticancer agents. Synth Commun. 2021;51(18):2782–95.
Abou-Seri SM, Taha AM, Mohamed MA, Abdelkader NM. New quinazoline-sulfonylurea conjugates: design, synthesis and hypoglycemic activity. Med Chem. 2019;15(6):634–47.
Shalaby AA, El-Khamry AMA, Shiba S, Ahmed AAAEA, Hanafi AA. Synthesis and antifungal activity of some new quinazoline and benzoxazinone derivatives. Archiv der Pharmazie Int J Pharm Med Chem. 2000;333(11):365–72.
Hassanzadeh F, Jafari E, Hakimelahi G, Khajouei MR, Jalali M, Khodarahmi G. Antibacterial, antifungal and cytotoxic evaluation of some new quinazolinone derivatives. Res Pharm Sci. 2012;7(2):87.
Khodarahmi G, Khajouei MR, Hakimelahi G, Abedi D, Jafari E, Hassanzadeh F. Antibacterial, antifungal and cytotoxic evaluation of some new 2, 3-disubstituted 4 (3H)-quinazolinone derivatives. Res Pharm Sci. 2012;7(3):151.
Honkanen E, Pippuri A, Kairisalo P, Nore P, Karppanen H, Paakkari I. Synthesis and antihypertensive activity of some new quinazoline derivatives. J Med Chem. 1983;26(10):1433–8.
Dutta A, Sarma D. Recent advances in the synthesis of Quinazoline analogues as Anti-TB agents. Tuberculosis. 2020;124: 101986.
Faraj FL, Zahedifard M, Paydar M, Looi CY, Abdul Majid N, Ali HM, et al. Synthesis, characterization, and anticancer activity of new quinazoline derivatives against MCF-7 cells. Sci World J. 2014;2014:1–15.
Kamal A, Bharathi EV, Ramaiah MJ, Dastagiri D, Reddy JS, Viswanath A, et al. Quinazolinone linked pyrrolo [2, 1-c][1, 4] benzodiazepine (PBD) conjugates: design, synthesis and biological evaluation as potential anticancer agents. Bioorg Med Chem. 2010;18(2):526–42.
Lee S, Kim J, Duggirala KB, Go A, Shin I, Cho BC, et al. Allosteric inhibitor TREA-0236 containing non-hydrolysable quinazoline-4-one for EGFR T790M/C797S mutants inhibition. Bull Korean Chem Soc. 2018;39(7):895–8.
Park H, Jung HY, Mah S, Hong S. Discovery of EGF receptor inhibitors that are selective for the d746–750/T790M/C797S mutant through structure-based de novo design. Angew Chem Int Ed. 2017;56(26):7634–8.
Ayati A, Moghimi S, Salarinejad S, Safavi M, Pouramiri B, Foroumadi A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg Chem. 2020;99: 103811.
Hameed A, Al-Rashida M, Uroos M, Ali SA, Arshia, Ishtiaq M, et al. Quinazoline and quinazolinone as important medicinal scaffolds: a comparative patent review (2011–2016). Expert Opin Ther Pat 2018; 28(4):281–97.
Alaa A-M, Abou-Zeid LA, ElTahir KEH, Ayyad RR, Magda A-A, El-Azab AS. Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibitory activities and molecular docking studies of substituted 2-mercapto-4 (3H)-quinazolinones. Eur J Med Chem. 2016;121:410–21.
Garofalo S, Rosa R, Bianco R, Tortora G. EGFR-targeting agents in oncology. Expert Opin Ther Pat. 2008;18(8):889–901.
Emami L, Faghih Z, Sakhteman A, Rezaei Z, Faghih Z, Salehi F, et al. Design, synthesis, molecular simulation, and biological activities of novel quinazolinone-pyrimidine hybrid derivatives as dipeptidyl peptidase-4 inhibitors and anticancer agents. New J Chem. 2020;44(45):19515–31.
Abd-El Gawad NM, Georgey HH, Ibrahim NA, Amin NH, Abdelsalam RM. Synthesis of novel pyrazole and dihydropyrazoles derivatives as potential anti-inflammatory and analgesic agents. Arch Pharmacal Res. 2012;35(5):807–21.
Ganesan A, Coote ML, Barakat K. Molecular dynamics-driven drug discovery: leaping forward with confidence. Drug Discovery Today. 2017;22(2):249–69.
Gholampour M, Seradj H, Pirhadi S, Khoshneviszadeh M. Novel 2-amino-1, 4-naphthoquinone hybrids: design, synthesis, cytotoxicity evaluation and in silico studies. Bioorg Med Chem. 2020;28(21): 115718.
Bharadwaj S, Rao AK, Dwivedi VD, Mishra SK, Yadava U. Structure-based screening and validation of bioactive compounds as Zika virus methyltransferase (MTase) inhibitors through first-principle density functional theory, classical molecular simulation and QM/MM affinity estimation. J Biomol Struct Dyn. 2021;39(7):2338–51.
Islam R, Parves MR, Paul AS, Uddin N, Rahman MS, Mamun AA, et al. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J Biomol Struct Dyn. 2021;39(9):3213–24.
Lobanov MY, Bogatyreva N, Galzitskaya O. Radius of gyration as an indicator of protein structure compactness. Mol Biol. 2008;42(4):623–8.
Faghih Z, Rahmannejadi N, Sabet R, Zomorodian K, Asad M, Khabnadideh S. Synthesis of some novel dibromo-2-arylquinazolinone derivatives as cytotoxic agents. Res Pharm Sci. 2019;14(2):115.
Hashemi S, Jassbi AR, Erfani N, Kiani R, Seradj H. Two new cytotoxic ursane triterpenoids from the aerial parts of Salvia urmiensis Bunge. Fitoterapia. 2021;154: 105030.
Emami L, Faghih Z, Khabnadideh S, Rezaei Z, Sabet R, Harigh E, et al. 2-(Chloromethyl)-3-phenylquinazolin-4 (3H)-ones as potent anticancer agents; cytotoxicity, molecular docking and in silico studies. J Iran Chem Soc 2021:1–13.
Sabet R, Khabnadideh S, Fathi D, Emami L. QSAR and docking study of isatin analogues as cytotoxic agents. J Pharm Res Int. 2019;27(5):1–22.
Fereidoonnezhad M, Faghih Z, Mojaddami A, Rezaei Z, Sakhteman A. A comparative QSAR analysis, molecular docking and PLIF studies of some N-arylphenyl-2, 2-dichloroacetamide analogues as anticancer agents. Iran J Pharm Res IJPR. 2017;16(3):981.
Gaussian09 RA. 1, mj frisch, gw trucks, hb schlegel, ge scuseria, ma robb, jr cheeseman, g. Scalmani, v. Barone, b. Mennucci, ga petersson et al., gaussian. Inc, Wallingford CT. 2009;121:150–66.
Kohnke B, Kutzner C, Grubmüller H. A GPU-accelerated fast multipole method for GROMACS: Performance and accuracy. J Chem Theory Comput. 2020;16(11):6938–49.
Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct Funct Bioinform. 2010;78(8):1950–8.
Jorgensen W, Chandrasekhar J, Madura J. lmpey, RW; Klein. ML J Chem Phys. 1983;79:926.
Gheshlaghi SZ, Ebrahimi A, Faghih Z. A detailed theoretical exploration on the THR-β binding affinities and antioxidant activity of some halogenated bisphenols. J Biomol Struct Dyn. 2021. https://doi.org/10.1080/07391102.2021.1950568.
Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–8.
Acknowledgements
Not applicable.
Funding
Financial assistance from the Shiraz University of Medical Sciences by way of grant number 16026 is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
LE prepare the manuscript, write the simulation section and performed docking study. SK. supervised the study and contribute to revise the manuscript. ZF supervised the biological tests. FF contributed to the synthesis of compounds. FZ performed the biological assay. SZG performed simulation study. SD edit the manuscript. AE supervised simulation study. ZF contributed to the preparation of the manuscript and supervise the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1
. The FT-IR spectrum of 7a. Figure S2. The 1H NMR spectrum of 7a. Figure S3. The 13C-NMR spectrum of 7a. Figure S4. The Mass spectrum of 7a. Figure S5. The FT-IR spectrum of 7b. Figure S6. The 1H NMR spectrum of 7b. Figure S7. The 13C-NMR spectrum of 7b. Figure S8. The Mass spectrum of 7b. Figure S9. The FT-IR spectrum of 7c. Figure S10. The 1H NMR spectrum of 7c. Figure S11. The 13C-NMR spectrum of 7c. Figure S12. The Mass spectrum of 7c. Figure S13. The FT-IR spectrum of 7d. Figure S14. The 1H NMR spectrum of 7d. Figure S15. The 13C-NMR spectrum of 7d. Figure S16. The Mass spectrum of 7d. Figure S17. The FT-IR spectrum of 7e. Figure S18. The 1H NMR spectrum of 7e. Figure S19. The 13C-NMR spectrum of 7e. Figure S20. The Mass spectrum of 7e. Figure S21. The FT-IR spectrum of 7f. Figure S22. The 1H NMR spectrum of 7f. Figure S23. The 13C-NMR spectrum of 7f. Figure S24. The Mass spectrum of 7f. Figure S25. The FT-IR spectrum of 7g. Figure S26. The 1H NMR spectrum of 7g. Figure S27. The 13C-NMR spectrum of 7g. Figure S28. The Mass spectrum of 7g. Figure S29. The FT-IR spectrum of 7h. Figure S30. The 1H NMR spectrum of 7h. Figure S31. The 13C-NMR spectrum of 7h. Figure S32. The Mass spectrum of 7h.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Emami, L., Khabnadideh, S., Faghih, Z. et al. Synthesis, biological evaluation, and computational studies of some novel quinazoline derivatives as anticancer agents. BMC Chemistry 16, 100 (2022). https://doi.org/10.1186/s13065-022-00893-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-022-00893-z