
Abstract
Background
A series of coumarin-indole hybrids was synthesized as the new α-glucosidase inhibitors. The title hybrids were considered as α-glucosidase inhibitors because had two active pharmacophores against α-glucosidase: coumarin and indole.
Methods
The thirteen various derivatives 4a–m were synthesized, purified, and fully characterized. These compounds were evaluated against α-glucosidase in vitro and in silico. In silico pharmacokinetic studies of the most potent compounds were also performed.
Results
Most of the title compounds exhibited high anti-α-glucosidase activity in comparison to standard drug acarbose. In particular, the phenoxy derivative 4d namely 3-((1H-indol-3-yl)(3-phenoxyphenyl)methyl)-4-hydroxy-2H-chromen-2-one showed promising activity. This compound is a competitive inhibitor against α-glucosidase and showed the lowest binding energy at the α-glucosidase active site in comparison to other potent synthesized compounds and acarbose.
Conclusion
Compound 4d can be a lead compound for further structural development to obtain effective and potent α-glucosidase inhibitors.
Similar content being viewed by others

Introduction
Type 2 diabetes (T2DM) is a metabolic disorder which is considered as a serious chronic health condition [1]. The prevalence of this disorder is increasing in worldwide due to false lifestyle patterns such as physical inactivity and incorrect nutrition [2]. This disease, if untreated, can lead to serious problems including kidney failure, blindness, cardiovascular diseases, and nerve damage [3]. Considering the limitations of current therapies such as adverse side effects and high secondary failure rates, there are a lot of demands for the design and development of new drugs for treatment of T2DM. Inhibition of carbohydrate degrading enzymes such as α-glucosidase is one of the therapeutic goals for T2DM treatment [4]. α-Glucosidase is an intestinal enzyme that convers carbohydrates to glucose and plays a key role in increasing postprandial blood glucose level [5]. α-Glucosidase inhibitors have been widely prescribed to treat of T2DM. These medications often increased secretion of undigested starch into the colon and thus their use is associated with a variety of undesirable gastrointestinal symptoms. For instance, acarbose as the most widely used drug in this category causes diarrhea, bloating, flatulence, and abdominal discomfort in nearly 20% of patients [6].
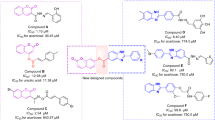
Coumarin ring has extensive utilization in design of new bioactive compounds [7]. Many natural and synthetic derivatives of coumarin with various remarkable bioactivities such as antibacterial, anticancer, anti-Parkinson, anti-HIV, and anti-proliferative activities have been reported [8,9,10]. This ring also found in the several series of synthetic potent α-glucosidase inhibitors such as compounds A–C (Fig. 1) [11,12,13]. Furthermore, interestingly, derivatives containing two coumarin rings such as biscoumarins D and E also exhibited high inhibitory activity against α-glucosidase (Fig. 1) [14, 15].

Rationale for the design of coumarin-indole hybrids as the new α-glucosidase inhibitors. Therefore, based on anti-α-glucosidase agents containing coumarin (compounds A–C), biscoumarin (compounds D–E), indole (compounds F–H), or bisindole (compounds I–J), we considered coumarin-indole hybrids as new α-glucosidase inhibitors. These compounds after synthesis, evaluated against α-glucosidase in vitro and in silico
Indole is a bicyclic heterocyclic ring with considerable applications in medicinal chemistry and crucial role in the biological systems [16]. Indole scaffold composed of a benzene ring fused to pyrrole ring. This ring is found in many natural derivatives such as plant alkaloids, fungal metabolites, and marine natural products [17]. Indole is also involved in the formation of amino acids, growth hormones, and alkaloids [18]. There are the several drugs containing indole ring with treatment applications such as anti-cancer, anti-hypertensive, and antimitotic activities in the pharmaceutical market [19, 20]. Recent studies showed that indole ring had attracted much attention for design of effective structures for targeting of α-glucosidase [21]. In this regards, several series of synthetic indole or bisindole based α-glucosidase inhibitors have been reported (Fig. 1, compounds F-J) [22,23,24,25,26].
Results and discussion
Chemistry
The coumarin-indole derivatives 4a–m were prepared according to Scheme 1 in the excellent yields (79–87%) [27,28,29,30]. These compounds were synthesized via a simple one-step reaction of 4-hydroxycoumarin 1, benzaldehyde derivatives 2a–m, and 1H-indole 3 in the solvent free condition at 50 °C.

Synthesis of coumarin-indole derivatives 4a–m
Anti-α-glucosidase activity and SAR discussion
The in vitro anti-α-glucosidase activity of the target compounds 4a–m was evaluated against yeast form of this enzyme, in comparison with acarbose as a positive control and the obtained IC50 values are listed in Table 1.
As listed in Table 1, the general structure of hybrid derivatives of coumarin and indole moieties was varied by substituents on pendant phenyl ring between the latter moieties. As evidenced from IC50 values, the most potent compounds were 3-phenoxyphenyl derivative 4d and un-substituted phenyl derivative 4a. These compounds were around sixfold more potent than acarbose. Evaluation on other derivatives with electron-donating substituents demonstrated that 4-methyl and 4-methoxy derivatives 4b–c also exhibited high inhibitory activity against α-glucosidase while introduction of hydroxy substituent on the pendant phenyl ring, as in case of compounds 4e and 4f, led to loss of effect. SAR evaluation of derivatives 4g–m with electron-withdrawing substituents revealed that the best effects obtained with fluoro and nitro substituents in 3-position of the pendant phenyl ring (compounds 4 g and 4m, respectively). Movement of fluoro substituent of 3- to 4-position led to a dramatically decrease in inhibitory activity (compound 4h) while movement of nitro substituent of 3- to 2-position completely abolished anti-α-glucosidase activity (compound 4l). The third potent compound among the compounds containing electron-withdrawing substituent was 4-chloro derivative 4j. Changing the position of this substituent to 3-position led to loss of effect as observed in 3-chloro derivative 4i. Like to 3-chloro derivative, 3-bromo derivative (compound 4k) also did not show activity against α-glucosidase (Additional file 1).
According to SAR study, in general, it should be mentioned that in addition to the type of substitution, the position of the substitutions has a significant effect on the observed inhibitory activities against α-glucosidase.
Kinetic study
To determine the mechanism of α-glucosidase inhibition of the newly synthesized compounds, the kinetic study was performed on compound 4d as representative compound. The relative velocity of the α-glucosidase was determined on four increasing concentrations of the p-nitrophenyl glucopyranoside as substrate. To construct the Lineweaver–Burk plot, the enzyme velocity was calculated in the presence of compound 4d as inhibitor at following concentrations: 0, 28, 58 and 116 µM. Then, the Lineweaver-Burke plot was depicted using the reciprocal of velocity and substrate concentration (Fig. 2a). Based on the obtained plot, a competitive type of inhibition by compound 4d was observed. Using by the Lineweaver–Burk secondary plot (Fig. 2b), a Ki value equal to 148 µM was determined for compound 4d.

Kinetic study of compound 4d into α-glucosidase. a The Lineweaver–Burk plot in the absence and presence of different concentrations of compound 4d; b The secondary plot between Km and various concentrations of compound 4d
Docking study
The molecular modeling was performed to gain insight into the binding modes of coumarin-indole derivatives to the conceivable target enzyme (α-glucosidase, modeled form) [31]. The most potent compounds 4a–d were docked at α-glucosidase active site and the best docked poses in terms of the binding energy (BE) were selected. The interaction modes of the latter compounds were shown in Fig. 3. BE values of compounds 4d, 4a, 4b, 4c, and acarbose in the α-glucosidase active site were − 9.08, − 8.65, − 8.61, − 8.26, and − 4.04 kcal/mol, respectively. These BE values suggested high affinities to the active site in the new α-glucosidase inhibitors 4d, 4a, 4b, and 4c in comparison to acarbose. The order of BEs in the selected compounds 4d, 4a, 4b, and 4c is in agreement with the obtained in vitro inhibitory activities of these compounds.

View of the two-dimensional structure of ligand binding cavity of the modeled α-glucosidase with the docked compounds 4d, 4a, 4b, and 4c visualized in the BIOVIA Discovery Studio v.3.5
Hydroxy and carbonyl units of coumarin ring in the most potent compound 4d attached to Pro309 and His279, respectively, through hydrogen bonds (Fig. 3). His279 and His239 formed two π-π stacking interactions with indole ring. Moreover, a π-anion interaction also observed between pendant phenyl ring of compound 4d and Glu304. Furthermore, several hydrophobic interactions with residues Pro309 and Arg312 and a none-classical hydrogen bond with the latter amino acid were observed in the binding mode of compound 4d.
The second potent compound 4a established three classical hydrogen bonds with Glu304 (hydroxy group), His279 (carbonyl unit), and Phe310 (NH unit) and two none-classical hydrogen bonds whit Arg312 and Pro309 (Fig. 3). Compound 4a also formed a π-anion interaction with Glu304 (coumarin ring) and a π-cation interaction with His279 (pendant phenyl ring). This compound also attached to residues Ser308, Ala279, Pro309, and Arg312 through hydrophobic interactions.
As can be seen in Fig. 3, the third potent compound 4b formed hydrogen bonds with residues Glu304 (hydroxy group) and Phe310 (NH unit). This compound formed several π-ion interactions with Glu304 (two π-anion interactions with coumarin ring and a π-anion interaction with pendant 4-methyphenyl ring), His239 (a π-cation interaction with indole ring), and His279 (a π-cation interaction with coumarin ring). Furthermore, hydrophobic and none-classical hydrogen bonds between this compound and residues Arg312 and Pro309 are also observed.
The fourth potent compound 4c established a hydrogen bond with Glu304 via hydroxy group, two π-cation interactions with His279 and His239 via coumarin and indole rings, respectively, and a π-anion interaction with Glu304 via pendant 4-methoxyphenyl group. This compound also formed three none-classical hydrogen bonds with Pro309, Thr307, and Glu304, and two hydrophobic interactions with Pro309 and Arg312, and an unfavorable interaction with Arg312.
In silico druglikeness, ADME, and toxicity studies
Druglikeness, ADME, and toxicity prediction of the most potent compounds 4a-d, 4g, and 4m were performed using by online software PreADMET [32]. The obtained results were showed in Table 2. This table demonstrated that all title compounds followed of Lipinski ‘Rule of five’. Therefore, presumably, compounds 4a–d, 4g, and 4m are orally active. These compounds have moderate (4a–d) to poor (4g and 4m) permeability to Caco-2 cell. Moreover, all the studied compounds have high human intestinal absorption (HIA). Permeability of the compounds 4a–d and 4g to blood brain barrier (BBB) is not in the acceptable range while permeability of compound 4m to BBB is in the acceptable range. Skin permeability of all the title compounds is in the acceptable range. All the studied compounds, with the exception of compound 4d, are mutagenic. Compounds 4a–d, 4g, and 4m have not carcinogenic effect on mouse. Moreover, compounds 4a-c and 4g have not carcinogenic effect on rat while compounds 4d and 4m are carcinogen on rat. In term of cardiotoxicity (hERG inhibition), all the title compounds have high risk.
Experimental
General procedure for the preparation of coumarin-indole derivatives 4a–m
A mixture of 4-hydroxycoumarin 1 (1.0 mmol), benzaldehyde derivatives 2a–m (1.0 mmol), and 1H-indole 3 (1.0 mmol) was heated at 50 °C for 24 h in solvent free condition. After that, the mixture was washed with petroleum ether and the obtained participate was purified using recrystallization from ethyl acetate to obtain pure products 4a–m.
3-((1H-indol-3-yl)(phenyl)methyl)-4-hydroxy-2H-chromen-2-one (4a)
Isolated yield: 87%, mp: 231–233 °C; IR (KBr) 3518, 1740, 1401, 1271, 1142 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 11.65 (s, 1H), 10.95 (s, 1H), 8.05 (d, J = 8.2 Hz, 1H), 7.61 (td, J = 7.9, 7.2, 1.5 Hz, 1H), 7.43–7.31 (m, 6H), 7.27 (t, J = 7.3 Hz, 2H), 7.22–7.12 (m, 2H), 7.12–7.02 (m, 1H), 6.98–6.89 (m, 1H), 6.15 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 162.24, 160.76, 152.65, 143.18, 136.46, 132.34, 128.63, 128.19, 127.77, 126.15, 124.81, 124.26, 123.92, 121.28, 118.93, 118.78, 116.75, 116.66, 114.64, 111.93, 109.00, 37.56 ppm. MS (EI): 367.1 m/z. Anal. Calcd. for C24H17NO3: C, 78.46; H, 4.66; N, 3.81. Found: C, 78.65; H, 4.81; N, 3.62.
3-((1H-indol-3-yl)(p-tolyl)methyl)-4-hydroxy-2H-chromen-2-one (4b)
Isolated yield: 79%, mp: 269–271 °C; IR (KBr) 3397, 1737, 1387, 1284, 1122 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 9.69 (s, 2H), 7.89 (d, J = 7.9 Hz, 2H), 7.63–7.52 (m, 2H), 7.42–7.24 (m, 4H), 7.02 (s, 5H), 6.31 (s, 1H), 2.23 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 165.63, 165.26, 152.63, 137.14, 134.87, 132.31, 129.11, 128.73, 128.52, 127.06, 124.32, 124.18, 123.89, 121.11, 118.36, 118.30, 116.39, 116.31, 114.23, 111.86, 104.69, 36.06, 20.97 ppm. MS (EI): 381.1 m/z. Anal. Calcd. for C25H19NO3: C, 78.72; H, 5.02; N, 3.67. Found: C, 78.95; H, 5.19; N, 3.37.
3-((1H-indol-3-yl)(4-methoxyphenyl)methyl)-4-hydroxy-2H-chromen-2-one (4c)
Isolated yield: 85%, mp: 246–248 °C; IR (KBr) 3406, 1736, 1387, 1243, 1123 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 11.86 (s, 2H), 7.90 (d, J = 8.0 Hz, 2H), 7.65–7.55 (m, 2H), 7.44–7.23 (m, 5H), 7.05 (d, J = 8.6 Hz, 2H), 6.79 (d, J = 7.2 Hz, 2H), 6.29 (s, 1H), 3.69 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 165.40, 165.25, 157.79, 152.60, 133.13, 132.37, 131.73, 128.21, 124.35, 124.30, 124.23, 123.63, 121.52, 118.22, 118.11, 116.80, 116.42, 114.89, 113.95, 111.99, 104.86, 55.40, 35.69 ppm. MS (EI): 397.1 m/z. Anal. Calcd. for C25H19NO4: C, 75.55; H, 4.82; N, 3.52. Found: C, 75.83; H, 5.03; N, 3.29.
3-((1H-indol-3-yl)(3-phenoxyphenyl)methyl)-4-hydroxy-2H-chromen-2-one (4d)
Isolated yield: 83%, mp: 183–185 °C; IR (KBr) 3489, 1729, 1401, 1231, 1112 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 10.89 (s, 2H), 7.88 (dd, J = 7.9, 1.6 Hz, 2H), 7.61–7.52 (m, 2H), 7.26 (dq, J = 37.6, 8.2 Hz, 9H), 6.94 (dq, J = 32.0, 6.5, 5.7 Hz, 3H), 6.82 (s, 1H), 6.75 (dd, J = 8.0, 2.2 Hz, 1H), 6.31 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 166.45, 165.08, 157.21, 156.53, 152.73, 143.55, 136.25, 132.12, 130.25, 130.19, 129.92, 127.25, 124.41, 123.96, 123.92, 123.32, 122.48, 121.27, 118.84, 118.38, 117.96, 116.27, 116.19, 114.08, 111.91, 109.99, 104.18, 36.49 ppm. MS (EI): 459.1 m/z. Anal. Calcd. for C30H21NO4: C, 78.42; H, 4.61; N, 3.05. Found: C, 78.25; H, 4.36; N, 3.24.
4-Hydroxy-3-((3-hydroxyphenyl)(1H-indol-3-yl)methyl)-2H-chromen-2-one (4e)
Isolated yield: 80%, mp: 200–202 °C; IR (KBr) 3509, 1735, 1399, 1226, 1110 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.04 (s, 3H), 7.90 (dd, J = 8.0, 1.8 Hz, 2H), 7.68–7.53 (m, 2H), 7.41 – 7.26 (m, 4H), 7.00 (t, J = 7.5 Hz, 1H), 6.61–6.45 (m, 3H), 6.28 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 165.73, 165.26, 157.67, 152.62, 141.85, 136.51, 132.35, 129.42, 126.68, 124.37, 124.21, 123.98, 121.22, 118.37, 118.34, 117.85, 116.42, 116.39, 114.03, 113.05, 109.95, 104.55, 36.28 ppm. MS (EI): 383.4 m/z. Anal. Calcd. for C24H17NO4: C, 75.19; H, 4.47; N, 3.65. Found: C, 74.93; H, 4.69; N, 3.81.
4-Hydroxy-3-((4-hydroxyphenyl)(1H-indol-3-yl)methyl)-2H-chromen-2-one (4f)
Isolated yield: 86%, mp: 286–288 °C; IR (KBr) 3513, 1732, 1378, 1241, 1089 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 12.88 (s, 3H), 7.82 (dd, J = 7.9, 1.6 Hz, 2H), 7.50 (ddd, J = 8.6, 7.2, 1.7 Hz, 2H), 7.30–7.14 (m, 5H), 7.08 (d, J = 8.6 Hz, 2H), 6.56 (d, J = 8.6 Hz, 2H), 6.17 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 168.00, 165.01, 155.08, 152.94, 140.00, 132.78, 131.21, 128.00, 124.55, 124.36, 123.68, 123.25, 121.45, 118.39, 118.27, 117.06, 116.85, 114.95, 114.70, 112.02, 104.28, 35.79 ppm. MS (EI): 383.2 m/z. Anal. Calcd. for C24H17NO4: C, 75.19; H, 4.47; N, 3.65. Found: C, 75.38; H, 4.57; N, 3.39.
3-((3-Fluorophenyl)(1H-indol-3-yl)methyl)-4-hydroxy-2H-chromen-2-one (4g)
Isolated yield: 87%, mp: 222–224 °C; IR (KBr) 3494, 1730, 1410, 1259, 1126 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 11.78 (s, 1H), 11.00 (s, 1H), 8.06 (d, J = 8.2 Hz, 1H), 7.63 (ddd, J = 8.6, 7.1, 1.5 Hz, 1H), 7.45–7.30 (m, 5H), 7.22–7.14 (m, 2H), 7.12–6.84 (m, 4H), 6.15 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 164.07 (1JCF = 240 Hz), 162.26, 161.02, 152.69, 146.51, 136.45 (3JCF = 6.75 Hz), 132.45, 130.01 (3JCF = 8.25 Hz), 127.59, 124.86, 124.68 (4JCF = 2.25 Hz), 124.29, 123.99, 121.39, 118.92, 118.87, 116.85, 116.71, 115.49 (2JCF = 21.75 Hz), 113.95, 113.07 (2JCF = 21 Hz), 112.01, 108.57, 37.30 ppm. MS (EI): 385.3 m/z. Anal. Calcd. for C24H16FNO3: C, 74.80; H, 4.18; N, 3.63. Found: C, 75.05; H, 3.96; N, 3.77.
3-((4-Fluorophenyl)(1H-indol-3-yl)methyl)-4-hydroxy-2H-chromen-2-one (4h)
Isolated yield: 85%, mp: 243–245 °C; IR (KBr) 3490, 1736, 1392, 1237, 1073, 912 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 11.65 (s, 1H), 10.92 (s, 1H), 8.03 (d, J = 7.9 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.45 – 7.18 (m, 6H), 7.11 (d, J = 2.4 Hz, 1H), 7.04 (dd, J = 10.3, 7.8 Hz, 3H), 6.90 (t, J = 7.5 Hz, 1H), 6.10 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 162.18, 161.93 (1JCF = 240 Hz), 160.78, 152.64, 139.22 (4JCF = 2.5 Hz), 136.49, 132.34, 130.43 (3JCF = 7.5 Hz), 127.56, 124.72, 124.23, 123.93, 121.30, 118.89, 118.81, 116.70, 116.64, 114.82 (2JCF = 20 Hz), 114.51, 111.93, 108.82, 36.94 ppm. MS (EI): 385.1 m/z. Anal. Calcd. for C24H16FNO3: C, 74.80; H, 4.18; N, 3.63. Found: C, 74.62; H, 4.41; N, 3.36.
3-((3-Chlorophenyl)(1H-indol-3-yl)methyl)-4-hydroxy-2H-chromen-2-one (4i)
Isolated yield: 80%, mp: 205–207 °C; IR (KBr) 3513, 1728, 1354, 1270, 1118 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 12.21 (s, 1H), 11.00 (s, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.84 (dd, J = 7.8, 1.4 Hz, 1H), 7.75–7.53 (m, 2H), 7.45–7.32 (m, 6H), 7.20 (d, J = 2.1 Hz, 1H), 7.09 (t, J = 7.2 Hz, 1H), 6.95 (t, J = 7.4 Hz, 1H), 6.15 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 162.29, 161.15, 152.71, 136.46, 133.13, 132.90, 132.43, 130.02, 128.37, 127.56, 127.38, 126.15, 124.88, 124.36, 123.67, 121.43, 118.95, 118.88, 116.75, 116.70, 113.81, 112.03, 108.47, 37.27 ppm. MS (EI): 401.0 m/z. Anal. Calcd. for C24H16ClNO3: C, 71.73; H, 4.01; N, 3.49. Found: C, 71.98; H, 4.16; N, 3.27.
3-((4-Chlorophenyl)(1H-indol-3-yl)methyl)-4-hydroxy-2H-chromen-2-one (4j)
Isolated yield: 82%, mp: 235–237 °C; IR (KBr) 3482, 1663, 1727, 1411, 1240, 1100 cm−1; 1H NMR (499 MHz, DMSO-d6) δ 11.71 (s, 1H), 10.95 (s, 1H), 8.04 (d, J = 7.8 Hz, 1H), 7.61 (td, J = 7.6, 7.0, 1.5 Hz, 1H), 7.41–7.22 (m, 8H), 7.14 (s, 1H), 7.06 (t, J = 7.4 Hz, 1H), 6.92 (t, J = 7.4 Hz, 1H), 6.09 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 162.26, 161.67, 152.58, 140.06, 136.57, 133.36, 132.16, 130.86, 127.22, 125.50, 124.69, 124.49, 123.89, 121.31, 118.69, 118.59, 116.72, 116.61, 114.32, 111.78, 104.82, 35.77 ppm. MS (EI): 401.0 m/z. Anal. Calcd. for C24H16ClNO3: C, 71.73; H, 4.01; N, 3.49. Found: C, 71.56; H, 3.87; N, 3.70.
3-((3-Bromophenyl)(1H-indol-3-yl)methyl)-4-hydroxy-2H-chromen-2-one (4k)
Isolated yield: 86%, mp: 212–214 °C; IR (KBr) 3505, 1737, 1412, 1272, 1010 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 11.74 (s, 1H), 10.95 (s, 1H), 8.02 (d, J = 7.9 Hz, 1H), 7.60 (t, J = 7.7 Hz, 1H), 7.40–7.26 (m, 7H), 7.20 (t, J = 7.9 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 7.05 (t, J = 7.6 Hz, 1H), 6.91 (t, J = 7.5 Hz, 1H), 6.09 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 162.21, 161.01, 152.66, 146.24, 136.41, 132.46, 131.15, 130.36, 129.03, 127.74, 127.49, 124.82, 124.29, 123.97, 121.58, 121.39, 118.91, 118.81, 116.70, 116.66, 113.71, 111.99, 108.47, 37.21 ppm. MS (EI): 445.0 m/z. Anal. Calcd. for C24H16BrNO3: C, 64.59; H, 3.61; N, 3.14. Found: C, 64.79; H, 3.44; N, 3.32.
3-((1H-indol-3-yl)(2-nitrophenyl)methyl)-4-hydroxy-2H-chromen-2-one (4l)
Isolated yield: 84%, mp: 240–242 °C; IR (KBr) 3488, 1726, 1551, 1357, 1239, 1101 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 11.71 (s, 2H), 7.84 (d, J = 7.9 Hz, 2H), 7.65 (d, J = 7.9 Hz, 1H), 7.59 – 7.46 (m, 4H), 7.40 (d, J = 7.8 Hz, 3H), 7.34–7.19 (m, 5H), 6.52 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 166.02, 163.74, 152.76, 149.92, 135.27, 132.32, 132.03, 130.28, 128.97, 127.42, 126.36, 124.42, 124.26, 123.93, 123.85, 121.70, 118.69, 118.52, 116.43, 116.30, 114.87, 112.00, 103.65, 34.67 ppm. MS (EI): 412.1 m/z. Anal. Calcd. for C24H16N2O5: C, 69.90; H, 3.91; N, 6.79. Found: C, 70.17; H, 4.16; N, 6.96.
3-((1H-indol-3-yl)(3-nitrophenyl)methyl)-4-hydroxy-2H-chromen-2-one (4m)
Isolated yield: 84%, mp: 198–200 °C; IR (KBr) 3521, 1729, 1557, 1353, 1150 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 7.98 (d, J = 8.2 Hz, 1H), 7.88 (s, 1H), 7.82 (d, J = 7.9 Hz, 2H), 7.62–7.41 (m, 5H), 7.29 (d, J = 8.2 Hz, 2H), 7.24 (t, J = 7.6 Hz, 2H), 6.35 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 167.93, 164.75, 152.96, 148.21, 145.28, 137.29, 134.30, 131.82, 129.85, 127.77, 124.58, 124.35, 123.63, 121.53, 120.86, 119.80, 118.91, 118.82, 116.12, 116.03, 114.68, 112.13, 103.22, 36.71 ppm. MS (EI): 412.1 m/z. Anal. Calcd. for C24H16N2O5: C, 69.90; H, 3.91; N, 6.79. Found: C, 70.11; H, 4.08; N, 6.58.
In vitro α‐glucosidase inhibition assay and kinetic study
The α‐glucosidase inhibition assays of the coumarin-indole derivatives 4a–m and kinetic study of the most potent compound 4d were performed into yeast α‐glucosidase according to the literature [31]. α-Glucosidase (EC3.2.1.20, Saccharomyces cerevisiae (S. cerevisiae), 20 U/mg) and substrate (p-nitrophenyl glucopyranoside) were prepared from Sigma-Aldrich. Appropriate enzyme concentration was obtained in potassium phosphate buffer (pH 6.8, 50 mM), and coumarin-indole hybrids 4a–m were dissolved in DMSO (10% final concentration). The potassium phosphate buffer (135 µL), various concentrations of the target compounds 4a–m (20 µL), and prepared enzyme solution (20 µL) were added to the 96-well plate and the later mixture was incubated for 10 min at 37 °C. Then, p-nitrophenyl glucopyranoside (substrate, 25 µL, 4 mM) was added to the incubated mixture and allowed to incubate at 37 °C for 20 min. Finally, the change in absorbance of the final mixture was measured at 405 nm by using spectrophotometer (Gen5, Power wave xs2, BioTek, America). DMSO (10% final concentration) as negative control and acarbose as positive control were used. The percentage of enzyme inhibition (% Inhibition) for each sample was calculated by using the following formula:
IC50 values were calculated from non-linear regression curve using by the Logit method.
Docking study
Docking study of the most potent compounds 4a–d in the modeled α-glucosidase active site was performed according to our previously described method [31]. S. cerevisiae α-glucosidase that was used in the experimental section had not any crystallographic structure in the protein data bank (PDB), thus, we constructed a modeled enzyme using SWISS-MODEL Repository [33]. For this purpose, our research team used of a method that was described by Imran et al. [34, 35]. After searching by using SWISS-MODEL to identify an appropriate protein with a high sequence similarity with S. cerevisiae α-glucosidase in PDB, we selected S. cerevisiae isomaltase with PDB code of 3A4A. The latter enzyme has 72% identical and 85% similarity with the S. cerevisiae α-glucosidase. Next, S. cerevisiae isomaltase was subjected through sequence alignment and homology model using by automated homology modeling pipeline SWISS-MODEL (managed by Swiss Institute of Bioinformatics) and the quality of the obtained model was verified using PROCHECK [33].
The 3D structures of the positive control acarbose and the most potent compounds 4d, 4a, 4b, and 4c were built by MarvineSketch 5.8.3, 2012, ChemAxon (http://www.chemaxon.com) and converted to pdbqt coordinate using Auto dock Tools. The pdbqt coordinate of the modeled α-glucosidase was created using the latter software by the following process: the polar hydrogen atoms were added and the Koullman charges were assigned. The obtained pdbqt file of enzyme was used as an input file for the AUTOGRID program. In AUTOGRID for each atom type in the studied compounds, maps were calculated with 0.375 Å spacing between grid points and the center of the grid box was placed at x = 12.5825, y = − 7.8955, and z = 12.519 Å. The appropriate dimensions for the active site box were determined by BIOVIA Discovery Studio v.3.5 (40 × 40 × 40 Å). Flexible ligand dockings were accomplished for the target compounds. Each docked system for these compounds was carried out by 50 runs of the AUTODOCK search by the Lamarckian genetic algorithm. The best poses of the title compounds were selected for analyzing the interactions between enzyme and ligands. The results were visualized using BIOVIA Discovery Studio v.3.5 and the obtained data showed in Fig. 3.
In silico druglikeness/ADME/T studies
In silico druglikeness/ADME/T prediction of the most potent compounds 4a–d, 4g, and 4m were performed using the preADMET online server [32].
Conclusion
Coumarin-indole hybrids 4a–m considered as new α-glucosidase inhibitors and synthesized by a one-step simple reaction. Enzymatic testing of the prepared compounds exhibited that most of the title compounds are potent inhibitor against α-glucosidase and the most potent entry (compound 4d) was a competitive inhibitor for this enzyme. SAR study of the title compounds revealed that in addition to the nature of substitution, the position of the substitutions play an important role in the observed anti-α-glucosidase activities. All the most potent compounds were docked at α-glucosidase active site. The latter study revealed that potent derivatives with coumarin-indole scaffold interacted with α-glucosidase active site with low BEs in comparison to standard inhibitor acarbose.
Availability of data and materials
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
References
Schulman AP, del Genio F, Sinha N, Rubino F. “Metabolic” surgery for treatment of type 2 diabetes mellitus. Endocr Pract. 2009;15:624–31.
Bellou V, Belbasis L, Tzoulaki I, Evangelou E. Risk factors for type 2 diabetes mellitus: an exposure-wide umbrella review of meta-analyses. PLoS ONE. 2018;13: e0194127.
Asif M. The prevention and control the type-2 diabetes by changing lifestyle and dietary pattern. J Educ Health Promot. 2014. https://doi.org/10.4103/2277-9531.127541.
Derosa G, Maffioli P. α-Glucosidase inhibitors and their use in clinical practice. Arch Med Sci. 2012;8:899.
Israili ZH. Advances in the treatment of type 2 diabetes mellitus. Am J Ther. 2011;18:117–52.
Hollander P. Safety profile of acarbose, an α-glucosidase inhibitor. Drugs. 1992;44:47–53.
Sandhu S, Bansal Y, Silakari O, Bansal G. Coumarin hybrids as novel therapeutic agents. Bioorg Med Chem. 2014;22:3806–14.
Hussain MI, Syed QA, Khattak MN, Hafez B, Reigosa MJ, El-Keblawy A. Natural product coumarins: biological and pharmacological perspectives. Biologia. 2019;74:863–88.
Peng XM, Damu LV, G, Zhou H. Current developments of coumarin compounds in medicinal chemistry. Curr Pharm Des. 2013. https://doi.org/10.2174/1381612811319210013.
Musa MA, Cooperwood JS, Khan MO. A review of coumarin derivatives in pharmacotherapy of breast cancer. Curr Med Chem. 2008;15:2664–79.
Taha M, Shah SA, Afifi M, Imran S, Sultan S, Rahim F, Khan KM. Synthesis, α-glucosidase inhibition and molecular docking study of coumarin based derivatives. Bioorg Chem. 2018;77:586–92.
Salar U, Taha M, Khan KM, Ismail NH, Imran S, Perveen S, Gul S, Wadood A. Syntheses of new 3-thiazolyl coumarin derivatives, in vitro α-glucosidase inhibitory activity, and molecular modeling studies. Eur J Med Chem. 2016;122:196–204.
Chaudhry F, Choudhry S, Huma R, Ashraf M, Al-Rashida M, Munir R, Sohail R, Jahan B, Munawar MA, Khan MA. Hetarylcoumarins: synthesis and biological evaluation as potent α-glucosidase inhibitors. Bioorg Chem. 2017;73:1–9.
Zawawi NK, Taha M, Ahmat N, Ismail NH, Wadood A, Rahim F, Rehman AU. Synthesis, in vitro evaluation and molecular docking studies of biscoumarin thiourea as a new inhibitor of α-glucosidases. Bioorg Chem. 2015;63:36–44.
Asgari MS, Mohammadi-Khanaposhtani M, Kiani M, Ranjbar PR, Zabihi E, Pourbagher R, Rahimi R, Faramarzi MA, Biglar M, Larijani B, Mahdavi M. Biscoumarin-1, 2, 3-triazole hybrids as novel anti-diabetic agents: design, synthesis, in vitro α-glucosidase inhibition, kinetic, and docking studies. Bioorg Chem. 2019;92: 103206.
Chadha N, Silakari O. Indoles as therapeutics of interest in medicinal chemistry: bird’s eye view. Eur J Med Chem. 2017;134:159–84.
Lunagariya J, Bhadja P, Zhong S, Vekariya R, Xu S. Marine natural product bis-indole alkaloid caulerpin: chemistry and biology. Mini-Rev Med Chem. 2019;19(9):751–61.
Ganapathi B, Kargi F. Recent advances in indole alkaloid production by Catharanthus roseus (Periwinkle). J Exp Bot. 1990;41:259–67.
Sravanthi TV, Manju SL. Indoles—a promising scaffold for drug development. Eur J Pharm Sci. 2016;91:1–10.
de Sa Alves FR, Barreiro EJ, Manssour Fraga CA. From nature to drug discovery: the indole scaffold as a ‘privileged structure.’ Mini-Rev Med Chem. 2009;9:782–93.
Wang J, Lu S, Sheng R, Fan J, Wu W, Guo R. Structure-activity relationships of natural and synthetic indole-derived scaffolds as α-glucosidase inhibitors: a mini-review. Mini-Rev Med Chem. 2020;20:1791–818.
Kawde AN, Taha M, Alansari RS, Almandil NB, Uddin N, Rahim F, Chigurupati S, Nawaz M, Hayat S, Ibrahim M, Elakurthy PK. Exploring efficacy of indole-based dual inhibitors for α-glucosidase and α-amylase enzymes: In silico, biochemical and kinetic studies. Int J Biol Macromol. 2020;154:217–32.
Solangi M, Khan KM, Saleem F, Hameed S, Iqbal J, Shafique Z, Qureshi U, Ul-Haq Z, Taha M, Perveen S. Indole acrylonitriles as potential anti-hyperglycemic agents: synthesis, α-glucosidase inhibitory activity and molecular docking studies. Bioorg Med Chem. 2020;28: 115605.
Sadat-Ebrahimi SE, Babania H, Mohammadi-Khanaposhtani M, Asgari MS, Mojtabavi S, Faramarzi MA, Yahya-Meymandi A, Zareie S, Larijani B, Biglar M, Rastgar H. Design, synthesis, and biological evaluation of new indole-acrylamide-1, 2, 3-triazole derivatives as potential α-glucosidase inhibitors. Polycycl Aromat Compd. 2022;42(6):3157–65.
Uddin I, Ullah H, Bibi A, Taha M, Khan F, Rahim F, Wadood A, Ahmad N, Khan AA, Ahmad F, Rehman ZU. Synthesis, in vitro alpha glucosidase, urease activities and molecular docking study of bis-indole bearing Schiff base analogs. Chem Data Collect. 2020;28:100396.
Gollapalli M, Taha M, Ullah H, Nawaz M, AlMuqarrabun LM, Rahim F, Qureshi F, Mosaddik A, Ahmat N, Khan KM. Synthesis of Bis-indolylmethane sulfonohydrazides derivatives as potent α-Glucosidase inhibitors. Bioorg Chem. 2018;80:112–20.
Yadav A, Patil P, Chandam D, Jadhav S, Ghule A, Hangirgekar S, Sankpal S. Fe3O4@ SiO2-SO3H-DABCO: a novel magnetically retrievable bifunctional catalyst for ecofriendly synthesis of diheteroarylmethanes. J Mol Struct. 2021;1245:130960.
Kumar A, Kumar S, Ahmed N. ‘Naked-eye’colorimetric/fluorimetric detection of F− ions by biologically active 3-((1 H-indol-3-yl) methyl)-4-hydroxy-2 H-chromen-2-one derivatives. RSC Adv. 2016;6:108105–12.
Brahmachari G, Das S. L-Proline catalyzed multicomponent one-pot synthesis of gem-diheteroarylmethane derivatives using facile grinding operation under solvent-free conditions at room temperature. RSC Adv. 2014;4:7380–8.
Mousavizadeh F, Hekmatshoar R, Beheshtiha SY, Rahnamafar R. Highly selective one-pot coupling reaction of indole, aromatic aldehydes, and 4-hydroxycoumarin using copper octoate as a homogeneous catalyst. Synth Commun. 2014;44:1483–91.
Adib M, Peytam F, Rahmanian-Jazi M, Mahernia S, Bijanzadeh HR, Jahani M, Mohammadi-Khanaposhtani M, Imanparast S, Faramarzi MA, Mahdavi M, Larijani B. New 6-amino-pyrido [2, 3-d] pyrimidine-2, 4-diones as novel agents to treat type 2 diabetes: a simple and efficient synthesis, α-glucosidase inhibition, molecular modeling and kinetic study. Eur J Med Chem. 2018;155:353–63.
Seul, South Corea: Bioinformatics and Molecular Design Research Center 2004. PreADMET program. http://preadmet.bmdrc.org.
Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–92.
Imran S, Taha M, Ismail NH, Kashif SM, Rahim F, Jamil W, Hariono M, Yusuf M, Wahab H. Synthesis of novel flavone hydrazones: in-vitro evaluation of α-glucosidase inhibition, QSAR analysis and docking studies. Eur J Med Chem. 2015;105:156–70.
Imran S, Taha M, Ismail NH, Kashif SM, Rahim F, Jamil W, Wahab H, Khan KM. Synthesis, in vitro and docking studies of new flavone ethers as α-glucosidase inhibitors. Chem Biol Drug Des. 2016;87:361–73.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
MM and MM-K designed the research work, performed the docking study, and wrote the manuscript. SB supervised the practical experiments. DR, MH, and AM synthesized and purified the compounds, and carried out 1H NMR and 13C NMR. MA supervised the biological tests. SM performed the biological tests. BL and HR supervised the research. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Images of 1H NMR and 13C NMR of the new synthesized compounds 5a-m and IC50 graphs of these compounds are available in the Supporting Information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Niri, D.R., Sayahi, M.H., Behrouz, S. et al. Design, synthesis, in vitro, and in silico biological evaluations of coumarin-indole hybrids as new anti-α-glucosidase agents. BMC Chemistry 16, 84 (2022). https://doi.org/10.1186/s13065-022-00882-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-022-00882-2