
Abstract
A new rapid, simple, and sensitive RP-HPLC method was carried out through applying Quality by Design approach for determination of xipamide and valsartan in Human plasma. Fractional factorial design was used for screening of four independent factors: pH, flow rate, detection wavelength, and % of MeOH. Analysis of variance (ANOVA) confirmed that flow rate and % of MeOH were only significant. Chromatographic conditions optimization was carried out through using central composite design. Method analysis was performed using BDS Hypersil C8 column (250 × 4.6 mm, 5 μm) and an isocratic mobile phase of MeOH and 0.05 M KH2PO4 buffer pH 3 (64.5:35.5, v/v) at 1.2 mL/min flow rate with UV detection at 240 nm and 10 μL injection volume. According to FDA guidelines, the method was then validated for the determination of the two drugs clinically in human plasma in respect of future pharmacokinetic and bioequivalence simulation studies. The standard curve was linear in the concentration range of 5–100 µg/mL for both drugs, with a determination coefficient (R2) of 0.999. Also, the average recoveries lied within the range from 99.89 to 100.03%. The proposed method showed good predictability and robustness.
Similar content being viewed by others

Introduction
Quality by design (QbD) is a modern and systematic approach for quality control of pharmaceuticals and product development. Pharmaceutical quality can be assured by understanding and controlling variable parameters for formulation and manufacturing processes through such structured context [1,2,3]. Now-a-days the concept of QbD can be extended to analytical and bioanalytical techniques. The application of QbD principles can help in clinical laboratories to develop a suitable analytical method providing a significant improvement better than the traditional and empirical methodology [4]. One of these QbD approaches is fractional factorial design (FFD) which is commonly used and effective tool in scientific research and industrial applications. The main advantage of FFD is that it allows building statistical models with a few numbers of runs. Using the models allows identification of the significant factors affecting certain responses during analytical method development. Central composite design (CCD) is an efficient tool in optimization of significant factors. CCD suggests the optimal variables value that gives the best and most desired response and defines process conditions which are robust to deliberate variations in factor settings. Also, it suggests a mathematical model relating the response with the critical variables, thus allowing to predict response with minimal error transmitted to that response (propagation of error or POE) [5].
Different classes are indicated for management of hypertension with concomitant disease. These classes include diuretics, beta-blockers, angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), and Aldosterone receptor antagonists. Diuretics have an initial decreasing effect on blood volume and consequently reduce blood pressure. ARBs have more complete blockade of angiotensin II actions compared with ACE inhibitors, so they are a substitute for the latter in treating patients with heart failure and noticeable ACE inhibitors side effects. Therefore, diuretics and ARBs can be considered as a rational drug combination for patients with hypertension associated with heart failure (HF). This combination is more effective than monotherapy with one of its components. It offers a remarkable reduction in blood pressure with lower doses and minimized adverse effects [6].
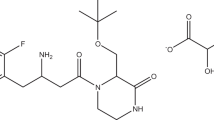
Xipamide (XIP) is a sulphonamide diuretic drug used in the treatment of hypertension either alone or in combination with other antihypertensives. It is also used in treatment of oedema including that related to HF [7]. Chemical structure of XIP, 5-(Arninosulphonyl) 4-chloro-N-(2,6-dimethylphenyl)-2-hydroxy-benzamide, is presented in Fig. 1. XIP acts mainly on both kidneys to reduce reabsorption of sodium in the distal convoluted tubule. The determination of XIP has been performed by HPLC [8,9,10], spectrophotometry [11, 12], spectroflourimetry [13], and voltammetry [14].

Structure of xipamide (XIP) and valsartan (VAL)
Valsartan (VAL) is an orally active and potent, non-peptide tetrazole derivative where it selectively inhibits Angiotensin II Receptor type 1 leading to reduction in blood pressure and so it can be used in hypertension treatment, to reduce mortality in patients with left ventricular dysfunction following myocardial infarction, and in HF management [7, 15] Chemically, it is 2(S)-3-Methyl-2-(pentanoyl{[2ʹ-(1H-tetrazol-5-yl)-4-biphenyl]methyl}amino) butanoic acid (Fig. 1). Literature review revealed that the determination of VAL has been carried out using HPLC [16,17,18,19,20,21,22,23,24,25,26,27,28], spectrophotometry [29,30,31,32,33] and spectroflourimetry [34, 35].
To the best of our comprehensive survey, XIP and VAL were not determined before as combined mixture (despite their synergistic action) by chromatographic techniques neither in biological nor pharmaceutical samples. As such, in line with keeping in mind the current FDA requirements while pursuing the study considering QbD based approach, the objective of our research is to develop a novel, accurate, robust, simple and specific HPLC method suitable for determination of XIP and VAL using FFD regarding pharmacokinetic and bioequivalence simulation studies and robustness testing. Among the different experimental designs, FFD as a response surface was preferrable for nonlinear response prediction in addition to its flexibility, in respect of experimental runs and information correlated with main and interaction factor effects.
Experimental
Apparatus
-
Agilent 1200® HPLC instrument (Germany) with a Thermo Scientific® BDS Hypersil C8 column (5 µm, 250 × 4.60 mm), DAD absorbance detector, in addition to HPLC QUAT pumps are connected to PC computer which is loaded with Agilent 1200 software [36, 37].
-
Labomed® Spectro (U6VD-2950) UV–VIS Double Beam Spectrophotometer (England) with 1 cm quartz cells and connected to PC computer loaded with UVWin5 Software v6 [36, 37].
-
HANNA® HI 8314 (Romania) membrane pH-meter for pH adjustment [37].
Materials and Reagents
-
All materials, chemicals, and solvents were of HPLC grade [37].
-
XIP (99.79%) and VAL (99.90%) were obtained from EIPICO (Tenth of Ramadan City, Egypt). Standard solutions of 200 µg/mL were prepared through dissolving 10 mg of each pure drug in 50 mL of the mobile phase [36].
-
Mobile phase was a binary mixture (freshly prepared) of MEOH: 0.05 M potassium dihydrogen phosphate (64.5: 35.5, v/v) adjusted to pH 3 by using ortho-phosphoric acid, filtered and degassed by using 0.45 µm membrane filters (Millipore, USA) [36].
-
MeOH (Fischer Scientific, Hampton, USA), Potassium dihydrogen phosphate (Techno Pharmchem, Delhi, India) and orthophosphoric acid (Merck, India) were all analytical grade assigned [33].
-
The human plasma was provided kindly by Zagazig University Hospital and was labeled to be disease and drug free. It was kept frozen at −20 °C before initial use and was then stored at −4 °C during usual uses [37].
Procedures
Construction of calibration curves
Appropriate mixed dilutions of XIP and VAL standard stock solutions were done in 10 mL volumetric flasks to get final concentrations of 5, 12.5, 25, 50 and 100 µg/mL for both drugs. A 10 μL of each mixture was injected then into the column while the chromatogram was monitored at 240 nm. A calibration graph was plotted as drug concentration against peak area response [37].
Human plasma samples procedure
All experimental protocols in the current study were approved by the EGYPTIAN NETWORK OF RESEARCH ETHICS COMMITTEES at the Faculty of Pharmacy, Zagazig University (Approved 2008). Calibration curves and validation QC samples in plasma at various concentrations of 2.50, 5, 15 and 20 µg/mL were prepared. Aliquots of 200 µL plasma samples and various drug mixture volumes ranging from 100–200 µL were added to 10 mL centrifuge tubes and then vortexed for 1 min. After that, the mixture was precipitated using methanol (total volume is 2 mL). After vortexing for 1 min, the samples were then centrifuged at 5000 rpm for 15 min. Aliquots of 10 µL of each supernatant was filtered using 0.45 µm PTFE syringe filters (Membrane solutions, USA) and directly injected into HPLC instrument for analysis [37].
Experimental design
Scouting step
Some trials were included in this step to find out a suitable mobile phase that can give an acceptable separation for both drugs. At the beginning, different concentrations containing either 0.025 or 0.05 M KH2PO4 buffer (as an aqueous part of the mobile phase) were tried. In addition, acetonitrile and MeOH were tested as organic modifiers. Finally, the variables that may clearly affect the selected responses were chosen [38].
Screening design
A resolution IV FFD with a minimum number of runs was used to identify the significant factors affecting the measured responses (Table 1). In this study, 4 independent factors were tested at 2 levels; pH at 3 & 4, flow rate at and 1.2 mL/min, detection wavelength at 230 and 250 nm, and also % of MeOH at 58 and 63%. The mathematical model related to the design consists of main effects and possible interaction effects (2 FI). In this case, 2 responses were taken into consideration: retention time (VAL) and resolution [39].
Optimization design
Central composite design (CCD) was commonly used due to its high efficiency and capability to reduce number of runs. A CCD with k factors should require 2 Table 2 k factorial runs, 2k axial experiments, symmetrically spaced at ± α along each variable axis, and one center point at least [40]. A rotatable CCD (α = 1.68) was built for the 4 significant factors to get the optimum level for desired responses using 5 levels of each factor (− α, − 1, 0, + 1, + α) with total number of 13 random runs which are including 5 center points (Table 2). The technique of numerical optimization and desirability function approach are used together usually to locate the optimized conditions through different trading off selected responses [41]. In this study, the numerical optimization was based on minimizing retention time (VAL) (+++ importance) and maximizing resolution (+ importance) between the analytes, obtaining a reasonable desirability function, and minimizing POE of both responses (+++ importance) to ensure that minimum error was transferred to responses.
Another tool was graphical optimization used to specify the design space (sweet spot) where desired CQAs meet. Graphical optimization goal was to minimize retention time (VAL) to be less than 6 min., and maximizing resolution with 3.6 as a lower limit, as well as, to minimize a POE of both responses by adjusting the highest acceptable upper limit. In addition, interval criteria were applied for CQAs and POE to understand the uncertainty impact on achieving the process goals. The sweet spot (sometimes called the bright yellow area) was obtained for each two variables, whilst the remaining factors were kept at a certain fixed value.
Finally, model predictability confirmation was checked through assuring that the predicted means of retention time (VAL), resolution and their POE lie within the low & high 95% values of prediction interval (PI low 95% and PI high 95%).
Investigation of model predictability was also achieved through prediction error calculation in accordance with the following equation [42]:
Results and discussion
Chromatographic conditions optimization
All chromatographic conditions are detailed in Table 3. Spectral analysis of both drugs in the range of 200–400 nm showed that XIP and VAL have λmax at 237 nm and 250 nm, respectively. As such, the chromatographic detection was set at 240 nm using a DAD detector as the appropriate wavelength. The method was carried out using a Thermo Scientific® BDS Hypersil C8 column (5 µm, 250 × 4.60 mm). The optimum mobile phase was determined as a MeOH: 0.05 M potassium dihydrogen phosphate mixture adjusted to pH 3 by using ortho-phosphoric acid (64.5: 35.5, v/v) at a flow rate of 1.2 mL/min. Under such conditions, XIP and VAL in human plasma can be completely separated at 3.23 and 4.34 min respectively as depicted in Fig. 2B, respectively. In addition, the mixture in plasma didn’t exhibit any matrix interference effect where human plasma chromatogram (Fig. 2A) showed no peaks at retention times of XIP and VAL.

HPLC chromatogram of (A) blank plasma (B) mixture of 12.50 µg/mL XIP and VAL in human plasma sample
The optimal mobile phase showed good symmetrical peaks (0.8 < T < 1.2), capacity factor (1 < k < 10), and resolution higher than 2 and theoretical plates more than 2000. Table 4 shows all system suitability parameters of the proposed HPLC method for simultaneous determination of those two drugs in pure and plasma matrices.
Scouting step
This step explains the effect of different mobile phases on analysis of the two analytes. In this step, four factors were chosen; pH, flow rate, detection wavelength, and % of MeOH to be tested in screening step.
Screening with FFD
Analysis of variance (ANOVA) for the studied factors is given in Table 5. The results indicated that only flow rate and % MeOH were the significant variables. Pareto charts, presented in Fig. 3, showed that flow rate had a significant effect only on the retention time (VAL), while % MeOH was a critical variable for both responses.

Pareto chart showing factors effect on: (A) retention time (VAL) and (B) resolution between XIP and valsartan (VAL)
Optimization with CCD
The results calculated by ANOVA of the significant factors are mentioned in detail in Table 6. Results confirmed the previous factors effects obtained by screening ANOVA. In addition, quadratic effects on retention time (VAL) were observed, while 2FI model was suggested for resolution.
Perturbation figure shows that % MeOH and flow rate had the most significant negative effect on retention time (VAL), Fig. 4A; increasing the variables was followed by a decrease in the response. The quadratic effect of % MeOH (factor A) is confirmed by the curvature of line A. On the other hand, % MeOH showed a similar effect on resolution (Fig. 4B). Contour and 3D plots (Fig. 5) show the interaction effect of the critical factors on retention time (VAL), and on resolution. Numerical optimization solution suggested those following optimal conditions: 64.5% MeOH, and a 1.2 mL/min flow rate. These optimal conditions have a desirability function of 0.716.

Perturbation plot for effect of factors on: (A) retention time (VAL) and (B) resolution, where line (A) is % MeOH and line (B) is flow rate

Contour (A) and 3D (B) plots showing the interaction effect of the % MeOH and flow rate on retention time (VAL) and resolution
The overlay plot represents the best desirable requirements of factors, responses and POE which are met in the sweet spot (S) as depicted in Fig. 6. Then, the variables optimum ranges were determined using the overlay contour plots as: % MeOH 63.95–64.99% and flow rate 1.12–1.2 mL/min. These ranges are representing the design space and confirm method robustness.

Overlay plot showing the sweet spot (S) where the desired responses met
The responses predicted means and their POE were reported within the low and high PI 95%, thus confirming predictability of the model. Additionally, the percentage prediction error was equal to −0.718 and 0.474 for retention time (VAL) and resolution, respectively (predicted retention time (VAL) = 4.734 and resolution = 4.998).
The following quadratic equation shows the relation between the significant factors and the selected responses (y):
Y = b0 + b1A + b2B + b3AB+ b4A2 where b0 is the intercept, b1–b5 represents the regression coefficients of quadratic polynomials and 2FI for both responses (Table 6).
Method validation
The method validation was performed according to food and drug administration [43,44,45].
Linearity
Five different concentrations of the drug mixture were specified for linearity studies in the range of 5–100 µg/mL for both drugs (Table 7). Linear regression equations of XIP and VAL were found to be y = 45.396x + 127.84 and y = 32.53x + 108.21, respectively and the regression coefficient values (r) were calculated to be 0.9999 for both drugs indicating a high degree of linearity (Fig. 7).

Calibration curves for authentic mixture of XIP and valsartan VAL using the proposed HPLC method
Accuracy
The accuracy of the proposed method was indicated by % recovery of the two different concentrations of XIP and VAL in human plasma.
precision
The method precision was evaluated in terms to intra-day and inter-day precision using the validation QC samples at concentrations of 12.50, 25 and 50 µg/ml. Intra-day precision was evaluated depending on standard deviation (SD) & coefficient of variation (CV%) where three replicates using the same solution of pure drugs were used. The SD values (ranged from 0.12 to 0.37) and CV% values (ranged from 0.12 to 0.38) indicated that the method is highly precise. Also, for inter-day reproducibility, SD & CV% values were in the acceptable range of 0.06–0.52 and 0.06–0.53, respectively (Table 8). These results show that the proposed method has an adequate precision in simultaneous determination of both drugs in either pharmaceutical or biological samples.
Selectivity and specificity
The method selectivity was checked by injecting XIP and VAL solutions separately into the column where 2 sharp peaks were eluted at retention times of 3.4, and 4.6 min, respectively, and these peaks were not monitored for the blank solution.
Limits of detection and limits of quantification
For estimating the limits of detection and quantification, the method reported by Bhaskaran et al. [46] was used based on equations: LOD = 3.3 σ/s and LOQ = 10 σ/s, where, σ is SD of y-intercepts of the regression line and s is the slope of the calibration line. LODs were reported to be 0.075 and 0.134, while LOQs were calculated to be 0.248 and 0.448 µg/mL for both XIP and VAL, respectively (Table 7) showing that the proposed method is highly sensitive and being applicable for future bioequivalence studies where it is mandatory to detect small drug concentrations in plasma.
Stability
Stability and precision studies were also conducted through application of plasma freeze–thaw cycles at −20 °C (over 3 days) using validation samples (5, 15 and 20 µg/mL of XIP and VAL) in plasma (Table 8). The recoveries for XIP and VAL were reported to be 93.09% and 89.17%, respectively as presented in Table 9.
Analysis of human plasma
XIP is well absorbed with maximum observed plasma concentration (Cmax) occurring 1 h of oral doses. Cmax after oral administration of 20 mg is 3 μg/mL [47]. VAL is rapidly absorbed after administration of tablets and oral solution with bioavailability of 23% and 39%, respectively. It is not significantly metabolized, so it is excreted mainly as unchanged form via the bile [7]. Following a single oral dose of 80 mg, Cmax is approximately 3.128 ng/mL with a tmax of 1.5 h for oral solution [48].
The proposed method was adopted for determination of XIP and VAL in human plasma by applying protein precipitation procedure. XIP and VAL retention times in plasma samples and the other system suitability parameters were pretty similar to those values in pure ones (Table 4). Also, the plasma chromatogram (Fig. 2A) confirms the method specificity in clinical studies as the plasma peak is not interfering with both XIP and VAL peaks.
Comparison with the reported method
Analytical parameters of the developed method were compared with some of the previously reported ones for estimation of VAL. The comparison presented in Table 10 shows that the developed procedure has the shortest run time. In addition, none the reported method used CCD for method optimization; CCD is superior to full factorial design (FFD) that is not generally advised in optimization procedures because of its incapability of examining quadratic models. Therefore, FFD can be used only for mapping linear relationships while CCD help obtaining more reliable models [49]. Moreover, the rotatable CCD applied in this study is better than FFD and other CCD; it uses five variable levels and consequently, can provide more accurate results. In term of greenness, the proposed mobile phase is the most eco-friendly. Therefore, this study could be considered as a promising would show a better performance. In addition, statistical analysis showed no significant difference between the two methods.
Conclusion
QbD strategy was adopted to develop a robust and an efficient RP-HPLC method for simultaneous estimation of xipamide and valsartan mixture in human plasma. Multivariate regression analysis was successfully carried out to study the main effects of 4 factors on both column efficiency and resolution. CCD was carried out to optimize of chromatographic conditions through studying the interaction and quadratic effects of significant factors on the two selected responses. The models which were used for screening and optimization steps were highly significant and confirmed the method predictability. The method is very simple, accurate, robust, and can be applied successfully to the analysis of XIP and VAL in human plasma with a high degree of selectivity.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
References
Lawrence XY, Gregory A, Mansoor AK, Stephen WH, James P, et al. Understanding pharmaceutical quality by design. AAPSJ. 2014;16(4):771–83. https://doi.org/10.1208/s12248-014-9598-3.
Rozet E, Pierre L, Jean-Francois M, Sondag P, Scherderac T, Bruno B. Analytical procedure validation and the quality by design paradigm. J Biopharm Stat. 2015;25(2):260–8. https://doi.org/10.1080/10543406.2014.971176.
Jaiprakash NS, Mrinmayee D, Zahid Z, Devanand BS, Rohidas A. Quality by design approach: regulatory need. Arab J Chem. 2017;10(2):S3412–25. https://doi.org/10.1016/j.arabjc.2014.01.025.
Baldelli S, Marrubini G, Cattaneo D, Clementi E, Cerea M. Application of quality by design approach to bioanalysis: development of a method for elvitegravir quantification in human plasma. Ther Drug Monit. 2017;39(5):531–42. https://doi.org/10.1097/FTD.0000000000000428.
Montgomery DC. Design and analysis of experiments. New York: Wiley; 2013.
Whalen K. Lippincott illustrated reviews: pharmacology. 7th ed. Philadelphia: Wolters Kluwer; 2019.
Sweetman SC. Martindale: the complete drug reference. 38th ed. London: Pharmaceutical Press; 2014.
Sebaiy MM, Abdellatef HE, Elmosallamy MAF, Alshuwaili MK. Isocratic HPLC method for simultaneous determination of amlodipine and xipamide in human plasma. Open J Anal Bioanal Chem. 2020;4(1):001–6. https://doi.org/10.17352/ojabc.000017.
Abd El-Hay SS, Hashem H, Gouda AA. High performance liquid chromatography for simultaneous determination of xipamide, triamterene and hydrochlorothiazide in bulk drug samples and dosage forms. Acta Pharm. 2016;66(1):109–18. https://doi.org/10.1515/acph-2016-0022.
Sane RT, Sadana GS, Bhounsule GJ, Gaonkar MV, Nadkarni AD, Nayak VG. High-performance liquid chromatographic determination of xipamide and clopamide in pharmaceuticals. J Chromatogr A. 1986;1(356):468–72. https://doi.org/10.1016/S0021-9673(00)91520-6.
Wagieh NE, Abbas SS, Abdelkawy M, Abdelrahman MM. Spectrophotometric and spectrodensitometric determination of triamterene and xipamide in pure form and in pharmaceutical formulation. Drug Test Anal. 2010;2(3):113–21. https://doi.org/10.1002/dta.92.
Gaber M, Khedr AM, El-Kady AS. New and sensitive spectrophotometric method for determination of xipamide in pure and dosage forms by complexation with Fe (III), Cu (II), La (III), UO2 (II), Th (IV) and ZrO (II) ions. Int Res J Pharm Pharmacol. 2011;1:215–20.
Walash MI, El-Enany N, Eid MI, Fathy ME. Stability—indicating spectrofluorimetric methods for the determination of metolazone and xipamide in their tablets. application to content uniformity testing. J Fluoresc. 2014;24(2):363–76. https://doi.org/10.1007/s10895-013-1301-z.
Legorburu MJ, Alonso RM, Jiménez RM. Voltammetric study of the diuretic xipamide. Bioelectrochem Bioenerg. 1993;32(1):57–66. https://doi.org/10.1016/0302-4598(93)80020-U.
Flesch G, Müller P, Lloyd P. Absolute bioavailability and pharmacokinetics of valsartan, an angiotensin II receptor antagonist, in man. Eur J Clin Pharmacol. 1997;52(2):115–20.
Zareh MM, Saad MZ, Hassan WS, Elhennawy ME, Soltan MK, Sebaiy MM. Gradient HPLC method for simultaneous determination of eight sartan and statin drugs in their pure and dosage forms. Pharmaceuticals. 2020;13(2):32. https://doi.org/10.3390/ph13020032.
Kumar L, Sreenivasa Reddy M, Managuli RS, Pai K. G Full factorial design for optimization, development and validation of HPLC method to determine valsartan in nanoparticles. Saudi Pharm J SPJ Pharm Soc. 2015;23:549–55. https://doi.org/10.1016/J.JSPS.2015.02.001.
Sreenivasa Reddy M, Kumar L, Attari Z, Verma R. Statistical optimization of extraction process for the quantification of valsartan in rabbit plasma by a HPLC method. Indian J Pharm Sci. 2017;79:16–28. https://doi.org/10.4172/PHARMACEUTICAL-SCIENCES.1000196.
Al Wassil O, Omer ME, Yassin AEB, Ahmad D. RP-HPLC method development for quantitation of valsartan in nano-structured lipid carrier formulation and in vitro release studies. Dig J Nanomater Biostruct. 2020;15:1017–102.
Patro SK, Kanungo SK, Patro VJ, Choudhury NS. Stability indicating RP-HPLC method for determination of valsartan in pure and pharmaceutical formulation. J Chem. 2010;7(1):246–52. https://doi.org/10.1155/2010/487197.
Vinzuda DU, Sailor GU, Sheth NR. RP-HPLC method for determination of valsartan in tablet dosage form. Int J Chem Tech Res. 2010;2(3):1461–7.
Kokil SU, Bhatia MS. Simultaneous estimation of nebivolol hydrochloride and valsartan using RP HPLC. Indian J Pharm Sci. 2009;71(2):111. https://doi.org/10.4103/0250-474X.54270.
Sharma M, Kothari C, Sherikar O, Mehta P. Concurrent estimation of amlodipine besylate, hydrochlorothiazide and valsartan by RP-HPLC, HPTLC and UV–spectrophotometry. J Chromatogr Sci. 2013;52(1):27–35. https://doi.org/10.1093/chromsci/bms200.
Ramadan NK, Mohamed HM, Moustafa AA. Rapid and highly sensitive HPLC and TLC methods for quantitation of amlodipine besilate and valsartan in bulk powder and in pharmaceutical dosage forms and in human plasma. Anal Lett. 2010;43(4):570–81. https://doi.org/10.1080/00032710903406953.
Sudesh BM, Uttamrao KS. Determination and validation of valsartan and its degradation products by isocratic HPLC. J Chem Metrol. 2009;3(1):1.
Macek J, Klima J, Ptáček P. Rapid determination of valsartan in human plasma by protein precipitation and high-performance liquid chromatography. J Chromatogr B. 2006;832(1):169–72. https://doi.org/10.1016/j.jchromb.2005.12.035.
Şatana E, Altınay Ş, Göğer NG, Özkan SA, Şentürk Z. Simultaneous determination of valsartan and hydrochlorothiazide in tablets by first-derivative ultraviolet spectrophotometry and LC. J Pharm Biomed Anal. 2001;25(5–6):1009–13. https://doi.org/10.1016/S0731-7085(01)00394-6.
Tatar S, Sağlık S. Comparison of UV-and second derivative-spectrophotometric and LC methods for the determination of valsartan in pharmaceutical formulation. J Pharm Biomed Anal. 2002;30(2):371–5. https://doi.org/10.1016/S0731-7085(02)00360-6.
Lakshmi K, Lakshmi S. Simultaneous spectrophotometric determination of valsartan and hydrochlorothiazide by H-point standard addition method and partial least squares regression. Acta Pharmaceutica. 2011;61(1):37–50. https://doi.org/10.2478/v10007-011-0007-5.
Erk N. Spectrophotometric analysis of valsartan and hydrochlorothiazide. Anal Lett. 2002;35(2):283–302. https://doi.org/10.1081/AL-120002530.
Anandakumar K, Jayamariappan M. Absorption correction method for the simultaneous estimation of amlodipine besylate, valsartan and hydrochlorothiazide in bulk and in combined tablet dosage form. Int J Pharm Pharm Sci. 2011;3(1):23–7.
Gupta KR, Mahapatra AD, Wadodkar AR, Wadodkar SG. Simultaneous UV spectrophotometric determination of valsartan and amlodipine in tablet. Int J Chem Tech Res. 2010;2(1):551–6.
Mohamed NG. Simultaneous determination of amlodipine and valsartan. Anal Chem Insights. 2011. https://doi.org/10.4137/ACI.S7282.
Shaalan RA, Belal TS. Simultaneous spectrofluorimetric determination of amlodipine besylate and valsartan in their combined tablets. Drug Test Anal. 2010;2(10):489–93. https://doi.org/10.1002/dta.160.
Cagigal E, Gonzalez L, Alonso RM, Jimenez RM. Experimental design methodologies to optimise the spectrofluorimetric determination of losartan and valsartan in human urine. Talanta. 2001;54(6):1121–33. https://doi.org/10.1016/S0039-9140(01)00379-4.
Sebaiy MM, Abdellatef HE, Elmosallamy MAF, Alshuwaili MK. Isocratic HPLC method for simultaneous determination of amlodipine and xipamide in human plasma. Open J Anal Bioanal Chem. 2020;4(1):001–6. https://doi.org/10.17352/ojabc.000017.
Sebaiy MM, El-Adl SM, Baraka MM, Hassan AA. Rapid RP-HPLC method for simultaneous estimation of some antidiabetics; metformin, gliclazide and glimepiride in tablets. Egy J Chem. 2019;62(3):1–12. https://doi.org/10.21608/ejchem.2018.4394.1388.
Hashem H, El-Sayed HM. Quality by design approach for development and validation of a RP-HPLC method for simultaneous determination of co-administered levetiracetam and pyridoxine HCl in prepared tablets. Microchem J. 2018;143:55–63. https://doi.org/10.1016/j.microc.2018.07.031.
Elhawi MM, Hassan WS, El-Sheikh R, El-Sayed HM. Multivariate analysis of perampanel in pharmaceutical formulations using RP-HPLC. Chromatographia. 2020;83:1335–43. https://doi.org/10.1007/s10337-020-03950-8.
Ficarra R, Calabrò ML, Cutroneo P, Tommasini S, Melardi S, et al. Validation of a LC method for the analysis of oxaliplatin in a pharmaceutical formulation using an experimental design. J Pharm Biomed Anal. 2002;29(6):1097–103. https://doi.org/10.1016/S0731-7085(02)00151-6.
Thakur D, Kaur A, Sharma S. Application of QbD based approach in method development of RP-HPLC for simultaneous estimation of antidiabetic drugs in pharmaceutical dosage form. J Pharm Investig. 2017;47(3):229–39. https://doi.org/10.1007/s40005-016-0256-x.
Guang W, Baraldo M, Furlanut M. Calculating percentage prediction error: a user’s note. Pharmacol Res. 1995;32:241–8. https://doi.org/10.1016/S1043-6618(05)80029-5.
US Food and drug administration, guidance for industry: bioanalytical method validation. 2001. (http://www.fda.go v/downloads/Drugs/Guidance/ucm070107.pdf).
Zimmer D, New US. FDA draft guidance on bioanalytical method validation versus current FDA and EMA guidelines: chromatographic methods and ISR. Bioanal. 2014;6(1):13–9. https://doi.org/10.4155/bio.13.298.
CDER Center for drug evaluation and research reviewer guidance validation of chromatographic methods. 1994. https://www.fda.gov/downloads/drugs/guidances/ucm134409.pdf.
Bhaskaran NA, Kumar L, Reddy MS, Pai GK. An analytical “quality by design” approach in RP-HPLC method development and validation for reliable and rapid estimation of irinotecan in an injectable formulation. Acta Pharm. 2021;71:57–79. https://doi.org/10.2478/acph-2021-0008.
Knauf H, Mutschler E. Pharmacodynamics and pharmacokinetics of xipamide in patients with normal and impaired kidney function. Eur J Clin Pharmacol. 1984;26:513–20. https://doi.org/10.1007/BF00542150.
Sunkara G, Bende G, Mendonza AE, Solar-Yohay S, Biswal S, Neelakantham S, Wagner R, Flarakos J, Zhang Y, Jarugula V. Bioavailability of valsartan oral dosage forms. Clin Pharmacol Drug Dev. 2014;3:132–8. https://doi.org/10.1002/CPDD.56.
Rakić T, Kasagić-Vujanović I, Jovanović M, Jančić-Stojanović B, Ivanović D. Comparison of full factorial design, central composite design, and Box-Behnken design in chromatographic method development for the determination of fluconazole and its impurities. Analyt Lett. 2014;47:1334–47. https://doi.org/10.1080/00032719.2013.867503.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
MMS, AA, H and HMEl-S designed and wrote the research work, SMEl-A and MMB revised the manuscript and supervised the research. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental protocols in the current study were approved by the Egyptian Network of Research Ethics Committees at the Faculty of Pharmacy, Zagazig University (Approved 2008). All methods were carried out in accordance with relevant regulations and guidelines. Zagazig University Hospital waived consent as the human plasma was provided kindly by Zagazig University Hospital.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sebaiy, M.M., El-Adl, S.M., Baraka, M.M. et al. Quality by design approach for development and validation of a RP-HPLC method for simultaneous estimation of xipamide and valsartan in human plasma. BMC Chemistry 16, 70 (2022). https://doi.org/10.1186/s13065-022-00864-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-022-00864-4