
Abstract
Background
Thiazoles, thiazolidinones and azetidinones are highly ranked amongst natural and synthetic heterocyclic derivatives due to their great pharmaceutical potential.
Results
New thiazolidinone and azetidinone class of bioactive agents based on 4-(2,7-dichloro-9H-fluoren-4-yl)thiazole moiety have been successfully synthesized. 4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-amine was synthesized and allowed to react with various aryl/heteroaryl aldehydes to afford the corresponding Schiff base intermediates. The target thiazolidinone and azetidinone analogues have derived from Schiff bases by their reactions with thioglycolic acid and chloroacetyl chloride, respectively. The newly synthesized compounds were then evaluated for their antimicrobial activity against some multidrug resistant strains and examined for cytotoxic activity against normal lung fibroblast (WI-38), human lung carcinoma (A549), and human breast carcinoma (MDA-MB-231) cell lines to develop a novel class of fluorene-based bioactive agents. The mode of action and the binding interaction of the synthesized compound with the active sites of dihydrofolate reductase enzyme were well identified by fluorescence-activated cell sorting (FACS) analysis and molecular docking study.
Conclusion
Some of the synthesized compounds showed remarkable activity against A-549 and MDA-MB-231 when compared to Taxol, which was used as a reference drug. 2,7-dichloro-9H-fluorene-based azetidinones are more efficient as antimicrobial and anticancer agents compared to dichloro-9H-fluorene-based thiazolidinones derivatives.

Similar content being viewed by others


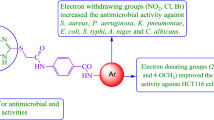
Introduction
In the last few years, fluorene derivatives exposed effective uses as precursors in broad ranging of synthetic and medical applications [1]. As example, 2,7-dichloro-7H-fluorene considered as a backbone moiety for the synthesis of a well-known antimalarial drug which known as Lumefantrine [2] (Fig. 1). On the other hand, heterocyclic compounds are highly ranked amongst natural and synthetic pharmaceutically significant agents. The fabulous ability of heterocyclic moiety to serve as both biomimetic and active pharmacophores has mainly contributed to their distinctive value as traditional key elements of various drugs. Due to their broad pharmacological profile, the nitrogen and sulfur-containing heterocycles demonstrate an imperative class in the biological research and drug industry areas [3,4,5,6,7,8]. Amongst them, the thiazole ring is a core structural moiety found in a wide range of biologically and medicinally active molecules. The thiazole derivatives are useful for treatment of several diseases such as allergies [9], hypertension [10], microbial [11], human immunodeficiency virus (HIV) infections [12], inflammation [13], and schizophrenia [14]. Moreover, 4-thiazolidinone and its derivatives have considerable attention for the last decades due to their pharmacological potential. These derivatives are known to acquire several promising chemotherapeutical activities such as antihistaminic [15], anti-inflammatory [16], hypolipidaemic [17], antimicrobial [18], anticonvulsant and antipsychotic [19], antimalarial [20], and anti-cancer [21] activities. Numerous drugs containing thiazole or 4-thiazolidinone moieties in their structure used in broad range in the pharmaceutical market such as Niridazole, Abafungin, Fanetinole, Ralitoline and Etozoline (Fig. 1). The traditional synthesis of 4-thiazolidinone derivatives involves cycloaddition of Schiff base with thioglycolic acid [22]. Additionally, the 2-azetidinone moiety is commonly show wide range of biological activities and exist in several β-lactam antibiotics such as penicillins, carbapenems and cephalosporins (Fig. 1) which are used as broad spectrum antibacterial agents. A large number of 3-chloro monocyclic β-lactam exhibits powerful antimicrobial, anticonvulsant, anti-inflammatory and antitubercular activities [23,24,25]. Conventional synthesis of 3-chloro-2-azetidinones involves [2 + 2] Staudinger’s ketene-imine cycloaddition reaction between chloroacetyl chloride and Schiff bases [26].

Representative examples of drugs containing 2,7-dichloro-9H-fluorene (I), thiazole (II), 4-thiazolidinone (III) and 2-azetidinone (IV) moieties
On the other hand, dihydrofolate reductase (DHFR) is an indispensable enzyme that catalyzes the NADPH-dependent reduction of 7,8-dihydrofolate (DHF) to 5,6,7,8-tetrahydrofolate (THF), which is the precursor of the cofactors compulsory for the biosynthesis of thymidine and purine nucleotides [27]. Accordingly, inhibition of dihydrofolate reductase lead to the disturbance of deoxyribonucleic acid (DNA) synthesis and the death of the proliferating cells [27, 28]. Furthermore, bacteria need DHFR to grow and multiply and consequently inhibitors discerning for bacterial in contradiction of host DHFR have found usage as antibacterial agents [29]. These two remarkable features render DHFR enzyme as one of the main targets for both antimicrobial and anticancer drug design [30, 31].
In the light of the previous findings, we predicted that the combining of 2,7-dichlorofluorene moiety with the versatile thiazole, thiazolidinone and azetidinone pharmacophores into a single chemical structure could be competent for antimicrobial and anticancer activities [30,31,32,33,34]. As part of our interest towards the development of novel bioactive organic molecules [30,31,32,33,34], a drug strategy has been planned to synthesis of some novel 2-(aryl/heteroaryl)-3-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)thiazolidin-4-ones and 3-chloro-4-(aryl/heteroaryl)-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)azetidin-2-ones with the anticipation to improve the antimicrobial activity against multidrug resistant strains and anticancer activity against human lung carcinoma (A549), and human breast carcinoma (MDA-MB-231) cell lines.
Results and discussion
Chemistry
As the inhibition of DHFR is commonly considered as one of the most prominent mechanism in elucidating antimicrobial and anticancer activities [35, 36], the compounds synthetic approaches were designed in order to achieve: (i) possess hydrophilic and hydrophobic parts that can interact with the hydrophilic and hydrophobic regions of the DHFR active site, respectively; (ii) comply with the pharmacophores that may interest as DHFR inhibitors, as presented in Fig. 2.

Structural fragments of DHFR inhibitors in the DHFR enzymatic active site
A distinctive synthetic approach employed to synthesize the target fluorene derivatives (5, 6) in good yields is described in Schemes 1 and 2. The synthetic strategy starts with a simple and convenient methodology to 2-chloro-1-(2,7-dichloro-9H-fluoren-4-yl)ethanone (2) involving direct chloroacetylation of 2,7-dichloro-9H-fluoren (1) is performed in excellent yield by adding a solution of 1 in dichloromethane (DCM) at 0–5 °C to a suspension of chloroacetyl chloride and aluminum chloride in dichloromethane according to our previously reported procedure [31]. Accordingly, 4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-amine (3) is attained in 97% yield via Hantzsch reaction of 2-chloro-1-(2,7-dichloro-9H-fluoren-4-yl)ethanone (2) with thiourea in refluxing ethanol (Scheme 1).

Synthesis of 4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-amine (3)

Synthesis of the target thiazolidinone derivatives 5a–n and azetidinone derivatives 6a–n
4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-amine (3) on condensation with different aryl/heteroaryl aldehydes in ethanol using catalytic amount of piperidine under reflux conditions afforded 4-(2,7-dichloro-9H-fluoren-4-yl)-N-(aryl/heteroaryl-methylene)thiazol-2-amine (4a–n) in 71–96% yields. Cyclocondensation of compounds (4a–n) with thioglycolic acid in tetrahydrofuran (THF) in presence of N,N′-dicyclohexylcarbodiimide (DCC) as a dehydrating agent under reflux conditions yielded the target 2-(aryl/heteroaryl)-3-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)thiazolidin-4-ones (5a–n) in 64–90% yields. Moreover, 4a–n when subjected to cyclocondensation with chloroacetyl chloride in dimethylformamide (DMF) at room temperature, 3-chloro-4-(aryl/heteroaryl)-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)azetidin-2-ones (6a–n) were obtained in moderate to excellent yields (51–98%) (Scheme 2).
The chemical structures of all synthesized compounds 5a–n and 6a–n were well-confirmed based on spectroscopic data such as Fourier transform infrared (FT-IR), proton nuclear magnetic resonance (1H-NMR), carbon-13 nuclear magnetic resonance (13C-NMR) and The distortionless enhancement by polarization transfer (DEPT-135) data (c.f. “Experimental” section and Additional file 1). The FT-IR spectra of compounds 5a–n revealed the presence of characteristic absorption bands at 1780–1680 cm−1 for (C=O) group, 1636–1600 cm−1 for (C=N) group. Furthermore, to fully establish the chemical structures of the products, intensive 1D (1H, 13C, and DEPT-135) NMR spectroscopic analysis were recorded. For example, analysis of the 13C and 13C-DEPT-135 NMR spectra of 5a indicated the presence of 23 signals representing the 23 of nonequivalent carbons (10 aromatic quaternary carbons, 9 aromatic CH’s, 2 methylene carbons, one methine carbon and one carbonyl carbon). Its 1H-NMR spectrum showed three singlet signals at 7.66, 7.60, 7.28 ppm and two doublets at 7.48 and 6.99 ppm (J = 8.0 Hz) for five protons of the fluorene moiety. A multiplet at 7.20, 6.94 ppm and doublet signals at 7.38, 6.27 ppm (J = 8.0 Hz) appeared for the protons of phenyl moiety. In addition to this, a singlet signal at 6.76 ppm for thiazole moiety. Three singlet signals at 3.98, 3.60 and 3.51 ppm corresponded to two methylene and one methine protons.
On the other hand, the FT-IR spectra of compounds 6a–n showed the presence of characteristic absorption bands at 1792–1697 cm−1 for (C=O) group, 1698–1598 cm−1 for (C=N) group. Indeed, the 13C and 13C-DEPT-135 NMR spectra of 6b indicated the presence of 24 signals representing the 24 of nonequivalent carbons (11 aromatic quaternary carbons, 8 aromatic CH’s, 2 methine carbons, one methylene carbon, one methyl carbon and one carbonyl carbon). Its 1H-NMR spectrum showed two doublets at 7.87 and 7.27 ppm (J = 8.0, 8.0 Hz), three singlet signals at 7.70, 7.50 and 7.21 ppm for five protons of the fluorene moiety. Two multiplets at 7.64 and 7.44 ppm appeared for the protons of 4-methoxyphenyl moiety. In addition, a singlet signal at 7.12 ppm for thiazole moiety. Four singlet signals at 4.45, 4.28, 4.20, and 4.01 ppm corresponded to the one methyl, two methine and one methylene protons.
Biological activity
Antimicrobial activity
Nowadays, the microbial resistance to currently found antibiotics is considered a precarious problem. Therefore, performing some more trials and efforts to identify novel targets for discovering new antibiotics is supposed to be a strong challenge [37]. The multidrug resistant bacteria have been reported with a diversity of nosocomial and community acquired infections as pneumonia, surgical site infections and urinary tract infections [38].
In the current study, the synthesized fluorene derivatives 5a–n and 6a–n were evaluated for their antimicrobial activity against multidrug resistant strains of Gram-positive bacteria such as staphylococcus aureus (S. aureus), methicillin-resistant Staphylococcus aureus (MRSA) and Streptococcus pneumoniae (S. pneumoniae) and Gram-negative bacteria such as Escherichia coli (E. coli), Klebsiella pneumoniae (K. pneumoniae), Pseudomonas aeruginosa (P. aeruginosa) and Acinetobacter baumannii (A. baumannii) as well as three fungal strains such as Aspergillus flavus (A. flavus), Aspergillus niger (A. niger) and Candida albicans (C. albicans). Screening the antimicrobial activity was done by agar well diffusion assay [39] using a concentration of 500 µg/mL of the tested fluorene compounds, the results of the antimicrobial assay are given in Tables 1, 2, 3. It is clearly observed that some of the newly synthesized fluorene derivatives exhibited comparatively high antimicrobial activity when compared to the positive reference drugs; vancomycin for Gram-positive bacteria, gentamicin for Gram-negative bacteria and fluconazole for fungi. It’s worthy to mention that, the thiazolidinone derivatives 5g, 5h, 5i and 5l produced relative high activity against S. aureus with a zone of inhibition (ZOI) value 10 mm, 11 mm, 10 mm, and 9 mm, respectively. While the compound 5j showed higher activity against E. coli and P. aeruginosa with a zone of inhibition (ZOI) value 10 mm and 8 mm, respectively (Table 1).
Furthermore, azetidinone derivatives 6a–n achieved relatively high antimicrobial activity against both Gram positive and Gram-negative bacteria, particularly 6h against S. aureus, MRSA, E. coli and P. aeruginosa with a ZOI value 12 mm, 15 mm, 22 mm, and 8 mm, respectively. However, a higher activity was shown against E. coli with ZOI value 27 mm for compound 6m. On the other hand, 6d showed moderate activity against Gram-negative bacteria E. coli, K. pneumoniae and P. aeruginosa with a ZOI value 15 mm, 6 mm, 10 mm, and 8 mm, respectively. However, low activity was shown against S. aureus and no activity was shown against both MRSA and S. pneumoniae. Moreover, the compound 6n showed moderate antimicrobial activity against E. coli and P. aeruginosa with a ZOI value 19 mm and 13 mm, respectively. The rest of the newly synthesized fluorene derivatives display low antimicrobial activity therefore these derivatives have potential for further comprehensive studies (Table 2).
The minimum inhibitory concentration (MIC) of the most active newly synthesized fluorene derivatives was determined and reported in Table 3. The MIC varied within the range (500 µg/mL–7.8 µg/mL). Compounds 5h and 6e were potent against Gram positive bacteria particularly S. aureus with an MIC value 62.5 µg/mL. Also, 6h was potent but against both S. aureus and MRSA with an MIC (62.5–31.25 µg/mL, respectively). Furthermore, a lower MIC was observed by the compound 6l against both S. aureus and MRSA as the MIC value was (31.25 µg/mL). On the other hand, the newly synthesized fluorene derivatives showed higher activity against Gram negative bacteria which is clearly achieved by the compound 6j, 6k, 6l and 6 m with MIC ranged from (31.25–15.6 µg/mL) specially against E. coli. All results were compared to vancomycin and Gentamicin as antibacterial reference drug (Table 3).
It’s worth to report that, the obtained biological activities make the newly synthesized novel fluorene derivatives 5a–n and 6a–n, interesting molecules for the synthesis of new antibiotics either alone or in combination with other compounds, and subsequently help in fighting the multidrug resistant superbugs.
In vitro anticancer activity
The synthesized new fluorene derivatives 5a–n and 6a–n were tested as anti-proliferative agents against WI-38 normal human lung fibroblast cells, A549 adenocarcinomic human alveolar basal epithelial cells, and MDA-MB-231 human breast cancer cells and they showed selectivity in their cytotoxic activity. A well-known chemotherapeutic agent, Taxol (IC50 = 41, 2.30, and 40 µg/mL for WI-38, A549, and MDA-MB-231, respectively) was used as reference control. The obtained results are presented in Tables 4, 5, 6, 7, 8, 9 and Figs. 3, 4, 5, 6, 7, 8.

IC50 of the tested compounds 5a–n against WI-38, A549, and MDA-MB-231 cancer cells after 24 h treatments

Apoptotic and necrotic cell death were assessed using Annexin V and Probidium Iodide (PI) staining and analyzed using flow cytometer after 24 h treatment with 5g. a WI-38 cells control (DMSO), b WI-38 cell treated with 1000 µg/mL of 5g, c A549 cell treated with 500 µg/mL of 5g, d MDA-MB-231 cell treated with 500 µg/mL of 5g, and (E) quantification of apoptotic and necrotic cell death for each drug on MDA-MB-231 cells

IC50 of the tested compounds 6a–n against WI-38, A549, and MDA-MB-231 cancer cells after 24 h treatments

Apoptotic and necrotic cell death were assessed using Annexin V and Probidium Iodide (PI) staining and analyzed using flow cytometer after 24 h treatment of azetidinone derivatives. a WI-38 cells control (DMSO), b WI-38 cell treated with 500 µg/mL of 6c, c WI-38 cell treated with 500 µg/mL of 6d, d WI-38 cell treated with 500 µg/mL of 6k, and e quantification of apoptotic and necrotic cell death for each drug on WI-38 cells

Apoptotic and necrotic cell death were assessed using Annexin V and Probidium Iodide (PI) staining and analyzed using flow cytometer after 24 h treatment with azetidinone derivatives. a A549 cells control in dimethyl sulphoxide (DMSO), b A549 cell treated with 85 µg/mL of 6c, c A549 cell treated with 117 µg/mL of 6d, d A549 cell treated with 200 µg/mL of 6k, and e quantification of apoptotic and necrotic cell death for each drug on A549 cells

Apoptotic and necrotic cell death were assessed using Annexin V and Probidium Iodide (PI) staining and analyzed using flow cytometer after 24 h treatment with azetidinone derivatives. a MDA-MB-231 cells control (DMSO), b MDA-MB-231 cell treated with 105 µg/mL of 6c, c MDA-MB-231 cell treated with 170 µg/mL of 6d, d MDA-MB-231 cell treated with 130 µg/mL of 6k, and e quantification of apoptotic and necrotic cell death for each drug on MDA-MB-231 cells
Quantitatively, 5m, 5n, 5l, 5d, and 5k showed moderate cytotoxic effect on normal cell lines with IC50 (92, 130, 223, 268, 288 µg/mL); respectively, as shown in Fig. 3 and Table 4 but less effective on A549 and MDA-MB-31 cancer cells. Briefly, 5m, 5n, 5b, 5a, 5c, 5d, and 5l with IC50 (357, 380, 402, 413, 415, 574, and 577 µg/mL) on A549 cells as shown in Fig. 3 and Table 5; 5m, 5n, 5b, 5a, 5c, 5f, 5g, 5d, 5l, and 5k with (357, 380, 402, 413, 415, 567, 572, 574, 577, and 613 µg/mL) on MDA-MB-231 cells, respectively, as shown in Fig. 3 and Table 6. In conclusion, 5g is the only compound that exerts a moderate anti-cancer activity on both lung and breast cancer cells.
Fluorescence-activated cell sorting (FACS) analysis for annexin V and PI staining to follow the mechanisms of cell death show that 5g induce necrotic cell death as the following (15.8%, 16.7%, and 14.1% of total cell number) on WI-38, A549, and MDA-MB-231 cells, respectively. On the other hand, induce insignificant apoptotic cell death with (1.5%, 1.25%, and 1.34% of total cell number) as shown in Fig. 4.
Moreover, in vitro anti-proliferative effect of azetidinone derivatives 6a–n on normal lung cells, lung and breast cancer cells and uncover the mechanisms of cell death in selected drugs which show anti-cancer activities.
Concisely, compounds 6e, 6f, and 6g bearing 4-(dimethylamino)phenyl, 4-nitrophenyl and 4-carboxyphenyl moieties, respectively, exerted an observed cytotoxic activity with IC50 (65.4, 29, and 40 µg/mL); correspondingly, against WI-38 normal lung cells compared with taxol which induce cell death with IC50 (41 µg/mL) as shown in Fig. 5 and Table 7. In the case of A549 lung cancer cells, compounds 6a, 6c, 6d, 6j, 6k, 6l, and 6n bearing phenyl, 4-cholorophenyl, 4-bromophenyl, furan-2-yl, thiophen-2-yl, 1H-pyrrol-2-yl, and quinolin-4-yl moieties, respectively, showed weak anti-proliferative activity with IC50 (185, 85, 117, 175, 203, 95, and 159 µg/mL); respectively, compared with taxol (IC50 2.3 µg/mL) as shown in Fig. 5, Table 8. Moving to MDA-MB-231 breast cancer cells, the screening result showed that compounds 6c, 6d, 6f, 6i, and 6k bearing 4-cholorophenyl, 4-bromophenyl, 4-nitrophenyl, styryl, and thiophen-2-yl moieties exhibited cytotoxicity with IC50 (104, 169, 188, 120, and 131 µg/mL); respectively, compared with (IC50 40 µg/mL) for Taxol as illustrated in Fig. 5 and Table 9. In conclusion, we can quantitatively conclude that, compounds 6c, 6d and 6k exerted ant-cancer activity on normal lung cells versus lung and breast cancer cells with IC50 (515, 759, and 528 µg/mL), (85, 117, and 203 µg/mL), and (104, 169, and 131 µg/mL), respectively.
Additional study using FACS analysis was done to expose the mechanism of cell death for compounds 6c, 6d and 6k. Flow cytometry using annexin V and propidium Iodide show that, 6c, 6d and 6k induced low necrotic cell death (14.5%, 14.1%, and 9.93%) of total cell number while inducing non-observed apoptotic cell death (1.22%, 1.34% and 0.61%) of total cell number as shown in Figs. 6, 7, 8, correspondingly. In the case of lung cancer cells, 6c, 6d and 6k induced markedly apoptotic cell death with (27.32%, 36.3%, and 32.67%) while inducing insignificant necrotic cell death with (2.1%, 2%, and 1.71%) of total cell populations. More interestingly, the selective compounds show a highly significant apoptotic cell death induction with (80.32%, 55.355, and 67.25) of total cell number while inducing in visible necrotic cell death (2.15%, 6.515%, and 4.56%); respectively.
Docking and molecular modeling study
Molecular Docking study of 28 new synthesized compounds 5a–n and 6a–n has been performed. The main idea was to build molecules that have the ability to intercalate between the DNA base pairs while in the same time be able to stabilize their intercalating complex through formation of different bonding with topoisomerase I amino acids. Molecular Docking study was done in order to comprehend the mechanism of interaction of the synthesized compounds with DNA topoisomerase I and to verify the difference in activity as antibacterial and anticancer between different synthesized analogues. Molecular Operating Environment (MOE®) version 2019.01, Chemical Computing Group (CCG) Inc., Montreal, Canada was used for this purpose [Molecular Operating Environment (MOE)], Version, Chemical Computing group Inc., Montreal, Quebec, Canada, 2016. http://www.chemcomp.com.].
The crystal structure of DNA topoisomerase I was obtained from Protein Data Bank [https://www.rcsb.org] at 3.0°A resolution (PDB code: 1T8I). It consists of 592 amino acid residues in one chain. After preparation of the enzyme, molecular docking of the cocrystallised Camptothecin ligand was done (Fig. 9) with different placement protocol in order to choose the best methodology for docking. The Triangle matcher placement method showed RMSD value of less than 2 (1.3581) which indicates the confidence in the produced docking results. As can be seen from the 2D and 3D interaction between Camptothecin and DNA topoisomerase I enzyme, Camptothecin acts mainly through intercalation between DNA base pairs which halts the ability division of DNA double strand.

2D and 3D interaction of folate and DNA topoisomerase I enzyme
Molecular docking of the conformation database of the 42 synthesized compounds into the active site of DNA topoisomerase I was carried out using the mentioned protocol with the results refinement using force-field based scoring function GBVI/WSA dG which estimates the free energy of binding of the ligand from a given pose. The functional form is a sum of terms:
C is represents the average gain/loss of rotational and translational entropy. α, β is constants which were determined during training (along with c) and are forcefield-dependent. Ecoul is the columbic electrostatic term, which is calculated using currently loaded charges, using a constant dielectric of 1. Esol is the solvation electrostatic term which is calculated using the GB/VI solvation model. Evdw is the Van der Waals contribution to binding. SAweighted is the surface area weighted by exposure.
The output docking results were arranged according to scoring function and explored using the browser function embedded in MOE software. Representation of 2D and 3D of the ligand interaction between all the synthesized compounds and DNA topoisomerase I enzymes is shown in Fig. 10. The synthesized compounds can be sorted into two different groups 5 and 6 according to the attachment to the (9H-fluoren-4-yl)thiazole; first the attachment is through thiazolino-4-one moiety and second the attachment is through β-lactam ring. Upon examining the scoring results, most of the highest active compounds showed better energy scores. So, compounds 5e, 5h, 5l, 6e and 6h showed high scores in comparison with other analogues. The scores were in the range of − 9.0685 to − 8.4903 kcal/mole.

Molecular docking of compounds 5e, 5h, 5l, 6e, 6h, and 6k
The details of the interactions are as the following: most of the compounds were able to intercalate between the DNA base pairs while forming hydrophobic interactions with the different nucleic acid skeleton and forming other types of interaction with the amino acid residues in the topoisomerase I enzyme. So, for all the compounds beside intercalation with DNA, the following binding interaction was present: Compound 5e interacts with the active site through formation of hydrogen bond between the sulfur of the thiazole ring and ASN352 with a distance of 4.03 Ǻ; beside hydrophobic interactions with different amino acid residues like LYS425 and TYR426. Compound 5h interacts with the active site through formation of 4 hydrogen bonds between the hydroxyl, chloro, carbonyl group and sulfur of the thiazolidine ring and PRO357, LYS354, LYS425 and GLU418, respectively. The distances of hydrogen bonds in order are 2.72, 2.92, 3.06 and 3.11 Ǻ. Compound 5l interacts with the active site through formation of 2 hydrogen bonds between sulfur of thiazolidinone ring and carbonyl group on one side and GLU356 and TRP426 on the other hand with distances of 3.00 and 3.15 Ǻ, respectively. Compound 6e interacts with the active site through formation of 2 hydrogen bonds between chloro groups on the fluorene moiety and on the β-lactam ring on one hand and MET428 and LYS425 on the other hand with distances of 3.76 and 3.72 Ǻ, respectively. Compound 6h interacts with the active site through formation of 2 hydrogen bonds between keto group on the β-lactam ring and sulfur of the thiazole ring on one side and LYS374 and one of the DNA nucleic acids with a distance of 3.32 and 3.72 Ǻ, respectively. The high activity of group three (β-lactam) against both cancer cell lines and bacteria may be attributed to the opening of the β-lactam ring and the increase in the flexibility of the molecules.
Conclusion
In this study, various 2,7-dichloro-9H-fluorene-based thiazolidinone and azetidinone derivatives were designed, synthesized, fully characterized and screened in vitro against various multidrug resistant microorganisms as well as against human lung carcinoma (A-549) and human breast carcinoma (MCF-7) cell lines. The results indicated that 2,7-dichloro-9H-fluorene-based azetidinones are more efficacious antimicrobial and anticancer agents compared to dichloro-9H-fluorene-based thiazolidinones analogues. Hence, there is adequate scope for further study in developing such compounds as a good lead activity.
Experimental
Chemistry
General methods
All Chemicals and solvents used purchased from Sigma-Aldrich are spectroscopic grade and used without further purifications. Melting points were determined on a Stuart SMP3 melting point apparatus and are uncorrected. FT-IR spectra were recorded on a Shimadzu IR-3600 FT-IR spectrometer in KBr pellets. NMR spectra were acquired on a Bruker Avance 400 instrument (400 MHz for 1H, 100 MHz for 13C) in DMSO-d6 solutions, using residual solvent signals as internal standards. Starting materials 2,7-dichloro-9H-fluorene (2) and 2-chloro-1-(2,7-dichloro-9H-fluoren-4-yl)ethanone (3) were prepared according to our previously reported method [31].
Synthesis of 4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-amine (3)
A mixture of chloroacetyl derivative 2 (15.55 g, 50 mmol) and thiourea (5.70 g, 75 mmol) in ethanol (250 mL) was refluxed for 3 h. The reaction mixture was cooled and neutralized with saturated aqueous solution of sodium biocarbonate. The obtained solid product was filtered off, washed with cold water (3 × 50 mL), then with cold ethanol (3 × 10 mL), dried and recrystallized from ethanol to afford 16.15 gm (97%) of pure 2-aminothiazole derivative 3 as pale yellow crystals, m.p. 199–200 °C. FT-IR (KBr): ν (cm−1) 3282, 3106 (NH2), 1639 (C=N); 1H-NMR (DMSO-d6): δ 7.66 (s, 1H, Flu-H), 7.63 (s, 1H, Flu-H), 7.55 (d, 1H, J = 5.5 Hz, Flu-H), 7.37 (s, 1H, Flu-H), 7.31 (d, 1H, J = 5.5 Hz, Flu-H), 7.18 (s, 2H, NH2), 6.77 (s, 1H, Thiazolyl-H), 4.00 (s, 2H, CH2); 13C-NMR (DMSO-d6): δ 168.8 (C=N), 148.9 (C), 146.7 (C), 146.3 (C), 139.1 (C), 136.9 (C), 133.5 (C), 131.9 (C), 131.2 (C), 128.7 (CH), 126.9 (CH), 125.3 (CH), 125.1 (CH), 124.9 (CH), 105.5 (Thiazole-CH), 36.7 (CH2).
Synthesis of 4-(2,7-dichloro-9H-fluoren-4-yl)-N-(aryl/heteroaryl-methylene)thiazol-2-amine 4a–n
A mixture of 3 (10 mmol, 3.33 g) and appropriate aromatic aldehyde (10 mmol) in absolute ethanol (50 mL) was heated under reflux for 4 h in the presence of two drops of dry piperidine. The reaction was concentrated and left to cool. The solid products were filtered and recrystallized from ethanol to give compounds 4a–n in 71–96% yields.
Synthesis of 2-(aryl/heteroaryl)-3-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)thiazolidin-4-ones (5a–n)
A mixture of Schiff base 4 (1.0 mmol) and thioglycolic acid (1.5 mmol) was stirred in THF with ice cooling for 5 min, followed by addition of DCC (308 mg, 1.5 mmol) was added to the reaction mixture at 0 °C, and the reaction mixture was stirred for an additional 50 min at room temperature. Dicyclohexylurea was filtered off and the filtrate was concentrated under reduced pressure. The solid product was collected, washed thoroughly with diluted sodium bicarbonate solution, dried and recrystallized from methanol to afford the thiazolidinone derivatives 5a–n.
3-(4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-yl)-2-phenylthiazolidin-4-one (5a)
Pale yellow crystals, yield (76%), m.p. 79–82 °C; FT-IR (KBr): ν (cm−1) 3064 (CH arom.), 2920 (CH aliph.), 1700 (C=O), 1621 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.66 (s, 1H, Flu-H), 7.60 (s, 1H, Flu-H), 7.48 (d, J = 8.0 Hz, 1H, Flu-H), 7.38 (d, J = 8.0 Hz, 2H, Ph-H), 7.28 (s, 1H, Flu-H), 7.20–7.17 (m, 2H, Ph-H), 6.99 (d, J = 8.0 Hz, 1H, Flu-H), 6.98–6.95 (m, 1H, Ph-H), 6.93 (s, 1H, Thiazole-H), 6.76 (s, 1H, CH), 3.98 (s, 2H, CH2), 3.60 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 171.5 (C=O), 166.5 (C=N), 148.8 (C), 146.7 (C), 146.1 (C), 140.1 (C), 138.9 (C), 133.3 (C), 133.1 (C), 131.8 (C), 131.5 (C), 129.9 (CH), 129.7 (CH), 129.1 (CH), 128.9 (CH), 128.4 (CH), 127.1 (CH), 126.9 (CH), 125.4 (CH), 106.9 (Thiazole-CH), 66.2 (CH), 37.0 (CH2), 35.7 (CH2).
3-(4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-yl)-2-(4-methoxyphenyl)thiazolidin-4-one (5b)
Pale yellow crystals, yield (80%), m.p. 74–76 °C; FT-IR (KBr): ν (cm−1) 3008 (CH arom.), 2930 (CH aliph.), 1694 (C=O), 1600 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.87 (d, J = 8.0 Hz, 1H, Flu-H), 7.67 (d, J = 8.0 Hz, 2H, Ph-H), 7.57 (s, 1H, Flu-H), 7.48 (s, 1H, Flu-H), 7.45 (d, J = 8.0 Hz, 1H, Flu-H), 7.32 (s, 1H, Flu-H), 7.11 (s, 1H, Thiazole-H), 6.93 (d, J = 8.0 Hz, 2H, Ph-H), 6.83 (s, 1H, CH), 4.00 (s, 2H, CH2), 3.87 (s, 3H, CH3), 3.76 (s, 2H, CH2); 13C NMR (100 MHz, DMSO- d6): δ 173.3 (C=O), 168.9 (C=N), 148.7 (C), 146.6 (C), 146.1 (C), 139.2 (C), 136.9 (C), 133.9 (C), 132.8 (C), 132.2 (C), 131.9 (C), 131.5 (C), 129.2 (CH), 128.6 (CH), 126.9 (CH), 125.4 (CH), 125.2 (CH), 124.9 (CH), 123.3 (CH), 105.7 (Thiazole-CH), 82.4 (CH), 48.4 (CH3) 35.9 (CH2), 32.9 (CH2).
2-(4-Chlorophenyl)-3-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)thiazolidin-4-one (5c)
Pale yellow crystals, yield (78%), m.p. 83–85 °C; FT-IR (KBr): ν (cm−1) 3025 (CH arom.), 2928 (CH aliph.), 1775 (C=O), 1694 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.64 (d, J = 8.0 Hz, 2H, Ph-H), 7.57 (s, 1H, Flu-H), 7.52 (d, J = 8.0 Hz, 1H, Flu-H), 7.44 (s, 1H, Flu-H), 7.32 (s, 1H, Flu-H), 7.18 (d, J = 8.0 Hz, 1H, Flu-H), 6.95 (s, 1H, Thiazole-H), 6.76 (d, J = 8.0 Hz, 2H, Ph-H), 6.53 (s, 1H, CH), 3.95 (s, 2H, CH2), 3.32 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 173.3 (C=O), 167.0 (C=N), 156.5 (C), 148.8 (C), 147.8 (C), 146.6 (C), 145.9 (C), 140.6 (C), 138.4 (C), 133.2 (C), 132.8 (C), 132.2 (C), 129.3 (CH), 129.1 (CH), 128.9 (CH), 127.7 (CH), 126.8 (CH), 123.8 (CH), 114.3 (CH), 105.7 (Thiazole-CH), 66.2 (CH), 36.8 (CH2), 33.5 (CH2).
2-(4-Bromophenyl)-3-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)thiazolidin-4-one (5d)
Orange crystals, yield (73%), m.p. 155–157 °C; FT-IR (KBr): ν (cm−1) 3018 (CH arom.), 2927 (CH aliph), 1776 (C=O), 1636 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.63 (s, 1H, Flu-H), 7.52 (d, J = 8.0 Hz, 2H, Flu-H), 7.24 (s, 1H, Flu-H), 7.18 (s, 1H, Flu-H), 7.08 (d, J = 8.0 Hz, 2H, Ph-H), 6.78 (s, 1H, Thiazole-H), 6.66 (s, 1H, CH), 6.56 (d, J = 8.0 Hz, 2H, Ph-H), 3.96 (s, 2H, CH2), 3.59 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 171.6 (C=O), 168.0 (C=N), 156.6 (C), 150.4 (C), 147.9 (C), 146.6 (C), 145.8 (C), 138.4 (C), 137.1 (C), 132.1 (C), 131.7 (C), 131.5 (C), 129.3 (CH), 128.1 (CH), 126.8 (CH), 125.5 (CH), 125.2 (CH), 124.1 (CH), 114.2 (CH), 112.4 (CH), 111.5 (Thiazole-CH), 68.1 (CH), 36.6 (CH2), 34.4 (CH2).
3-(4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-yl)-2-(4-(dimethylamino)phenyl)thiazolidin-4-one (5e)
Pale yellow crystals, yield (84%), m.p. 94–96 °C; FT-IR (KBr): ν (cm−1) 3074 (CH arom.), 2926 (CH aliph.), 1689 (C=O), 1607 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 8.23 (d, J = 8.0 Hz, 2H, Ph-H), 7.72 (d, J = 8.0 Hz, 1H, Flu-H), 7.64 (s, 1H, Flu-H), 7.56 (s, 1H, Flu-H), 7.33 (s, 1H, Flu-H), 7.16 (d, J = 8.0 Hz, 1H, Flu-H), 6.96 (s, 1H, Thiazole-H), 6.89 (d, J = 8.0 Hz, 2H, Ph-H), 6.35 (s, 1H, CH), 3.95 (s, 2H,CH2), 3.42 (s, 2H, CH2), 1.74 (s, 6H, 2CH3); 13C NMR (100 MHz, DMSO-d6): δ 171.3 (C=O), 166.8 (C=N), 148.8 (C), 147.7 (C), 147.38 (C), 146.6 (C), 146.0 (C), 138.8 (C), 137.1 (C), 132.9 (C), 131.8 (C), 131.5 (C), 128.6 (CH), 128.4 (CH), 127.9 (CH), 126.6 (CH), 125.2 (CH), 124.5 (CH), 124.2 (CH), 107.4 (Thiazole-CH), 68.8 (CH), 61.3 (CH3), 36.9 (CH2), 33.8 (CH2).
3-(4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-yl)-2-(4-nitrophenyl)thiazolidin-4-one (5f)
Yellow crystals, yield (76%), m.p. 90–92 °C; FT-IR (KBr): ν (cm−1) 3074 (CH arom.), 2930 (CH aliph.), 1707 (C=O), 1600 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.76 (d, J = 8.0 Hz, 1H, Flu-H), 7.67 (s, 1H, Flu-H), 7.58 (s, 1H, Flu-H), 7.35 (s, 1H, Flu-H), 7.24 (d, J = 8.0 Hz, 2H, Ph-H), 7.18 (s, 1H, Flu-H), 7.10 (d, J = 8.0 Hz, 2H, Ph-H), 6.86 (s, 1H, Thiazole-H), 6.68 (s, 1H, CH), 3.96 (s, 2H,CH2), 3.60 (s, 2H,CH2); 13C NMR (100 MHz, DMSO-d6): δ 168.9 (C=O), 166.8 (C=N), 148.7 (C), 146.6 (C), 146.2 (C), 139.2 (C), 136.9 (C), 133.6 (C), 132.7 (C), 132.5 (C), 131.9 (C), 131.5 (C), 128.5 (CH), 127.4 (CH), 127.4 (CH), 126.9 (CH), 125.4 (CH), 125.1 (CH), 124.9 (CH), 105.7 (Thiazole-CH), 102.6 (CH), 36.9 (CH2), 33.6 (CH2).
4-(3-(4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-oxothiazolidin-2-yl)benzoic acid (5g)
Yellow crystals, yield (90%), m.p. 103–105 °C; FT-IR (KBr): ν (cm−1) 3326 (OH), 3065 (CH arom.), 2930 (CH aliph.), 1778 (C=O), 1696 (C=O), 1628 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 9.10 (m, 1H, OH), 7.64–7.58 (m, 3H, Ph-H & Flu-H), 7.54 (s, 1H, Flu-H), 7.42 (d, J = 8.0 Hz, 1H, Flu-H), 7.38 (d, J = 8.0 Hz, 1H, Flu-H), 7.24 (s, 1H, Flu-H), 6.91–6.83 (m, 3H, Ph-H & Thiazole-H), 6.68 (s, 1H, CH), 3.92 (s, 2H, CH2), 3.69 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 191.4 (C=O), 171.6 (C=O), 157.1 (C=N), 156.6 (C), 147.9 (C), 147.7 (C), 147.2 (C), 146.7 (C), 146.3 (C), 138.5 (C), 136.8 (C), 132.1 (C), 131.5 (C), 129.2 (CH), 126.8 (CH), 125.5 (CH), 125.3 (CH), 124.3 (CH), 117.9 (CH), 115.7 (CH), 114.3 (CH), 111.2 (Thiazole-CH), 68.1 (CH), 36.8 (CH2), 34.7 (CH2).
3-(4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-yl)-2-(4-hydroxy-3-methoxyphenyl)thiazolidin-4-one (5h)
Yellow crystals, yield (76%), m.p. 85–87 °C; FT-IR (KBr): ν (cm−1) 3328 (OH), 3069 (CH arom.), 2930 (CH aliph.), 1689 (C=O), 1600 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.66–7.61 (m, 3H, Flu-H & Ph-H), 7.51 (d, J = 8.0 Hz, 1H, Flu-H), 7.35 (s, 1H, Flu-H), 7.31 (d, J = 8.0 Hz, 1H, Flu-H), 7.16 (s, 1H, Flu-H), 7.01–6.99 (m, 2H, Ph-H & Thiazole-H), 6.77 (s, 1H, CH), 4.38 (s, 2H, CH2), 4.03 (s, 2H, CH2), 3.98 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 168.9 (C=O), 157.1 (C=N), 148.7 (C), 147.7 (C), 146.63 (C), 146.2 (C), 146.0 (C), 139.2 (C), 137.2 (C), 136.9 (C), 133.6 (C), 131.9 (C), 131.5 (C), 129.0 (CH), 128.5 (CH), 127.0 (CH), 126.9 (CH), 125.4 (CH), 125.2 (CH), 124.9 (CH), 105.7 (Thiazole-CH), 62.2 (CH), 47.9 (CH3), 37.5 (CH2), 33.8 (CH2).
3-(4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-yl)-2-styrylthiazolidin-4-one (5i)
Pale yellow crystals, yield (72%), m.p. 114–116 °C; FT-IR (KBr): ν (cm−1): 3096 (CH arom.), 2930 (CH aliph.), 1689 (C=O), 1625 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.66 (s, 1H, Flu-H), 7.60–7.53 (m, 2H, Flu-H), 7.49 (d, J = 4.0 Hz, 1H, Flu-H), 7.34 (s, 1H, Flu-H), 7.30–7.27 (m, 3H, Ph-H), 7.19–7.17 (m, 3H, Ph-H & Thiazole-H), 6.83–6.75 (m, 2H, CH=CH), 6.32 (dd, J = 8.0, 4.0 Hz, 1H, CH), 3.98 (s, 2H, CH2), 3.45 (dd, J = 12.0, 4.0 Hz, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 168.9 (C=O), 166.2 (C=N), 153.7 (C), 149.5 (C), 148.7 (C), 147.0 (C), 146.1 (C), 137.0 (C), 135.5 (C), 133.0 (C), 131.5 (C), 129.5 (CH), 129.2 (CH), 128.7 (CH), 127.7 (CH), 127.1 (CH), 126.9 (CH), 125.7 (CH), 125.4 (CH), 124.5 (CH), 118.4 (CH), 105.7 (Thiazole-CH), 61.8 (CH), 36.9 (CH2), 33.8 (CH2).
3-(4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-yl)-2-(furan-2-yl)thiazolidin-4-one (5j)
Pale yellow crystals, yield (64%), m.p. 139–141 °C; FT-IR (KBr): ν (cm−1) 3099 (CH arom.), 2927 (CH aliph.), 1689 (C=O), 1636 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.88 (d, J = 8.0 Hz, 1H, Flu-H), 7.62 (d, J = 8.0 Hz, 1H, Furyl-H), 7.51 (d, J = 8.0 Hz, 1H, Flu-H), 7.45 (s, 1H, Flu-H), 7.41 (d, J = 8.0 Hz, 1H, Furyl-H), 7.35 (s, 1H, Flu-H), 7.31 (s, 1H, Flu-H), 7.29–7.17 (m, 2H, Furyl-H & Thiazole-H), 4.06 (s, 2H, CH2), 3.92 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 172.6 (C=O), 168.9 (C=N), 155.3 (C), 148.7 (C), 146.6 (C), 146.1 (C), 139.2 (C), 136.9 (C), 133.5 (C), 131.9 (C), 131.5 (C), 128.5 (CH), 126.9 (CH), 125.3 (CH), 125.1 (CH), 124.9 (CH), 122.9 (CH), 121.7 (CH), 113.2 (CH), 105.7 (Thiazole-CH), 69.0 (CH), 36.9 (CH2), 33.8 (CH2).
3-(4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-yl)-2-(thiophen-2-yl)thiazolidin-4-one (5k)
Pale yellow crystals, yield (67%), m.p. 165–167 °C; FT-IR (KBr): ν (cm−1) 3029 (CH arom.), 2926 (CH aliph.), 1688 (C=O), 1636 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.62 (d, J = 4.0 Hz, 1H, Flu-H), 7.51 (d, J = 8.0 Hz, 1H, Thienyl-H), 7.35 (d, J = 4.0 Hz, 1H, Flu-H), 7.31–7.29 (m, 2H, Flu-H & Thienyl-H), 7.22 (s, 1H, Flu-H), 7.17 (s, 1H, Flu-H), 7.00–6.98 (s, 1H, Thiazole-H), 6.90–6.87 (m, 1H, Thienyl-H), 6.76 (s, 1H, CH), 4.01 (s, 2H, CH2), 3.93 (dd, J = 8.0, 4.0 Hz, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 168.9 (C=O), 166.0 (C=N), 148.6 (C), 146.6 (C), 146.1 (C), 143.5 (C), 139.1 (C), 136.9 (C), 133.5 (C), 131.9 (C), 131.5 (C), 128.9 (CH), 128.5 (CH), 126.9 (CH), 125.8 (CH), 125.4 (CH), 125.1 (CH), 124.9 (CH), 119.6 (CH), 105.7 (Thiazole-CH), 62.0 (CH), 44.4 (CH2), 36.9 (CH2).
3-(4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-yl)-2-(1H-pyrrol-2-yl)thiazolidin-4-one (5l)
Yellow crystals, yield (75%), m.p. 124–126 °C; FT-IR (KBr): ν (cm−1) 3459 (NH), 3099 (CH arom.), 2926 (CH aliph.), 1680 (C=O), 1636 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.81 (d, J = 8.0 Hz, 1H, Flu-H), 7.61 (d, J = 8.0 Hz, 1H, Flu-H), 7.52 (s, 1H, Flu-H), 7.43 (s, 1H, Flu-H), 7.34–7.28 (m, 2H, Thiazole-H & Pyrrole-H), 7.18 (s, 1H, Flu-H), 6.97 (d, J = 8.0 Hz, 1H, Pyrrole-H), 6.76–7.64 (m, 2H, Pyrrole-H & CH), 6.07 (s, 1H, NH), 4.00–3.90 (m, 4H, 2CH2); 13C NMR (100 MHz, DMSO-d6): δ 171.9 (C=O), 168.9 (C=N), 150.6 (C), 149.9 (C), 148.7 (C), 146.7 (C), 146.1 (C), 136.9 (C), 133.5 (C), 131.9 (C), 131.5 (C), 128.5 (CH), 126.9 (CH), 126.7 (CH), 125.3 (CH), 124.9 (CH), 122.1 (CH), 121.3 (CH), 120.6 (CH), 105.6 (Thiazole-CH), 66.9 (CH), 36.9 (CH2), 34.4 (CH2).
3-(4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-yl)-2-(pyridin-4-yl)thiazolidin-4-one (5m)
Orange crystals, yield (72%), m.p. 110–112 °C; FT-IR (KBr): ν (cm−1) 3097 (CH arom.), 2928 (CH aliph.), 1702 (C=O), 1636 (C=N), 1600 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 8.86 (d, J = 8.0 Hz, 2H, Py-H), 8.07 (s, 1H, Flu-H), 7.71 (d, J = 8.0 Hz, 1H, Flu-H), 7.37 (d, J = 8.0 Hz, 1H, Flu-H), 7.26 (s, 2H, Flu-H), 6.96 (s, 1H, Thiazole-H), 6.70 (d, J = 8.0 Hz, 2H, Py-H), 6.18 (s, 1H, CH), 4.11 (s, 2H, CH2), 3.59 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 168.7 (C=O), 166.5 (C=N), 153.9 (C), 150.8 (C), 143.9 (C), 143.4 (C), 134.0 (C), 131.4 (C), 130.9 (C), 130.2 (C), 130.0 (C), 128.2 (CH), 127.8 (CH), 127.5 (CH), 126.8 (CH), 126.2 (CH), 124.8 (CH), 124.5 (CH), 112.7 (CH), 106.3 (Thiazole-CH), 65.7 (CH), 36.9 (CH2), 34.4 (CH2).
3-(4-(2,7-Dichloro-9H-fluoren-4-yl)thiazol-2-yl)-2-(quinolin-4-yl)thiazolidin-4-one (5n)
Pale yellow crystals, yield (90%), m.p. 204–206 °C; FT-IR (KBr): ν (cm−1) 3099 (CH arom.), 2924 (CH aliph.), 1636 (C=O), 1583 (C=N), 1538 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.90–7.87 (m, 2H, Quinoline-H), 7.81–7.79 (m, 3H, Flu-H & Quinoline-H), 7.73–7.71 (m, 2H, Flu-H), 7.37–7.34 (m, 2H, Flu-H & Quinoline-H), 7.26–7.23 (m, 2H, Flu-H & Quinoline-H), 7.07 (s, 1H, Thiazole-H), 7.00 (s, 1H, CH), 4.34 (s, 2H, CH2), 3.61 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 168.7 (C=O), 165.9 (C=N), 160.1 (CH=N), 154.3 (C), 153.9 (C), 152.65 (C), 150.28 (C), 143.79 (C), 143.3 (C), 139.3 (C), 136.9 (C), 135.4 (C), 132.9 (C), 129.6 (CH), 129.4 (CH), 129.13 (CH), 128.91 (CH), 128.65 (CH), 127.63 (CH), 125.99 (CH), 120.2 (CH), 119.5 (CH), 113.3 (CH), 101.9 (Thiazole-CH), 65.9 (CH), 36.9 (CH2), 34.2 (CH2).
Synthesis of 3-chloro-4-(aryl/heteroaryl)-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)azetidin-2-ones 6a–n
To Schiff s base 4a–n (1 mmol) in dry DMF (10 mL), chloroacetyl chloride (1.2 mmol) was added with stirring at room temperature during 15 min. The mixture was further stirred at room temperature for 5 h. The mixture was poured onto crushed ice. The obtained product was filtered, washed with water and recrystallized from ethanol to get pure azetidinone derivatives 6a–n.
3-Chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-phenylazetidin-2-one (6a)
Yellow crystals, yield (68%), m.p. 118–120 °C; FT-IR (KBr): ν (cm−1) 3062 (CH arom.), 2927 (CH aliph.), 1695 (C=O), 1650 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.96 (s, 1H, Flu-H), 7.90 (d, J = 8.0 Hz, 1H, Flu-H), 7.84 (s, 1H, Flu-H), 7.78 (s, 1H, Flu-H), 7.72–7.65 (m, 2H, Ph-H), 7.54–7.51 (m, 2H, Ph-H), 7.41 (d, J = 8.0 Hz, 1H, Flu-H), 7.29–7.26 (m, 1H, Ph-H), 7.22 (s, 1H, Thiazole-H), 7.14 (d, J = 12.0 Hz, 1H, CH–N), 4.45 (d, J = 12.0 Hz, 1H, CH–Cl), 4.00 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 169.1 (C=O), 165.8 (C=N), 158.0 (C), 147.8 (C), 146.7 (C), 146.2 (C), 138.8 (C), 137.0 (C), 132.4 (C), 132.1 (C), 131.7 (C), 129.6 (CH), 128.7 (CH), 127.5 (CH), 126.6 (CH), 125.5 (CH), 124.6 (CH), 122.9 (CH), 113.3 (CH), 107.0 (Thiazole-CH), 69.0 (CH–N), 57.3 (CH–Cl), 36.9 (CH2).
3-Chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-(4-methoxyphenyl)azetidin-2-one (6b)
Yellow crystals, yield (98%), m.p. 110–112 °C; FT-IR (KBr): ν (cm−1) 3062 (CH arom.), 2951 (CH aliph.), 1691 (C=O), 1598 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.87 (d, J = 5.0 Hz, 1H, Flu-H), 7.70 (s, 1H, Flu-H), 7.64 (d, J = 8.0 Hz, 2H, Ph-H), 7.50 (s, 1H, Flu-H), 7.40 (s, 1H, Flu-H), 7.28–7.26 (m, 3H, Flu-H & Ph-H), 7.21 (s, 1H, Thiazole-H), 7.13 (d, J = 8.0 Hz, 1H, CH–N), 4.45 (s, 3H, CH3), 4.21 (d, J = 8.0 Hz, 1H, CH–Cl), 4.01 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 169.1 (C=O), 165.7 (C=N), 158.0 (C), 147.9 (C), 146.8 (C), 146.2 (C), 138.8 (C), 137.0 (C), 132.3 (CH), 131.6 (C), 130.1 (C), 128.9 (CH), 127.1 (CH), 125.9 (CH), 125.5 (CH), 124.6 (CH), 123.4 (C), 122.1 (C), 114.9 (CH), 113.30 (Thiazole-CH), 69.0 (CH–N), 57.3 (CH–Cl), 40.1 (CH3), 36.9 (CH2).
3-Chloro-4-(4-chlorophenyl)-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)azetidin-2-one (6c)
Yellow crystals, yield (96%), m.p. 105–106 °C; FT-IR (KBr): ν (cm−1) 3063 (CH arom.), 2929 (CH aliph.), 1697 (C=O), 1593 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 7.95 (d, J = 5.0 Hz, 1H, Flu-H), 7.72 (s, 1H, Flu-H), 7.66 (s, 1H, Flu-H), 7.51 (d, J = 8.0 Hz, 2H, Ph-H), 7.40 (m, 3H, Flu-H & Ph-H), 7.28 (s, 1H, Flu-H), 7.21 (s, 1H, Thiazole-H), 7.12 (d, J = 8.0 Hz, 1H, CH–N), 4.46 (d, J = 8.0 Hz, 1H, CH–Cl), 4.03 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 169.1 (C=O), 165.8 (C=N), 158.0 (C), 147.8 (C), 146.8 (C), 146.3 (C), 138.8 (C), 137.04 (C), 132.5 (C), 132.1 (C), 131.6 (C), 129.2 (CH), 129.0 (C), 127.7 (CH), 127.1 (CH), 126.0 (CH), 125.6 (CH), 124.6 (CH), 114.9 (CH), 105.4 (Thiazole-CH), 67.2 (CH–N), 61.1 (CH–Cl), 37.3 (CH2).
4-(4-Bromophenyl)-3-chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)azetidin-2-one (6d)
Pale yellow crystals, yield (87%), m.p. 112–114 °C; FT-IR (KBr): ν (cm−1) 3099 (CH arom.), 2955 (CH aliph.), 1792 (C=O), 1665 (C=N); 1H NMR (DMSO-d6): δ 7.83 (s, 1H, Flu-H), 7.76–7.65 (m, 4H, Flu-H & Ph-H), 7.50 (s, 1H, Flu-H), 7.40 (s, 1H, Flu-H), 7.27–7.22 (m, 3H, Flu-H & Thiazole-H), 7.12 (d, J = 8.0 Hz, 1H, CH–N), 4.27 (d, J = 8.0 Hz, 1H, CH–Cl), 4.02 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 170.9 (C=O), 165.7 (C=N), 158.0 (C), 147.8 (C), 146.7 (C), 146.1 (C), 138.8 (C), 136.6 (C), 135.0 (C), 132.7 (CH), 132.6 (CH), 131.5 (C), 129.3 (C), 129.0 (CH), 127.4 (C), 127.1 (CH), 124.6 (CH), 124.5 (CH), 113.3 (CH), 107.4 (Thiazole-CH), 66.1 (CH–N), 54.1 (CH–Cl), 37.2 (CH2).
3-Chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-(4-(dimethylamino)phenyl)-azetidin-2-one (6e)
Red crystals, yield (65%), m.p. 110–111 °C; FT-IR (KBr): ν (cm−1) 3069 (CH arom.), 2949 (CH aliph.), 1695(C=O), 1551 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 7.93 (d, J = 8.0 Hz, 1H, Flu-H), 7.77–7.60 (m, 4H, Flu-H & Ph-H), 7.51 (s, 1H, Flu-H), 7.40 (s, 1H, Flu-H), 7.28 (d, J = 8.0 Hz, 2H, Ph-H), 7.22 (s, 1H, Thiazole-H), 6.80 (d, J = 4.0 Hz, 1H, CH–N), 4.19 (d, J = 4.0 Hz, 1H, CH), 4.02 (s, 2H, CH2), 3.02 (s, 6H, 2CH3); 13C NMR (100 MHz, DMSO-d6): δ 169.1 (C=O), 165.7 (C=O), 158.0 (C), 147.9 (C), 146.8 (C), 146.3 (C), 138.8 (C), 137.0 (C), 134.1 (C), 132.5 (C), 131.7 (C), 129.0 (CH), 127.1 (CH), 125.5 (CH), 124.7 (CH), 122.9 (CH), 122.2 (CH), 119.9 (CH), 113.31 (CH), 112.3 (CH), 112.0 (Thiazole-CH), 66.3 (CH–N), 62.1 (CH–Cl), 41.9 (CH3), 36.7 (CH2).
3-Chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-(4-nitrophenyl)azetidin-2-one (6f)
Pale brown crystals, yield (89%), m.p. 114–116 °C; FT-IR (KBr): ν (cm−1) 3071 (CH arom.), 2954 (CH aliph.), 1691 (C=O), 1591 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 8.17 (d, J = 8.0 Hz, 2H, Ph-H), 7.72 (d, J = 8.0 Hz, 1H, Flu-H), 7.66 (s, 1H, Flu-H), 7.51 (s, 1H, Flu-H), 7.44–7.40 (m, 3H, Ph-H & Flu-H), 7.27 (s, 1H, Flu-H), 7.21 (s, 1H, Thiazole-H), 7.02 (d, J = 8.0 Hz, 1H, CH–N), 4.28 (d, J = 8.0 Hz, 1H, CH–Cl), 4.03 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 169.1 (C=O), 165.8 (C=N), 158.0 (C), 147.8 (C), 146.8 (C), 146.3 (C), 140.5 (C), 138.8 (C), 137.0 (C), 132.5 (C), 132.1 (C), 129.3 (C), 129.0 (CH), 127.5 (C), 127.10 (CH), 125.8 (CH), 125.5 (CH), 124.7 (CH), 124.5 (CH), 124.1 (CH), 113.3 (Thiazole-CH), 66.8 (CH–N), 61.6 (CH–Cl), 36.9 (CH2).
4-(3-Chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-oxoazetidin-2-yl)benzoic acid (6g)
Pale yellow crystals, yield (76%), m.p. 125–127 °C; FT-IR (KBr): ν (cm−1) 3366 (OH), 3099 (CH arom.), 2956 (CH aliph.), 1691 (C=O), 1546 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 12.73 (s, 1H, OH), 8.03 (d, J = 8.0 Hz, 2H, Ph-H), 7.75 (d, J = 8.0 Hz, 1H, Flu-H), 7.70 (s, 1H, Flu-H), 7.64 (s, 1H, Flu-H), 7.52 (m, 2H, Flu-H), 7.43–7.39 (m, 2H, Ph-H), 7.26 (s, 1H, Thiazole-H), 7.21 (d, J = 12.0 Hz, 1H, CH–N), 4.26 (d, J = 12.0 Hz, 1H, CH–Cl), 4.01 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 169.1 (C=O), 165.8 (C=O), 158.7 (C=N), 148.2 (C), 146.8 (C), 146.2 (C), 138.8 (C), 137.0 (C), 132.5 (C), 132.1 (C), 131.7 (C), 130.4 (C), 130.0 (C), 128.9 (CH), 127.5 (CH), 127.08 (CH), 125.5 (CH), 124.6 (CH), 122.9 (CH), 122.1 (CH), 113.3 (Thiazole-CH), 65.9 (CH–N), 60.1 (CH–Cl), 36.9 (CH2).
3-Chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-(4-hydroxy-3-methoxyphenyl)-azetidin-2-one (8h)
Pale yellow crystals, yield (84%), m.p. 95–97 °C; FT-IR (KBr): ν (cm−1) 3365 (OH), 3067 (CH arom.), 2954 (CH aliph.), 1691 (C=O), 1546 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.90 (d, J = 8.0 Hz, 1H, Flu-H), 7.77–7.65 (m, 3H, Flu-H & Ph-H), 7.51 (s, 1H, Flu-H), 7.45 (d, J = 8.0 Hz, 1H, Flu-H), 7.40 (s, 1H, Ph-H), 7.27 (m, 2H, Ph-H & Thiazole-H), 7.21 (d, J = 12.0 Hz, 1H, CH–N), 6.97 (s, 1H, OH), 4.45 (s, 3H, CH3), 4.28 (d, J = 12.0 Hz, 1H, CH–Cl), 4.02 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 169.1 (C=O), 165.8 (C=N), 158.0 (C), 157.1 (C), 147.9 (C), 146.8 (C), 146.3 (C), 138.8 (C), 137.0 (C), 132.5 (C), 132.1 (C), 131.7 (C), 129.0 (CH), 127.7 (CH), 127.1 (CH), 125.8 (CH), 125.5 (CH), 124.6 (CH), 122.9 (CH), 122.1 (CH), 113.3 (Thiazole-CH), 67.2 (CH–N), 61.0 (CH–Cl), 56.8 (CH3), 36.9 (CH2).
3-Chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-styrylazetidin-2-one (6i)
Pale brown crystals, yield (60%), m.p. 110–112 °C; FT-IR (KBr): ν (cm−1) 3062 (CH arom.), 2951 (CH aliph.), 1702 (C=O), 1542 (C=N); 1H NMR (400 MHz, DMS-d6) δ 7.80 (s, 1H, Flu-H), 7.65–7.60 (m, 2H, Flu-H), 7.52 (d, J = 4.0 Hz, 1H, Flu-H), 7.34 (s, 1H, Flu-H), 7.30–7.27 (m, 3H, Ph-H), 7.21–7.16 (m, 3H, Ph-H & Thiazole-H), 7.12 (d, J = 12.0 Hz, 1H, CH–N), 6.80–6.73 (m, 2H, CH=CH), 4.40 (d, J = 12.0 Hz, 1H, CH–Cl), 4.01 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 169.1 (C=O), 165.8 (C=N), 158.0 (C), 147.8 (C), 146.8 (C), 146.3 (C), 138.8 (C), 137.0 (C), 132.5 (C), 132.1 (C), 131.6 (C), 129.57 (CH), 129.0 (CH), 128.4 (CH), 127.7 (CH), 127.1 (CH), 125.5 (CH), 124.6 (CH), 123.1 (CH), 122.8 (CH), 121.4 (CH), 113.3 (Thiazole-CH), 68.0 (CH–N), 61.3 (CH–Cl), 36.4 (CH2).
3-Chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-(furan-2-yl)azetidin-2-one (6j)
Pale yellow crystals, yield (95%), m.p. 95–98 °C; FT-IR (KBr): ν (cm−1) 3056 (CH arom.), 2952 (CH aliph.), 1695 (C=O), 1544 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.90 (d, J = 8.0 Hz, 1H, Flu-H), 7.77 (s, 1H, Flu-H), 7.66 (s, 1H, Flu-H), 7.51 (d, J = 4.0 Hz, 1H, Furan-H), 7.46–7.40 (m, 3H, Flu-H & Furan-H), 7.28 (d, J = 8.0 Hz, 1H, Flu-H), 7.20 (s, 1H, Thiazole-H), 7.11 (d, J = 12.0 Hz, 1H, CH–N), 4.22 (d, J = 12.0 Hz, 1H, CH–Cl), 4.02 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 168.4 (C=O), 165.7 (C=N), 158.9 (C), 148.4 (C), 146.8 (C), 146.3 (C), 138.8 (C), 137.0 (C), 132.5 (C), 132.1 (C), 131.7 (C), 129.0 (CH), 127.7 (CH), 127.10 (CH), 125.9 (CH), 125.5 (CH), 124.6 (CH), 122.9 (CH), 122.1 (CH), 113.3 (Thiazole-CH), 66.7 (CH–N), 62.0 (CH–Cl), 37.3 (CH2).
3-Chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-(thiophen-2-yl)azetidin-2-one (6k)
Yellow crystals, yield (51%), m.p. 109–111 °C; FT-IR (KBr): ν (cm−1) 3109 (CH arom.), 2951 (CH aliph.), 1691 (C=O), 1646 (C=N); 1H NMR (400 MHz, DMSO-d6) δ 7.92 (s, 1H, Flu-H), 7.72–7.55 (m, 2H, Flu-H & Thienyl-H), 7.51 (s, 1H, Flu-H), 7.44 (s, 1H, Flu-H), 7.40–7.32 (m, 2H, Thienyl-H & Thiazole-H), 7.28 (d, J = 4.0 Hz, 1H, Flu-H), 7.21 (d, J = 4.0 Hz, 1H, Thienyl-H), 7.06 (d, J = 12.0 Hz, 1H, CH–N), 4.28 (d, J = 12.0 Hz, 1H, CH–Cl), 4.03 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 169.1 (C=O), 165.8 (C=N), 158.9 (C), 147.8 (C), 146.8 (C), 146.3 (C), 142.1 (C), 138.8 (C), 137.0 (C), 132.3 (C), 131.7 (C), 129.0 (C), 128.9 (CH), 128.3 (CH), 127.1 (CH), 125.6 (CH), 124.6 (CH), 123.4 (CH), 121.2 (CH), 113.6 (CH), 112.4 (Thiazole-CH), 71.1 (CH–N), 61.0 (CH–Cl), 37.3 (CH2).
3-Chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-(1H-pyrrol-2-yl)azetidin-2-one (6l)
Green crystals, yield (93%), m.p. 105–107 °C; FT-IR (KBr): ν (cm−1) 3046 (CH arom.), 2954 (CH aliph.), 1705 (C=O), 1695 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.78 (s, 1H, Flu-H), 7.73–7.66 (m, 2H, Flu-H), 7.55–7.51 (m, 2H, Flu-H & Pyrrole-H), 7.46 (d, J = 8.0 Hz, 1H, Flu-H), 7.41 (s, 1H, Thiazole-H), 7.29–7.27 (m, 2H, Pyrrole-H), 7.21 (d, J = 8.0 Hz, 1H, CH–N), 4.45 (s, 1H, NH), 4.28 (d, J = 8.0 Hz, 1H, CH–Cl), 4.03 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 169.1 (C=O), 166.6 (C=N), 158.0 (C), 157.1 (C), 147.8 (C), 146.8 (C), 146.3 (C), 138.8 (C), 137.0 (C), 132.5 (C), 131.7 (C), 129.0 (CH), 127.7 (CH), 127.11 (CH), 125.9 (CH), 125.6 (CH), 124.6 (CH), 122.9 (CH), 122.2 (CH), 113.3 (Thiazole-CH), 67.0 (CH–N), 61.9 (CH–Cl), 36.9 (CH2).
3-Chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-(pyridin-4-yl)azetidin-2-one (6m)
Pale yellow crystals, yield (56%), m.p. 249–250 °C; FT-IR (KBr): ν (cm−1) 3096 (CH arom.), 2927 (CH aliph.),1772 (C=O), 1686 (C=N), 1597 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 7.89 (s, 1H, Flu-H), 7.65 (d, J = 4.0 Hz, 2H, Pyridine-H), 7.44 (d, J = 8.0 Hz, 1H, Flu-H), 7.40 (m, 1H, Flu-H), 7.32 (s, 1H, Flu-H), 7.27–7.20 (m, 3H, Flu-H & Pyridine-H), 7.11 (s, 1H, Thiazole-H), 7.02 (d, J = 12.0 Hz, 1H, CH–N), 4.27 (d, J = 12.0 Hz, 1H, CH–Cl), 4.01 (s, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 169.1 (C=O), 167.9 (C=N), 165.8 (C=N), 158.0 (C), 147.8 (CH), 146.5 (C), 146.3 (C), 138.8 (C), 137.0 (C), 132.2 (C), 132.0 (C), 131.6 (C), 128.9 (CH), 127.1 (CH), 125.6 (CH), 125.4 (CH), 124.6 (CH), 118.8 (CH), 116.8 (CH), 113.3 (Thiazole-CH), 70.1 (CH–N), 65.8 (CH–Cl), 36.9 (CH2).
3-Chloro-1-(4-(2,7-dichloro-9H-fluoren-4-yl)thiazol-2-yl)-4-(quinolin-4-yl)azetidin-2-one (5n)
Orange crystals, yield (58%), m.p. 175–177 °C; FT-IR (KBr): ν (cm−1) 3068 (CH arom.), 2852 (CH aliph.), 1771 (C=O), 1683 (C=N), 1584 (C=N); 1H NMR (400 MHz, DMSO-d6): δ 9.39–9.30 (m, 1H, Quinoline-H), 8.59 (d, J = 8.0 Hz, 1H, Quinoline-H), 8.45 (d, J = 8.0 Hz, 1H, Quinolin-H), 8.38 (d, J = 8.0 Hz, 1H, Quinolin-H), 8.27 (d, J = 12.0 Hz, 1H, Quinolin-H), 8.21–8.19 (m, 1H, Quinolin-H), 7.93 (s, 1H, Flu-H), 7.66–7.53 (m, 2H, Flu-H), 7.41 (s, 1H, Flu-H), 7.29–7.14 (m, 2H, Flu-H & Thiazole-H), 7.06 (m, 1H, CH–N), 4.27 (m, 1H, CH–Cl), 4.02 (s, 2H,CH2); 13C NMR (100 MHz, DMSO-d6): δ 169.1 (C=O), 167.9 (C=N), 165.8 (C=N), 158.2 (CH), 158.0 (C), 147.1 (C), 146.9 (C), 146.3 (C), 141.6 (C), 140.5 (C), 138.7 (C), 134.7 (C), 133.0 (C), 128.7 (CH), 127.1 (CH), 126.8 (CH), 125.5 (CH), 125.1 (CH), 124.6 (CH), 124.5 (CH), 123.7 (CH), 123.5 (CH), 121.8 (CH), 113.9 (Thiazole-CH), 67.0 (CH–N), 62.5 (CH–Cl), 34.5 (CH2).
Antimicrobial screening
Used microorganisms
All microbial strains were kindly provided from the department of Medical Microbiology and Immunology faculty of Medicine Assiut University, these clinical isolates were obtained from clinical cases of infections admitted to Assiut University hospital as urinary tract infections, corneal ulcers, bacterial and fungal pneumonia, otomycosis, oral thrush and wound infections. The clinical isolates were proved by using the VITEK 2 automated microbiology system (BioMérieux).
The clinical isolates used were multidrug resistant strains, they were resistant to β lactam (penicillin, amoxacillin, oxacillin), cephalosporins (cefazolin, cefaclor and cefepime) and macrolides (erythromycin and clarithromycin), they included Gram positive bacteria as Staphylococcus aureus (S. aureus), Methicillin-resistant Staphylococcus aureus (MRSA), Streptococcus pneumoniae (S. pneumoniae), and Gram negative bacteria as Escherichia coli (E. coli), Klebsiella pneumoniae (K. pneumoniae), Pseudomonas aeruginosa (P. aeruginosa), and Acinetobacter baumannii (A. baumannii). The fungal strains that were tested are Aspergillus flavus (A. flavus), A. niger (A. niger) and Candida albicans (C. albicans).
Initial evaluation of the fluorene derivatives antibacterial and antifungal activity
The antimicrobial activity of the fluorene derivatives was initially evaluated by agar well diffusion assay [40]. Mueller–Hinton agar (CM0337) was poured into Petri dishes at 50–60 °C and left to solidify for 15 min. Subsequently, overnight microbial suspensions of tested strains was adjusted to turbidity of 0.5 McFarland Standard, which equals to 1–2 × 108 CFU/mL for bacteria and 1–5 × 106 for fungi. The microbial inoculums were then diluted in 1:100 ratio in case of bacteria and 1:10 ratio in case of fungi in order to get 1–5 × 105 CFU/mL. a sterile cotton swab was dipped into the adjusted microbial suspension and the Mueller–Hinton agar plates were inoculated by evenly streaking cotton swab over the agar medium. Then wells with a diameter of 0.5 cm were cut in the medium with a sterile cork borer. Stock solutions of the flourene derivatives were diluted in DMSO 1% to get 500 μg/mL concentrations. The tested flourene derivatives and controls (50 μL) were dispensed into the wells. The plates were incubated for 24 h at 37 °C for bacteria and C. albicans while at 25 °C for A. falvus and A. niger. The diameters of zones of inhibition (ZOI) around the wells were measured in mm. Following control agents were used: positive control agents—vancomycin (50 μg/mL) for Gram positive bacteria, gentamicin (10 μg/mL) (for Gram negative bacteria) and fluconazole 25 μg/mL for fungi and negative control agent is 1% DMSO.
Determination of MIC values for the most active fluorene derivatives
Determination of Minimum inhibitory concentrations (MIC) of flourene derivatives was done using broth microdilution method [41]. The procedure involved preparation of twofold dilutions of the fluorene derivatives ranging from (500–7.8 μg/mL) in sterile Mueller–Hinton broth inside the wells of 96-well microplate (Sarstedt, Germany). The inoculums of test strains prepared from fresh overnight cultures were adjusted to 0.5 McFarland standards, which equals to 1–2 × 108 CFU/mL for bacteria, the procedure was done according to CLSI 2012 [42]. The highest dilution of samples (flourene derivatives) without visible growth after 24 h incubation at 37 °C was considered as MIC. For this assay the positive control agents were vancomycin (range: 0.7–50 μg/mL), gentamicin (range: 0.15–10 μg/mL) and the negative control was 1% DMSO.
For proper determination of the MIC end point resazurin dye has been used. A stock solution of resazurin sodium salt powder (Titan Biotech) was prepared at 0.02% (wt/vol) in distilled water, sterilized by filtration through a 0–2 µm filter into a sterile light protected container then stored protected from light at 4 °C for up to 1 week, or at − 20% for long term use, then 10–15% resazurin solution of the total volume in wells was added to each well and incubation for 1–4 h at 37 °C was done. A change in color from blue to pink indicates the growth of bacteria, and MIC was defined as the lowest concentration of the drug that prevented this change in color.
Data processing
All experiments were independently repeated three times. Obtained data were processed; standard deviations were calculated using GraphPad Prism 5.03 (GraphPad Software, Inc.; USA) software.
Media and reagents:
Muller Hinton agar oxoid code: CM0337
Muller Hinton broth oxoid code: CM 0405
Mannitol salt agar oxoid code: CM 0151
Columbia agar oxoid code: CM 0331
Orsab oxoid code CM 1008
Nutrient agar oxoid code: CM0003
Eosin methylene Blue Himedia M317
Equipment:
Petri dishes
Crock borer
Sterile syringe needle and swabs
Microtitre plates
Micropipette
Sterile tips
Cytotoxicity screening
Cell culture
WI-38 normal lung fibroblast cells, A549 lung cancer cells, and MDA-MB-231 breast cancer cells were obtained from VACSERA—Cell Culture Unit, Cairo, Egypt. The cell lines were originally obtained from the American Tissue Culture Collection (ATCC). WI-38, A549, and MDA-MB-231 cell lines were cultured in RPMI-1640 medium supplemented with 10% inactivate fetal bovine serum (FBS) and 1% penicillin/streptomycin were bought (Gibco, Invitrogen, CA).
Cell viability assay
WI-38, A549, and MDA-MB-231 cells were seeded into 96-well plates (at a density of 5000 cells/well). On the following day, cells were treated with different concentrations (0, 1, 10, 31.25, 62.5, 125, 250, 500 µg/mL) of 16 fluorene derivatives in fresh medium and incubated for another 24 h. Cell viability was then assessed using the MTT assay (Sigma Aldrich, St. Louis, MO, USA), and the absorbance was read at 570 nM using an ELISA microplate reader (Molecular Devices, Downingtown, PA, USA).
FACS analysis
To uncover the mechanism of cell death for the compounds 5h, 6c, 6d and 6k on WI-38, A549, and MDA-MB-231 cells; Annexin v and propedium Iodide (PI) were used. In brief, WI-38, A549, MDA-MB-231 cells were cultured in 10 tissue culture dish with initial number 4 × 105 cell/Ml in RPMI growth media. In the following day, cells were treated with 6c, 6d and 6k as the following; (0.0, 500 µg/mL form each drug for WI-38 treatment, 0.0, 85, 117 and 200 µg/mL; respectively, for A5489 and 0.0, 250 from each for MDA-MB-231 cells treatment). After 24 h incubations, cells were washed and trypsinized and suspended in 50 µL 1X Annexin v binding buffer followed by adding 5 µL FITC Annexin V and incubated for 15 min at room temperature then 5 µL of PI were added to each tube. Finally, 400 µL of 1X Annexin v binding buffer were added to each tube and analyzed using Becton–Dickinson FACS Caliber.
Availability of data and materials
Additional information with NMR spectra and molecular docking are attached.
Abbreviations
- FACS:
-
Fluorescence-activated cell sorting
- HIV:
-
Human immunodeficiency virus
- DHFR:
-
Dihydrofolate reductase
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- DHF:
-
7,8-Dihydrofolate
- THF:
-
5,6,7,8-Tetrahydrofolate
- DNA:
-
Deoxyribonucleic acid
- DCM:
-
Dichloromethane
- THF:
-
Tetrahydrofuran
- DCC:
-
N,N′-Dicyclohexylcarbodiimide
- DMF:
-
Dimethylformamide
- FT-IR:
-
Fourier transform infrared
- 1H NMR:
-
Proton nuclear magnetic resonance
- 13C NMR:
-
Carbon-13 nuclear magnetic resonance
- DEPT-135:
-
Distortionless enhancement by polarization transfer
- ZOI:
-
Zone of inhibition
- S. aureus :
-
Staphylococcus aureus
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- S. pneumoniae :
-
Streptococcus pneumoniae
- E. coli :
-
Escherichia coli
- K. pneumoniae :
-
Klebsiella pneumoniae
- P. aeruginosa :
-
Pseudomonas aeruginosa
- A. baumannii :
-
Acinetobacter baumannii
- A. flavus :
-
Aspergillus flavus
- A. niger :
-
Aspergillus niger
- C. albicans :
-
Candida albicans
- MIC:
-
The minimum inhibitory concentration
- DMSO:
-
Dimethyl sulphoxide
- MOE:
-
Molecular Operating Environment
- CCG:
-
Chemical Computing Group
- RMSD:
-
The root-mean-square deviation
References
Gualtieri F, Teodori E, Bellucci C, Pesce E, Piacenza G (1985) Structure-activity relationship studies in the field of calcium (II) antagonists. Effect of modifications at the tetrasubstituted carbon of verapamil-like compounds. J Med Chem 28:1621–1628
Fun H-K, Yeap CS, Vijesh AM, Isloor AM, Vasudeva PK (2010) 2,7-Dichloro-4-(chloro-acet-yl)fluorine. Acta Cryst E66:2624–2625
Abdella AM, Moatasim Y, Ali MA, Elwahy AHM, Abdelhamid IA (2017) Synthesis and Anti-influenza Virus Activity of Novel bis(4H-chromene-3-carbonitrile) Derivatives. J Heterocycl Chem 54:1854–1862
Ibrahim NS, Mohamed MF, Elwahy AHM, Abdelhamid IA (2018) Biological activities and docking studies on novel Bis 1,4-DHPS linked to arene core via ether or ester linkage. Lett Drug Des Discov 15:1036–1045
Salama SK, Mohamed MF, Darweesh AF, Elwahy AHM, Abdelhamid IA (2017) Molecular docking simulation and anticancer assessment on human breast carcinoma cell line using novel bis(1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile) and bis(1,4-dihydropyrazolo-[4′,3′:5,6]pyrano[2,3-b]pyridine-6-carbonitrile) derivatives. Bioorg Chem 71:19–29
Mohamed MF, Darweesh AF, Elwahy AHM, Abdelhamid IA (2016) Synthesis, characterization and antitumor activity of novel tetrapodal 1,4-dihydropyridines: p53 induction, cell cycle arrest and low damage effect on normal cells induced by genotoxic factor H2O2. RSC Adv 6:40900–40910
Mohamed MF, Abdelmoniem AM, Elwahy AHM, Abdelhamid IA (2018) DNA fragmentation, cell cycle arrest, and Docking study Of novel Bis Spiro-cyclic 2-oxindole of Pyrimido[4,5-b]quinoline-4,6-dione derivatives against breast carcinoma. Curr Cancer Drug Targets 18:372–381
Mohamed MF, Hassaneen HM, Abdelhamid IA (2018) Cytotoxicity, molecular modeling, cell cycle arrest, and apoptotic induction induced by novel tetrahydro-[1,2,4]triazolo[3,4-a]isoquinoline chalcones. Eur J Med Chem 143:532–541
Salar U, Taha M, Khan KM, Ismail NH, Imran S, Perveen S, Gul S, Wadood A (2016) Syntheses of new 3-thiazolyl coumarin derivatives, in vitro α-glucosidase inhibitory activity, and molecular modeling studies. Eur J Med Chem 122:196–204
Aggarwal R, Kumar S, Kaushik P, Kaushik D, Gupta GK (2013) Synthesis and pharmacological evaluation of some novel 2-(5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazol-1-yl)-4-(coumarin-3-yl)thiazoles. Eur J Med Chem 62:508–514
Xu ZL, Guo J, Yang Y, Zhang M, Ba M, Li ZZ, Cao Y, He R, Yu M, Zhou H, Li X, Huang X, Guo Y, Guo C (2016) 2,4,5-Trisubstituted thiazole derivatives as HIV-1 NNRTIs effective on both wild-type and mutant HIV-1 reverse transcriptase: optimization of the substitution of positions 4 and 5. Eur J Med Chem 123:309–316
Satish K, Deepika RKC, Sreenivas A, Jayaram Reddy K, Srigiridhar K, Rambabu Y (2014) Synthesis and anticancer evaluation of 3-aryl-6-phenylimidazo[2,1-b]thiazoles. Bioorg Med Chem Lett 24:5428–5431
Karuvalam RP, Haridas KR, Nayak SK, Guru Rowb TN, Rajeesh P, Rishikesan R, Suchetha Kumari N (2012) Design, synthesis of some new (2-aminothiazol-4-yl)methylester derivatives as possible antimicrobial and antitubercular agents. Eur J Med Chem 49:172–182
Lu X, Liu X, Wan B, Franzblau SG, Chen L, Zhou C, You O (2012) Synthesis and evaluation of anti-tubercular and antibacterial activities of new 4-(2,6-dichlorobenzyloxy)phenyl thiazole, oxazole and imidazole derivatives. Part 2. Eur J Med Chem 49:164–171
Diurno MV, Mazzoni O, Piscopo E, Calignano A, Giordano F, Bolognesell A (1992) Synthesis and antihistaminic activity of some thiazolidin-4-one. J Med Chem 35:2910–2912
Kumar A, Rajput C, Bhati S (2007) Synthesis of 3-[4′-(p-chlorophenyl)-thiazol-2′-yl]-2-[(substituted azetidinone/thiazolidinone)-aminomethyl]-6-bromo-quinazolin-4-ones as anti-inflammatory agent. Bioorg Med Chem 15:3089–3096
Nampurath GK, Mathew SP, Khanna V, Zachariah RT, Kanji S, Chamallamudi MR (2008) Assessment of hypolipidaemic activity of three thiazolidin-4-ones in mice given high-fat diet and fructose. Chem Biol Interact 171:363–368
Patel NB, Patel SD (2009) Synthesis and antimicrobial activity of 2-phenyl-3-{1-cyclopropyl-6-fluoro-7-[4-methylpiperazin-1-yl]-4-quinolone}carboxamido-3-thiazolidin-4-ones. Pharm Chem J 43:305
Kaur H, Kumar S, Vishwakarma P, Sharma M, Saxena K, Kumar A (2010) Synthesis and antipsychotic and anticonvulsant activity of some new substituted oxa/thiadiazolylazetidinonyl/thiazolidinonylcarbazoles. Eur J Med Chem 45:2777–2783
Rojas Ruiz FA, Garcia-Sanchez RN, Estupinan SV, Barrio AG, Torres Amado DF, Perez-Solorzano BM, Nogal-Ruiz JJ, Martinez-Fernandez AR, Kouznetsov VV (2011) Synthesis and antimalarial activity of new heterocyclic hybrids based on chloroquine and thiazolidinone scaffolds. Bioorg Med Chem 19:4562–4573
Appalanaidu K, Kotcherlakota R, Dadmal TL, Bollu VS, Kumbhare RM, Patra CR (2016) Synthesis and biological evaluation of novel 2-imino-4-thiazolidinone derivatives as potent anti-cancer agents. Bioorg Med Chem Lett 26:5361–5368
Solankee AN, Patel KP, Patel RB (2012) A facile synthesis and studies of some new 4-thiazolidinones and 5-arylidenes. Pelagia Res Lib 3:117–122
Udupi RH, Kasinath N, Bhat AR (1998) Synthesis and biological activity of Mannich bases of certain 1,2-pyrazolines. Indian J Heterocycl Chem 17:221–224
Singh GS, Mbukwa E, Pheko T (2007) Synthesis and antimicrobial activity of new 2-azetidinones from N-(salicylidene) amines and 2-diazo-1, 2-diarylethanones. Arkivoc 9:80–90
Chavan S, Zangade S, Vibhute A, Vibhute Y (2013) Synthesis and antimicrobial activity of some novel 2-azetidinones and 4-thiazolidinones derivatives. Eur J Chem 4:98–101
Patel RB, Desai PS, Desai KR, Chikhalia KH (2006) Synthesis of pyrimidine based thiazolidinones and azetidinones: antimicrobial and antitubercular agents. Indian J Chem B 45:773–778
Blakley RL (1984) Dihydrofolate reductase. In: Blakley RL, Benkovic SJ (eds) Folates and pteridines. Wiley, New York, pp 191–253
Brown KA, Kraut J (1992) Exploring the molecular mechanism of dihydrofolate reductase. Faraday Discuss 93:217–224
Hawser S, Lociuro S, Islam K (2006) Dihydrofolate reductase inhibitors as antibacterial agents. Biochem Pharmacol 71:941–948
Hussein EM, Al-Rooqi MM, Abd El-Galil SM, Ahmed SA (2019) Design, synthesis, and biological evaluation of novel N4-substituted sulfonamides: acetamides derivatives as dihydrofolate reductase (DHFR) inhibitors. BMC Chem 13:91
Hussein EM, Alsantali RI, Abd El-Galil SM, Obaid RJ, Alharbi A, Abourehab MAS, Ahmed SA (2019) Bioactive fluorenes. part I. Synthesis, pharmacological study and molecular docking of novel dihydrofolate reductase inhibitors based-2,7-dichlorofluorene. Heliyon 5:e01982
Hussein EM, Al-Shareef HF, Aboellil AH, Elhady HA (2015) Synthesis of some novel 6′-(4-chlorophenyl)-3,4′-bipyridine-3′-carbonitriles: assessment of their antimicrobial and cytotoxic activity. Z Naturforsch 70B:783–795
Hussein EM, Masaret GS, Khairou KS (2015) Efficient synthesis and antimicrobial evaluation of some Mannich bases from 2-arylidine-1-thia-4-azaspiro[4.5]decan-3-ones. Chem Cent J 9:25
Al-Shareef HF, Elhady HA, Aboellil AH, Hussein EM (2016) Ammonium chloride catalyzed synthesis of novel Schiff bases from spiro[indoline-3,4′-pyran]-3′-carbonitriles and evaluation of their antimicrobial and anti-breast cancer activities. SpringerPlus 5:887
Bush K, Freudenberger JS, Slusarchyk DS, Sykes RB, Meyers E (1982) Activity of sulfa drugs and dihydrofolate reductase inhibitors against Candida albicans. Experientia 38:436–437
Rao KN, Venkatachalam SR (1999) Dihydrofolate reductase and cell growth activity inhibition by the β-carboline-benzoquinolizidine plant alkaloid deoxytubulosine from Alangium lamarckii: its potential as an antimicrobial and anticancer agent. Bioorg Med Chem 7:1105–1110
Tse-Dinh Y-C (2015) Targeting bacterial topoisomerase I to meet the challenge of finding new antibiotics. Future Med Chem 7:459–471
Vivas R, Barbosa AAT, Dolabela SS, Jain S (2019) Multidrug-resistant bacteria and alternative methods to control them: an overview. Microbial Drug Resistance 25:890–908
Ginovyan M, Keryan A, Bazukyan I, Ghazaryan P, Trchounian A (2015) The large scale antibacterial, antifungal and anti-phage efficiency of Petamcin-A: new multicomponent preparation for skin diseases treatment. Ann Clin Microbiol Antimicrob 14:28
Wiegand I, Hilpert K, Hancock RE (2008) Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc 3:163–175
CLSI (2012) Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standards—ninth edition
Khalifa RA, Nasser MS, Gomaa AA, Osman NM, Salem HM (2013) Resazurin Microtiter Assay Plate method for detection of susceptibility of multidrug resistant Mycobacterium tuberculosis to second-line anti-tuberculous drugs. Egypt J Chest Dis Tuberc 62:241–247
Acknowledgements
S. A. Ahmed is highly indebted to Alexander von Humboldt Foundation (AvH) and Prof. Dr. Karola Rück-Braun, Technical University Berlin (TU-Berlin), Germany for their non-stopping help and support.
Funding
Funding from the Deanship of the Scientific Research (DSR), Umm Al-Qura University through the project number 18-SCI-1-01-0009 is acknowledged. The funding body used in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
EMH and SAA effectively contributed in designing of the experiments, methodology, data analysis and curation, writing and publishing the manuscript. RIA has contributed in data collections and analysis. MM, RJO, HMA, AS and MASA have contributed in reagents, materials, analysis tools or data. AAE and ASMA, performed the anticancer, antimicrobial activities and molecular docking measurements. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing of interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
NMR spectra, docking and molecular modeling calculations of invetigated bioactive fluorenes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hussein, E.M., Alsantali, R.I., Morad, M. et al. Bioactive fluorenes. Part III: 2,7-dichloro-9H-fluorene-based thiazolidinone and azetidinone analogues as anticancer and antimicrobial against multidrug resistant strains agents. BMC Chemistry 14, 42 (2020). https://doi.org/10.1186/s13065-020-00694-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-020-00694-2