
Abstract
A new series of indole-based-sulfonamide derivatives (A1–A8) was synthesized by treating 5-fluoro-1H-indole-3-carbohydrazide with different aryl-sulfonyl chloride in the presence of pyridine. All synthesized derivatives (A1–A8) were characterized by different analytical methods. The electrochemical behavior of these compounds (A1–A8) was investigated in detail using cyclic voltammetry (CV) and square wave voltammetry (SWV) at the pencil graphite electrode (PGE). In the present study, the redox behavior of all derivatives varies due to the nature of substitutions in the indole sulfonamide moiety. Various fundamental electrochemical parameters, including the standard heterogeneous rate constants (ks), and the electroactive surface coverage (Г) were calculated from the obtained CVs. The obtained results shed light on the understanding of structure–activity relationships of this class of compounds.

Similar content being viewed by others


Introduction
Indole ring exists in several naturally occurring alkaloids [1]. Indole derivatives were reported to have biological effects [2], anti-inflammatory [3], antitubercular [4], and antimicrobial [5] activities. Thio-indole based analogs act as antifungal [6], antimicrobial [7], antibacterial, analgesic [8, 9], anticonvulsant [10], antioxidant [11], antidepressant [12], antihypertensive [13], and antiviral agents [14]. Indole sulfonamide undergoes substitution, mainly at the C3 position, possess hydrophilic features like sulfonyl group, and is regarded as an appropriate pharmacophore equivalent for replacing active sites in drug design [15,16,17]. Consequently, the redox properties of the indole sulfonamide may be very useful for their efficient uses. It is well known that the electroactive indole derivatives are easily oxidized at carbon-based electrodes [18]. Accordingly, the knowledge of the electrochemical oxidation of these compounds is helpful to understand their action mechanism as well as pharmacokinetic and pharmacodynamic purposes in chemical and biological processes, if they find use as drugs [19]. The electrochemical techniques have been effectively applied to study the redox properties of several biologically important compounds [20, 21]. As reported previously, the redox behavior of indole derivatives was investigated using various electrodes: pyrolytic carbon [22], glassy carbon [17, 23], platinum [24], lead dioxide [25] boron-doped diamond [26, 27], and other electrodes [28,29,30]. Among the studied indole compounds with a substituent at C3, the results of electrochemical oxidation of indole-3-acetic acid, indole-3-propionic acid, indole-3-butyric acid, indol-3-acetamide, tryptamine, gramine and tryptophan were reported for the measurements done with the usage of a glassy carbon electrode as the working electrode [23, 31]. The oxidation behavior of indole derivatives in aqueous solutions is more complex and suggested an irreversible pH-dependent process. Two oxidation peaks were found, the first peak corresponds to the oxidation at position C2 on the pyrrole ring and the second peak to electrochemical hydroxylation at the position C7 on the benzene moiety. Herein, we report the synthesis of eight biologically important indole-based-sulfonamide compounds with a different aryl-sulfonyl substituent at C3. Furthermore, the electrochemical properties for these new compounds were investigated using CV and SWV techniques at PGE. Based on the obtained results, the redox behavior was proposed to highlight important aspects of structure–activity relationships of this class of compounds.
Result and discussion
Synthesis of indole-based-sulfonamide derivatives
The synthesis of indole-based-sulfonamide derivatives (A1–A8) began with the synthesis of 5-fluoro-1H-indole-3-carbohydrazide (I) which was synthesized by heating under reflux methyl 5-fluoro-1H-indole-3-carboxylate with mixture of hydrazine and methanol. 5-Fluoro-1H-indole-3-carbohydrazide (I) reacted with different aryl sulfonyl chloride in the presence of pyridine. All synthesized compounds (A1–A8) were fully characterized by different spectroscopic methods Scheme 1.

Synthesis of indole-based-sulfonamide derivatives A1–A8
Electrochemical properties of Indole-Based-Sulfonamide Derivatives
Cyclic voltammetric studies
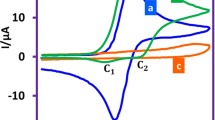
The electrochemical properties of the indole sulfonamide derivatives (A1–A8) were studied in comparison to the parent (A1) using CV. The CVs of 3 × 10−5 M of the investigated compounds (A1–A8) recorded in 10% aqueous ethanol (as optimum ethanol/H2O ratio) at pH 7.4 using pencil graphite electrode (PGE) as working electrode, Ag/AgCl as reference electrode and Pt wire as counter electrode at ambient temperature (Figs. 1a, b). The overlapping CV behavior of all compounds have come through the existence of the only one well-defined oxidation peak within the investigated potential range (from 0.0 to + 0.8 V). A well-known anodic peak (Pa) was developed from the oxidation of the analyte species in the positive scan for all compounds however; no cathodic peak/s was observed in the negative potential scan revealed the stability of the analyte species to reduction. The presence of a sole oxidation peak for all compounds suggests an irreversible electrochemical process. However, the values of Epa − Epa/2 between 50 and 70 mV (Table 1) confirmed the irreversibility of the electrochemical oxidation process.

CVs of 3 × 10−5 M of indole sulfonamide compounds (a) A1–A4 and (b) A5–A8 in PBS of pH 7.4 obtained at PGE scan rate, 0.10 Vs−1
The electrochemical oxidation active center of indole derivatives is the indole moiety, however the reduction of the sulfonyl group (–SO2–) is very difficult to attain [23, 32]. Figure 2 recorded the successive CV scans of A4 in the same analyte solution without changing the PGE surface. A significant decrease in peak height was observed with subsequent scans indicating that the oxidized products are strong adsorbed on the surface of PGE, hence blocking it for further sensing of the analyte. Similar electrochemical behavior was also observed for the other compounds (Additional file 1: Fig. S1). To support the working hypothesis the indole moiety in A1–A8 that undergoes oxidation, the oxidation behavior of A1 was compared with a model compound. As model substances for the oxidation of indole ring, the 5-fluoro-1H-indole-3-carbohydrazide (I) was used. The potential range, in which the model compound (I) is oxidized is comparable to that observed for oxidation in sulfonamide compound (A1) (Fig. 3). A small couple appears at 275 mV for (I), may be due to adsorption complications. By comparing the chemical structure of (I) with that of A1, it is observed that A1 contains additional substitution by benzosulfone moiety. Such substitution should not influence the mechanism of the electrochemical oxidation process. These results indicated that the potential at which the oxidation occurs is strongly dependent on the nature, position and number of substituents on benzene ring of the investigated compounds (Table 1). As reported previously [33,34,35], the oxidation of indole compounds, with a substituent at C3 position, in aqueous solutions is more complex and suggested an irreversible pH-dependent process. Also, it is well known the benzene ring of indoles is less reactive than the pyrrole ring, since the oxidation reaction for all indole derivatives (A1–A8), which present mainly one oxidation peak at the potential range from 240 mV to 300 mV, corresponds to the oxidation at C2 position on the pyrrole ring [23, 31, 36].

CVs (scan 1–10) of 3.0 × 10-5 M of A4 in PBS of pH 7.4 obtained at PGE scan rate, 0.10 Vs−1

CVs of 3x10-5 M of 5-fluoro-1H-indole-3-carbohydrazide (1) and compound A1 (2) in PBS of pH 7.4 obtained at PGE scan rate, 0.10 Vs−1
Effect of pH
CVs of the compounds under investigation were also recorded in phosphate buffer solution (PBS) at different pH values (from pH 3 to pH 11). The results indicated that, in acidic buffer solution the oxidation peak potential appeared at higher positive values and shifted to lower values with an increase in pH, which indicating that the electron transfer process is more easily in neutral and basic buffer solution. It is apparent that below pH 7 the -NH group of indole moiety is protonated to a great extent and therefore presumably the oxidation of compounds A is more difficult. These results suggest the involvement of H+ ion in the electron transfer process (Additional file 1: Fig. S2) [37]. Many studies reported that organic compounds showing pH-dependent oxidation undergo deprotonation reaction through oxidation [37, 38]. As shown in Table 2, the shifts of peak potential (Epa) with pH for all indole sulfonamide derivatives are linear with slopes in the range from 54 mV to 66 mV (R from 0.993 to 0.999). These data (the slope of ∼ 59 mV per pH unit) suggested that the electrochemical oxidation processes of the investigated compounds include the same number of protons and electrons [38]. Depend on the equation dEp/dpH = 0.059x/αn, the number of proton (x) was calculated to be ~ 1. In this manner, the oxidation reaction of the investigated compounds on the surface of PGE is a one-proton and one-electron irreversible process.
Effect of substitutions
The redox data for the compounds A2–A8 were compared to those of parent A1 and summarized in Table 1. It was found that, the oxidation peak potential of these compounds A2–A8 varies, because of the different substituent in the indole moiety. The appearance of the oxidation potentials of compounds A2–A8 at more positive potential values than the parent A1 (240 mV), indicated the most difficult electron transfer process due to the electron-withdrawing nature of substituents. It can be observed that oxidation potentials of the compounds A2–A8 increase with the increasing degree of electron-withdrawing groups, as noticed between A3 (3-NO2) and A4 (4-F), or between A4 (4-F), and A5 (4-OCF3). Compared to the parent compound A1, compound A8 (4-CH(CH3)2) has the lowest Epa (234 mV) among the all derivatives listed, and compound A7 (3,4-diOCH3) has the highest Epa (302 mV) due to its two electron-withdrawing substituents. Easily oxidation of A8 is attributed to the electron-donating effect of the isopropyl group (4-CH(CH3)2). This is as expected because electron-withdrawing groups increase Epa of a compound by removing electron density from the π system and thus more energy is needed to remove an electron, whereas electron-donating substituents decrease the oxidation potential. The electrochemical behavior of these compounds indicated that by varying the electronic properties of the substitution the anodic peak potential of the electrophore can be modified.
Effect of scan rate
The dependence of the anodic electrochemical process for all indole sulfonamide derivatives on the scan rate was examined at pH 7.4 at various sweep rates (Fig. 4). As shown in Fig. 4a by increasing the scan rate (v), the anodic peak potential (Epa) shifted toward the positive direction which confirms the irreversibility of the electrochemical process of indole sulfonamide derivatives [39]. Experimental plots of IPa vs. ν are shown in Figs. 4b. It was found that the peak currents increased linearly with the scan rate, proposing the electrochemical reactions were an adsorption-controlled step rather than a diffusion-controlled process [40]. This result was also confirmed from the data analysis through a plot of log Ipa vs. log ν, which reveals slope values in the range from 0.62 to 0.78 (Additional file 1: Fig. S3 and Table 1). These data propose a mainly adsorption-controlled mass transfer process [41]. As for an irreversible electrode process, the Laviron equation [42], was used to calculate αn and ks values as follows:
where ks is the standard heterogeneous electron transfer rate constant of the surface reaction, α is the electron transfer coefficient, ν is the scan rate, Eo is the formal redox potential and n is the electron transfer numbers. Other symbols have their common meanings. The Eo value at PGE can be deduced from the intercept of Epa vs. ν plot on the ordinate by extrapolating the line to ν = 0. Thus, the values of αn and ks (s−1) for all investigated compounds were obtained from the slope and intercept of the linear plot of Epa vs. ln ν, respectively and tabulated in Table 1 (Additional file 1: Fig. S4). Where, α was supposed as 0.5, for an irreversible electron transfer, the n was calculated to be ~ 1 which suggested that one electron was involved in the oxidation process. These results indicate the direct electron transfer on the surface of the PGE. The ks values with an order of 103 s−1 characterize the indole sulfonamide oxidation process to be irreversible. Furthermore, the higher values of ks showing faster electron transfer kinetics at pH 7.4 can be ascribed to the closest approach of indole sulfonamide at the surface of PGE due to the probable compression of the electrical double layer under these conditions. In this context, the electrochemical reaction process for the indole sulfonamide derivatives in aqueous solution (pH 7.4) at PGE occurs with the transfer of one electron and one proton and may be written as shown in scheme 2. This reaction mechanism in agreement with the previous studies [23, 31, 36]. These results displayed that the electrochemical oxidation of indole compounds with a substituent at C3 position occurs with a similar mechanism, with the only differences in the currents and the oxidation potentials due to the different substituent in C3 position of the pyrrole ring which does not change the oxidation pathway. Moreover, indole containing compounds are oxidizable derivatives, explanation of their oxidation pathways can introduce a model for the mechanism study of electron exchange between indole-containing enzymes or proteins and solid surfaces, and can be used for constructing new biomedical and biosensors devices.

a CVs of 6.5 × 10−5 M of A1 on surface of PGE at various scan rates; (1) 10, (2) 20, (3) 50, (4) 100, (5) 200, (6) 300 and (7) 500 m Vs−1 in PBS of pH 7.4. b Plot of Ipa (µA) vs. ν (m V s−1)

Proposed oxidation mechanism of indole sulfonamide derivatives at PGE
The surface coverage for all sulfonamide compounds (Г, mol cm−2) was further estimated from the calculated charge (Q) under the reductive CV peak. Where the adsorbed amount of the investigated compounds is proportional to the total charge (Q) consumed during the oxidation of adsorbed molecules according to equation [43]:
where Q is the charge involved in the electrooxidation process, n is the number of electrons, F is the Faraday constant and A is working electrode surface area. The Γ values of the investigated compounds at PGE were estimated and listed in the Table 1. The results showed that the 4-substiuted sulfonamide derivatives exhibit the largest tendency for specific adsorption.
The magnitude of the Г decreases in the order A5 > A4 > A7 > A6 > A2 > A1 > A3 > A8. This can be easily expected by the effect of the specific adsorption of 4-substituted withdrawing groups (A2, A4–A7) compared to 3-substituted withdrawing group (A3) or 3-substituted donating groups (A8). On the other hand, the Г value for compound A1 (R = H) was found to be less than 4-substituted compounds and greater than 3-substituted compounds. The large value of Γ for A5 confirmed that the presence of 4-OCF3 strongly affected the adsorption character of this sulfonamide compound compared to other substitutions, which proposing that the A5 molecules assume a perpendicular orientation at the electrode surface due to the enhance of the strength of stacking interactions between adjacent molecules. The results also indicate that the degree of adsorption of sulfonamide derivatives is influenced not only by the nature of the substitution but also by the position of this substitution.
Square wave voltammetry
The SWV behavior of the compounds under investigation was also studied in PBS (10% ethanol) of pH 7.4 at PGE (Fig. 5). Like a CV, an oxidation peak of A1–A8 was registered which attributed to the oxidation of carbohydrazide group. It was found that, the SWV oxidation peak potential of these compounds A1–A8 shifted to more positive potential in the sequence: A8 > A1 > A4 > A6 > A3 > A2 > A5 > A7. As expected, these results indicate that the electron-withdrawing groups shifted the SWV oxidation peak to a more positive potential, whereas the electron-donating substituents decrease the oxidation potential of these compounds. A broad oxidation peak was observed for A5 which confirm a multi-steps of oxidation process. Whereas, SWV of A3 and A8 showed two anodic peaks due to two-steps oxidation, thus complementing CV results which appear as a broad oxidation peak. Moreover, Ipcvs concentration plot is linear in lower concentration range (Example for A7: Ipa = 0.965 + 1.05 × 105 X, R = 0.995) which reflects diffusion-controlled nature of the oxidation process, and at higher concentration plot shows a tendency to limit, which indicates the involvement of adsorption complication (Fig. 6).

SWV of 3 × 10−5 M of A1, A2, A3, A6 and A7 (a) and A4, A5 and A8 (b) at PGE in PBS at pH 7.4. Accumulation potential, 0.0 V; accumulation time, 60 s; scan increment, 6 mV; frequency, 50 Hz and pulse height, 25 mVpp

a SW voltammograms of A7 at PGE in PBS at pH 7.4. [A7]: (1) blank, (2) 10, (3) 20, (4) 40, (5) 50, (6) 60, (7) 70 and (8) 90 μM. b Calibration plot of Ipa (μA) vs. [A7] in PBS of pH 7.4. Other conditions as in Fig. 5
Conclusion
In conclusion, a series of indole-based-sulfonamide analogs (A1–A8) were synthesized and characterized by 1HNMR and HR-EI-MS. These compounds (A1–A8) can be obtained directly from the reactions of 5-fluoro-1H-indole-3-carbohydrazide (I) with different aryl sulfonyl chloride in the presence of pyridine. The electrochemical behavior of these new compounds (A1–A8) has been studied using CV and SWV at ambient temperature on a PGE. The obtained electrochemical results provide useful information such as redox behavior, electron affinity, oxidation potential, electrochemical parameters, mechanism and the stability of these electroactive derivatives. Structure electrochemical activity relationship has been also reviewed for all compounds, which shows that the nature, position and number of substituents on benzene ring play an important role.
Experimental section
General experimental
NMR experiments were performed on Ultra Sheild Bruker FT NMR 500 MHz. IR experiments were performed on Perkin Elmer FT-IR and UV, Perkin Elmer Lambda 35 UV–VIS Spectrometer. CHN analysis was performed on a Carlo Erba Strumentazione-Mod-1106, Italy. Ultraviolet Electron impact mass spectra (EI MS) were recorded on a Finnigan MAT-311A, Germany. Thin layer chromatography (TLC) was performed on pre-coated silica gel aluminum plates (Kieselgel 60, 254, E. Merck, Germany). Chromatograms were visualized by UV at 254 and 365 nm.
Procedure for the synthesis 5-fluoro-1H-indole-3-carbohydrazide
The methyl 5-fluoro-1H-indole-3-carboxylate (10 g) was heated under reflux with hydrazine hydrated (10 mL) and methanol (25 mL) mixture for 6 h. The hydrazine and methanol were evaporated to get crude product that was recrystallized in ethanol and obtained pure 5-fluoro-1H-indole-3-carbohydrazide. Yield: 89%; 1HNMR (500 MHZ, DMSO-d6): δ 12.2 (s, 1H, NH), 11.24 (s, 1H, NH), 7.43 (d, J = 7.0 Hz, 1H, Ar), 7.35 (t, J = 7.0 Hz, 1H, Ar) 7.29 (dd, J = 7.0, 6.5 Hz, 1H, Ar), 7.22 (s, 1H, Ar), 4.26 (s, 2H, NH2); 13CNMR (125 MHZ, DMSO-d6): δ 163.9, 157.4, 131.2, 130.5, 127.1, 113.2, 112.3, 112.0, 111.9; HREI-MS: m/z calcd for C9H8FN3O, [M]+ 193.0651; Found; 193.0640.
General procedure for the synthesis indole-based-sulfonamide derivatives
The indole-based-sulfonamide derivatives were synthesized by heating under reflux a mixture of 1 mmol each 5-fluoro-1H-indole-3-carbohydrazide and aryl-sulfonyl chloride in 10 mL Pyridine 2 h. The development of reaction checked by TLC. After accomplishment of reaction, the solvent was evaporated by vacuum to afford crude products which were further recrystallized in ethanol and got pure product in 85–78 yields.
N′-(5-Fluoro-1H-indole-3-carbonyl)benzenesulfonohydrazide (A1)
Yield: 82%; 1HNMR (500 MHZ, DMSO-d6): δ 12.19 (s, 1H, NH), 11.40 (s, 1H, NH), 11.10 (s, 1H, NH), 7.92–7.89 (m, 2H, Ar), 7.46–7.42 (m, 3H, Ar), 7.42 (d, J = 7.0 Hz, 1H, Ar), 7.31 (t, J = 7.0 Hz, 1H, Ar) 7.29 (dd, J = 7.5, 6.0 Hz, 1H, Ar), 7.26 (s, 1H, Ar); 13CNMR (125 MHZ, DMSO-d6): δ 164.5, 157.5, 139.4, 132.2, 131.0, 130.4, 129.0, 129.0, 127.5, 127.0, 127.0, 113.4, 112.5, 112.0, 111.8; HREI-MS: m/z calcd for C15H12FN3O3S, [M] + 333.0583; Found; 333.0565; Anal. Calcd for, C15H12FN3O3S; C, 54.05; H, 3.63; N, 12.61; Found: C, 54.01; H, 3.59; N, 12.54.
N′-(5-Fluoro-1H-indole-3-carbonyl)-4-nitrobenzenesulfonohydrazide (A2)
Yield:80%; 1HNMR (500 MHZ, DMSO-d6): δ 12.10 (s, 1H, NH), 11.20 (s, 1H, NH), 11.09 (s, 1H, NH), 8.42 (d, J = 8.0 Hz, 2H, Ar), 8.09 (d, J = 8.0 Hz, 2H, Ar), 7.41 (d, J = 7.0 Hz, 1H, Ar), 7.32 (t, J = 7.0 Hz, 1H, Ar) 7.27 (dd, J = 7.5, 6.0 Hz, 1H, Ar), 7.24 (s, 1H, Ar); 13CNMR (125 MHZ, DMSO-d6): δ 164.5, 157.6, 151.4, 145.5, 131.0, 130.5, 128.0, 128.0, 127.5, 127.5, 121.2, 113.5, 112.5, 112.1, 111.9; HREI-MS: m/z calcd for C15H11FN4O5S, [M]+ 378.0434; Found; 378.0422; Anal. Calcd for, C15H11FN4O5S; C, 43.91; H, 2.70; N, 13.65; Found: C, 43.84; H, 2.66; N, 13.61.
N′-(5-Fluoro-1H-indole-3-carbonyl)-3-nitrobenzenesulfonohydrazide (A3)
Yield:80%; 1HNMR (500 MHZ, DMSO-d6): δ δ 12.42 (s, 1H, NH), 11.26 (s, 1H, NH), 11.30 (s, 1H, NH), 8.60 (d, J = 2.0 Hz, 1H, Ar), 8.29 (d,d J = 8.0, 2.0 Hz, 1H, Ar), 8.19 (d,d J = 7.0, 2.0 Hz, 1H, Ar), 7.80 (t, J = 7.5 Hz, 1H, Ar), 7.43 (d, J = 7.0 Hz, 1H, Ar), 7.31 (t, J = 7.0 Hz, 1H, Ar) 7.26 (dd, J = 7.5, 6.0 Hz, 1H, Ar), 7.22 (s, 1H, Ar); 13CNMR (125 MHZ, DMSO-d6): δ 164.6, 157.3, 148.4, 140.3, 133.2, 131.1, 130.6, 130.1, 127.60, 124.1, 120.4, 113.6, 112.4, 112.0, 111.9; HREI-MS: m/z calcd for C15H11FN4O5S, [M] + 378.0434; Found; 378.0440; Anal. Calcd for, C15H11FN4O5S; C, 43.91; H, 2.70; N, 13.65; Found: C, 43.83; H, 2.64; N, 13.59.
4-Fluoro-N′-(5-fluoro-1H-indole-3-carbonyl)benzenesulfonohydrazide (A4)
Yield:80%; 1HNMR (500 MHZ, DMSO-d6): δ 12.30 (s, 1H, NH), 11.55 (s, 1H, NH), 11.19 (s, 1H, NH), 7.89 (d, J = 8.5 Hz, 2H, Ar), 7.40 (d, J = 7.0 Hz, 1H, Ar), 7.31 (t, J = 7.0 Hz, 1H, Ar) 7.28 (dd, J = 7.5, 6.0 Hz, 1H, Ar), 7.25 (t, J = 8.5 Hz, 2H, Ar), 7.22 (s, 1H, Ar); 13CNMR (125 MHZ, DMSO-d6): δ 166.0, 164.5, 157.5, 135.2, 131.2, 130.5, 128.6, 128.6, 127.5, 115.7, 115.7, 113.7, 12.2, 112.0, 111.9; HREI-MS: m/z calcd for C15H11F2N3O3S, [M]+ 351.0489; Found; 351.0470; Anal. Calcd for, C15H11F2N3O3S; C, 51.28; H, 3.16; N, 11.96; Found: C, 51.22; H, 3.12; N, 11.91.
N′-(5-Fluoro-1H-indole-3-carbonyl)-4-methoxybenzenesulfonohydrazide (A5)
Yield: 84%; 1HNMR (500 MHZ, DMSO-d6): δ 12.60 (s, 1H, NH), 11.50 (s, 1H, NH), 11.30 (s, 1H, NH), 7.81 (d, J = 8.0 Hz, 2H, Ar), 7.41 (d, J = 7.0 Hz, 1H, Ar), 7.33 (t, J = 7.0 Hz, 1H, Ar) 7.27 (dd, J = 7.5, 6.0 Hz, 1H, Ar), 7.20 (s, 1H, Ar), 7.02 (d, J = 8.0 Hz, 2H, Ar), 3.82 (s, 3H, CH3); 13CNMR (125 MHZ, DMSO-d6): δ 164.7, 163.8, 157.4, 132.0, 131.2, 130.6, 128.0, 128.0, 127.5, 114.2, 114.2, 113.6, 112.4, 112.1, 111.9, 55.8; HREI-MS: m/z calcd for C16H14FN3O4S, [M] + 363.0689; Found; 363.0662; Anal. Calcd for, C16H14FN3O4S; C, 52.89; H, 3.88; N, 11.56; Found: C, C, 52.82; H, 3.83; N, 11.52.
4-Cyano-N’-(5-fluoro-1H-indole-3-carbonyl)benzenesulfonohydrazide (A6)
Yield: 84%; 1HNMR (500 MHZ, DMSO-d6): δ 12.20 (s, 1H, NH), 11.70 (s, 1H, NH), 11.16 (s, 1H, NH), 8.10 (d, J = 7.5 Hz, 2H, Ar), 7.78 (d, J = 7.5 Hz, 2H, Ar), 7.42 (d, J = 7.5 Hz, 1H, Ar), 7.31 (t, J = 7.5 Hz, 1H, Ar) 7.28 (dd, J = 70, 6.0 Hz, 1H, Ar), 7.20 (s, 1H, Ar); 13CNMR (125 MHZ, DMSO-d6): δ 164.8, 157.4, 144.1, 132.3, 132.3, 131.0, 130.5, 128.1 128.1, 127.4, 115.6, 114.9, 114.9, 113.6, 112.6, 112.2, 111.8; HREI-MS: m/z calcd for C16H11FN4O3S, [M]+ 358.0536; Found; 358.0522; Anal. Calcd for, C16H11FN4O3S; C, 55.32; H, 4.06; N, 12.10; Found: C, 55.27; H, 4.01; N, 12.04.
N’-(5-Fluoro-1H-indole-3-carbonyl)-3,4-dimethoxybenzenesulfonohydrazide (A7)
Yield: 84%; 1HNMR (500 MHZ, DMSO-d6): δ 12.82 (s, 1H, NH), 11.90 (s, 1H, NH), 11.28 (s, 1H, NH), (d, J = 7.5 Hz, 2H, Ar), 7.42 (d, J = 7.5 Hz, 1H, Ar),7.36 (d, J = 7.0 Hz, 2H, Ar), 7.32 (d, J = 2.0 Hz, 2H, Ar), 7.29 (t, J = 7.5 Hz, 1H, Ar) 7.26 (dd, J = 70, 6.0 Hz, 1H, Ar), 7.21 (s, 1H, Ar), 6.91 (d, J = 8.0 Hz, 2H, Ar); 13CNMR (125 MHZ, DMSO-d6): δ 164.6, 157.4, 153.1, 150.0, 133.2, 131.5, 130.4, 127.5, 120.5, 115.5, 113.6, 1128, 112.5, 112.0, 111.9, 56.4, 56.2; HREI-MS: m/z calcd for C17H16FN3O5S, [M]+ 93.0795; Found; 393.0778; Anal. Calcd for, C17H16FN3O5S; C, C, 51.90; H, 4.10; N, 10.68; Found: C, 51.83; H, 4.06; N, 10.63.
N’-(5-Fluoro-1H-indole-3-carbonyl)-4-isopropylbenzenesulfonohydrazide (A8)
Yield: 84%; 1HNMR (500 MHZ, DMSO-d6): δ 12.92 (s, 1H, NH), 11.86 (s, 1H, NH), 11.53 (s, 1H, NH), 7.82 (d, J = 8.0 Hz, 2H, Ar), 7.41 (d, J = 7.5 Hz, 1H, Ar), 7.38 (d, J = 8.0 Hz, 2H, Ar), 7.33 (t, J = 7.5 Hz, 1H, Ar) 7.29 (dd, J = 70, 6.0 Hz, 1H, Ar), 7.22 (s, 1H, Ar), 2.72 (m, 1H, CH), 1.26 (d,, J = 10 Hz, 6H, 2XCH3); 13CNMR (125 MHZ, DMSO-d6): δ 164.7, 157.6, 151.2, 136.7, 131.3, 130.7, 127.6, 127.1 127.1, 126.5, 126.5, 113.6, 112.7, 112.4, 111.9, 36.2, 23.8, 23.8; HREI-MS: m/z calcd for C18H18FN3O3S, [M]+ 75.1053; Found; 375.1040; Anal. Calcd for, C18H18FN3O3S; C, C, 57.59; H, 4.83; N, 11.19; Found: C, 57.53; H, 4.79; N, 11.14.
Voltammetric parameters and electrochemical cells
Voltammetric experiments, including cyclic voltammetry (CV) and square-wave voltammetry (SWV), were performed using an interface 1010E Potentiostat/Galvanostat/ZRA model (Gamry Instruments, Warminster, USA). The electrode system consisted of a pencil graphite electrode (PGE) as the working electrode, Ag/AgCl (saturated KCl) reference electrode, and a Pt wire auxiliary electrode. Operating conditions for SWV were frequency 50 Hz, pulse height 25 mVpp, scan increment 6 mV, and accumulation time 60 s. A Roca mechanical pencil Model M205R (Korea) was used as a holder for the pencil lead. Electrical contact with the lead was achieved by soldering a copper wire to the metallic part that holds the lead in place inside the pencil. The tested pencil leads were from Faber-Castell and named Super-polymer 9065S (black lead) of types 2B. All leads had a total length of 60 mm and a diameter of 0.5 mm. Immersing 10 mm of the pencil lead into a solution resulted in an active electrode area of 15.9 mm2. The pencil leads were used as received.
Voltammetric reagents and materials
A stock solution of sulfonamide compounds was prepared by dissolving an appropriate amount of each compound in ethanol and storing the solution in the dark at 4 °C. Phosphate buffer solution (PBS) was prepared using the adequate amount of Na2HPO4 and NaH2PO4 salts. The pH of the aqueous solutions was measured using a pH-meter (Model pH-M-31114 Molequle-on) with accurate to ± 0.02 unit. All the chemicals were of reagent grade (Merck, Darmstadt). All solutions were prepared using water purified in a Millipore Milli-Q direct 8/16 system. All the experiments were carried out at room temperature (24 ± 2 °C) and, to eliminate oxygen, N2 was bubbled into the solution for 5 min.
Availability of data and materials
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Abbreviations
- CV:
-
Cyclic voltammetry
- SWV:
-
Square wave voltammetry
- PGE:
-
Pencil graphite electrode
- ks :
-
Standard heterogeneous rate constants
- Г:
-
Electroactive surface coverage
- PBS:
-
Phosphate buffer solution
- ν:
-
Scan rate
- α:
-
Electron transfer coefficient
- Eo :
-
Formal redox potential
- Q :
-
Charge involved in the electrooxidation process
- n:
-
Number of electrons
- F:
-
Faraday constant
- A:
-
Working electrode surface area
- EI MS:
-
Electron impact mass spectra
- TLC:
-
Thin layer chromatography
References
Denhart DJ, Deskus JA, Ditta JL, Gao Q, Dalton King H, Kozlowski ES, Meng Z, Paglia MA, Mattson GK, Molski TF, Taber MT, Lodge NJ, Mattson RJ, Macor JE (2009) Conformationally restricted homotryptamines. Part 5: 3-(trans-2-aminomethylcyclopentyl) indoles as potent selective serotonin reuptake inhibitors. Bioorg Med Chem Lett 19(15):4031–4033. https://doi.org/10.1016/j.bmcl.2009.06.026
Singh P (2014) Structural optimization of indole based compounds for highly promising anti-cancer activities: structure activity relationship studies and identification of lead molecules. Euro J Med Chem 74(3):440–450. https://doi.org/10.1016/j.ejmech.2013.12.047
Sharma SK, Kumar P, Narasimhan B, Ramasamy K, Mani V, Mishra RK, Majeed AB (2012) Synthesis, antimicrobial, anticancer evaluation and QSAR studies of 6-methyl-4-[1-(2-substituted-phenylamino-acetyl)-1H-indol-3-yl]-2-oxo/thioxo-1, 2, 3, 4-tetrahydro-pyrimidine-5-carboxylic acid ethyl esters. Euro J Med Chem 48(1):16–25
Mehndiratta S, Hsieh YL, Liu YM, Wang AW, Lee HY, Liang LY, Kumar S, Teng CM, Yang CR, Liou JP (2014) Indole-3-ethylsulfamoylphenylacrylamides: potent histone deacetylase inhibitors with anti-inflammatory activity. Euro J Med Chem 85(6):468–479
Yamuna E, Kumar RA, Zeller M, Prasad KJ (2012) Synthesis, antimicrobial, antimycobacterial and structure–activity relationship of substituted pyrazolo-, isoxazolo-, pyrimido-and mercaptopyrimidocyclohepta [b] indoles. Euro J Med Chem 47(1):228–238
Zoumpoulakis P, Camoutsis C, Pairas G, Soković M, Glamočlija J, Potamitis C, Pitsas A (2012) Synthesis of novel sulfonamide-1,2,4-triazoles, 1,3,4-thiadiazoles and 1,3,4-oxadiazoles, as potential antibacterial and antifungal agents. Biological evaluation and conformational analysis studies. Bioorg Med Chem 20(4):1569–1583. https://doi.org/10.1016/j.bmc.2011.12.031
El-Gohary NS, Shaaban MI (2013) Synthesis, antimicrobial, antiquorum-sensing, antitumor and cytotoxic activities of new series of fused [1, 3, 4] thiadiazoles. Euro J Med Chem 63(1):185–195
Li P, Shi L, Gao MN, Yang X, Xue W, Jin LH, Hu DY, Song BA (2015) Antibacterial activities against rice bacterial leaf blight and tomato bacterial wilt of 2-mercapto-5-substituted-1, 3, 4-oxadiazole/thiadiazole derivatives. Bioorg Med Chem Lett 25(3):481–484
Ragab FA, Heiba HI, El-Gazzar MG, Abou-Seri SM, El-Sabbagh WA, El-Hazek RM (2017) Anti-inflammatory, analgesic and COX-2 inhibitory activity of novel thiadiazoles in irradiated rats. J Photochem Photobiol B Biol 166(1):285–300. https://doi.org/10.1016/j.jphotobiol.2016.12.007
Luszczki JJ, Karpińska M, Matysiak J, Niewiadomy A (2015) Characterization and preliminary anticonvulsant assessment of some 1,3,4-thiadiazole derivatives. Pharmacol Rep 67(3):588–592
Gür M, Muğlu H, Çavuş MS, Güder A, Sayıner HS, Kandemirli F (2017) Synthesis, characterization, quantum chemical calculations and evaluation of antioxidant properties of 1,3,4-thiadiazole derivatives including 2-and 3-methoxy cinnamic acids. J Mol Struct 1134(15):40–50
Kadi AA, Al-Abdullah ES, Shehata IA, Habib EE, Ibrahim TM, El-Emam AA (2010) Synthesis, antimicrobial and anti-inflammatory activities of novel 5-(1-adamantyl)-1,3,4-thiadiazole derivatives. Euro J Med Chem 45(11):5006–5011
Turner S, Myers M, Gadie B, Nelson AJ, Pape R, Saville JF, Berridge TL (1988) Antihypertensive thiadiazoles. 1. Synthesis of some 2-aryl-5-hydrazino-1,3,4-thiadiazoles with vasodilator activity. J Med Chem 31(5):902–906
Chen Z, Xu W, Liu K, Yang S, Fan H, Bhadury PS, Zhang Y (2010) Synthesis and antiviral activity of 5-(4-chlorophenyl)-1,3,4-thiadiazole sulfonamides. Molecules 15(12):9046–9056. https://doi.org/10.3390/molecules15129046
Alyar S, Özbek N, Kuzukıran K, Karacan N (2011) Quantitative structure–activity relationships studies for prediction of antimicrobial activity of synthesized disulfonamide derivatives. Med Chem Rese 20(2):175–183
Alyar S, Zengin H, Özbek N, Karacan N (2011) Synthesis, characterization, spectroscopic properties, theoretical calculation, and antimicrobial activity of new aryldisulfonamides. J Mol Struct 992(1–3):27–32
Gündüzalp AB, Özmen ÜÖ, Çevrimli BS, Mamaş S, Cete S (2014) Synthesis, characterization, electrochemical behavior, and antimicrobial activities of aromatic/heteroaromatic sulfonylhydrazone derivatives. Med Chem Res 23(7):3255–3268
Suzen S, Ates-Alagoz Z, Demircigil BT, Ozkan SA (2001) Synthesis and analytical evaluation by voltammetric studies of some new indole-3-proplonamide derivatives. Farmaco 56(11):835–840
DÖzel A, Durmuş Z, Yılmaz İ, Çukurovalı A, Kılıç E (2009) Electroreduction of some substituted hydrazones on platinum electrode in dimethylformamide. Acta Chim Slov 56:797–806
Sarhan AA, Ibrahim MS, Kamal MM, Mitobe K, Izumi T (2009) Synthesis, cyclic voltammetry, and UV–Vis studies of ferrocene-dithiafulvalenes as anticipated electron-donor materials. Monats für Chem Chem Monthly 140(3):315–323
Odewunmi NA, Kawde AN, Ibrahim M (2018) Electrochemically inspired copper (II) complex on disposable graphite pencil electrode for effective simultaneous detection of hypoxanthine, xanthine, and uric acid. Electroanalysis 30(10):2311–2320. https://doi.org/10.1002/elan.201800397
Goyal RN, Sangal A (2005) Oxidation chemistry of indole-2-carboxylic acid: mechanism and products formed in neutral aqueous solution. Electrochim Acta 50(10):2135–2143
Enache TA, Oliveira-Brett AM (2011) Pathways of electrochemical oxidation of indolic compounds. Electroanalysis 23(6):1337–1344
Keech PG, Chartrand MM, Bunce NJ (2002) Oxidation of simple indoles at a platinum anode. J Electroanal Chem 534(1):75–78
Keech PG, Bunce NJ (2003) Electrochemical oxidation of simple indoles at a PbO2 anode. J Appl Electrochem 33(1):79–83
Uslu B, Canbaz D (2010) Anodic voltammetry of zolmitriptan at boron-doped diamond electrode and its analytical applications. Die Pharmazie Int J Pharma Sci 65(4):245–250
Fabiańska A, Białk-Bielińska A, Stepnowski P, Stolte S, Siedlecka EM (2014) Electrochemical degradation of sulfonamides at BDD electrode: kinetics, reaction pathway and eco-toxicity evaluation. J Hazard Mater 280:579–587
Karaaslan C, Suzen S (2011) Electrochemical behavior of biologically important indole derivatives. Int J Electrochem ID 154804:2011
Mayuri P, Huang ST, Mani V, Kumar AS (2018) A new organic redox species-indole tetraone trapped MWCNT modified electrode prepared by in situ electrochemical oxidation of indole for a bifunctional electrocatalysis and simultaneous flow injection electroanalysis of hydrazine and hydrogen peroxide. Electrochim Acta 268:150–162
Wu J, Dou Y, Guillot R, Kouklovsky C, Vincent G (2019) Electrochemical dearomative 2, 3-difunctionalization of indoles. J Am Chem Soc 141(7):2832–2837
Wu K, Sun Y, Hu S (2003) Development of an amperometric indole-3-acetic acid sensor based on carbon nanotubes film coated glassy carbon electrode. Sens Actutat B Chem 96(3):658–662
Souza CD, Braga OC, Vieira IC, Spinelli A (2008) Electroanalytical determination of sulfadiazine and sulfamethoxazole in pharmaceuticals using a boron-doped diamond electrode. Sens Actutat B Chem 135(1):66–73
Rao TN, Sarada BV, Tryk DA, Fujishima A (2000) Electroanalytical study of sulfa drugs at diamond electrodes and their determination by HPLC with amperometric detection. J Electroanal Chem 491(1–2):175–181
Momberg A, von Baer D, Bruhn C, Smyth MR (1984) The oxidative voltammetric behaviour of some sulphonamides at the glassy carbon electrode. Anal Chim Acta 159(1):119–127
Kotoucek M, Skopalova J, Michalkova D, Kotouček M, Skopalová J, Michálková D (1997) Electroanalytical study of salazosulfapyridine and biseptol components at the mercury electrode. Anal Chim Acta 353(1):61–69
Brabec V, Mornstein V (1980) Electrochemical behaviour of proteins at graphite electrodes: II. Electrooxidation of amino acids. Biophy Chem 12(2):159–165
Alemu H, Khoabane NM, Tseki PF (2003) Electrochemical oxidation of niclosamide at a glassy carbon electrode and its determination by voltammetry. Bull Chem Soc Ethiol 17(1):95–106
Sun W, Han J, Ren Y, Jiao K (2006) Voltammetric studies on the interaction of orange G with proteins: analytical applications. J Braz Chem Soc 17(3):510–517
Mabrouk PA (1996) Direct electrochemistry for the imidazole complex of microperoxidase-11 in dimethyl sulfoxide solution at naked electrode substrates including glassy carbon, gold, and platinum. Anal Chem 68(1):189–191
Bard AJ, Faulkner LR, Leddy J, Zoski CG (1980) Electrochemical methods: fundamentals and applications (Vol. 2). Wiley, New York, p. 1
Gosser DK (1993) Cyclic voltammetry: simulation and analysis of reaction mechanisms. VCH, New York
Laviron E (1974) Adsorption, autoinhibition and autocatalysis in polarography and in linear potential sweep voltammetry. J Electroanal Chem Interf Electrochem 52(3):355–393
Bard AJ, Faulkner LR (2001) Fundamentals and applications. Electrochem Meth 2(482):580–632
Acknowledgements
The authors gratefully acknowledge the Institute for Research and Medical Consultations (IRMC), Imam Abdulrahman Bin Faisal University, for providing excellent lab facilities for research.
Funding
This research was supported by Deanship of Scientific Research, Imam Abdulrahman Bin Faisal University (Project No. 2018-086-IRMC).
Author information
Authors and Affiliations
Contributions
Conceptualization and supervision, MI; Interpretation of data, MI and MT; Methodology, NBA; Synthesis, MT and MN; Writing-review and editing, NBA; Data curation and characterization, A-NK; Formal analysis, MT. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Fig. S1.
CVs (scan 1–10) of 3.0 x 10−5 M of A5 (a) and A3 (b) in PBS of pH 7.4 obtained at PGE scan rate, 0.10 Vs−1. Fig. S2. CVs of 3.0 x 10−5 M of A1–A8 in PBS of pH 3.0 obtained at PGE scan rate, 0.10 Vs−1. Fig. S3. Plot of log Ipa vs. log ν of A1–A8 in PBS of pH 7.4 obtained from CV at PGE. Fig. S4. Plot of Ep (V) vs ln ν (mV/s) of A1–A8 in PBS of pH 7.40 obtained from CV at PGE.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ibrahim, M., Taha, M., Almandil, N.B. et al. Synthesis, characterization and electrochemical properties of some biologically important indole-based-sulfonamide derivatives. BMC Chemistry 14, 38 (2020). https://doi.org/10.1186/s13065-020-00691-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-020-00691-5