
Abstract
Background
Curcumin is one of the leading compound extracted from the dry powder of Curcuma longa (Zingiberaceae family), which possess several pharmacological properties. However, in vivo administration exhibited limited applications in cancer therapies.
Results
Twenty-four curcumin derivatives have synthesized, which comprises cyclohexanone 1–10, acetone 11–17 and cyclopentanone 18–24 series. All the curcuminoids were synthesized by the acid or base catalyzed Claisen Schmidt condenstion reactions, in which β-diketone moiety of curcumin was modified with mono-ketone. These curcuminoids 1–24 were screened against HeLa, K562, MCF-7 (an estrogen-dependent) and MDA-MB-231 (an estrogen-independent) cancer cell lines. Among them, acetone series 11–17 were found to be more selective and potential cytotoxic agents. The compound 14 was exhibited (IC50 = 3.02 ± 1.20 and 1.52 ± 0.60 µg/mL) against MCF-7 and MDA-MB-231 breast cancer cell lines. Among the cyclohexanone series, the compound 4 exhibited (IC50 = 11.04 ± 2.80, 6.50 ± 01.80, 8.70 ± 3.10 and 2.30 ± 1.60 µg/mL) potential cytotoxicity against four proposed cancer cell lines, respectively. All the curcucminoids were characterized with the detailed 1H NMR, IR, UV–Vis, and mass spectroscopic techniques. The structure of compound 4 was confirmed by using the single X-ray crystallography. Additionally, we are going to report the first time spectral data of (2E,6E)-2,6-bis(2-methoxybenzylidene)cyclohexanone (1). Structure–activity relationships revealed that the mono-carbonyl with 2,5-dimethoxy substituted curcuminoids could be an essential for the future drugs against cancer diseases.
Conclusions
Curcuminoids with diferuloyl(4-hydroxy-3-methoxycinnamoyl) moiety with mono carbonyl exhibiting potential cytotoxic properties. The compound 14 was exhibited (IC50 = 3.02 ± 1.20 and 1.52 ± 0.60 µg/mL) against MCF-7 and MDA-MB-231 breast cancer cell lines.

Similar content being viewed by others
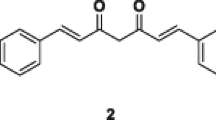
Introduction
Cancer is one of the leading causes of death worldwide, with approximately 14 million new cases in 2012 [1]. The number of new cases is expected to rise by about 70% over the next two decades. Cancer causes of death globally and was responsible for 8.8 million deaths in 2015. Globally, nearly 1 in 6 deaths is due to cancer [2]. In 2016, 1,685,210 new cancer cases and 595,690 cancer deaths are projected to occur in the United States [3]. Breast cancer was the commonest cancer in women amongst all races from the age of 20 years in Malaysia for 2003 to 2005. According to the National Cancer Institute, 232,340 female breast cancers and 2240 male breast cancers are reported in the USA. It accounts for 16% of all female cancers and 22.9% of invasive cancers in women [4,5,6]. Curcumin (1,7-bis-(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione) is a natural diarylheptanoid extracted from the rhizome of Curcuma longa [7, 8]. Curcumin is a fascinating symmetrical molecules possessing interesting skeleton of β-diketone with diferuloyl (4-hydroxy-3-methoxycinnamic acid) moieties [9]. It exhibited remarkable biological activities mainly anticancer [10,11,12], anti-inflammatory [13,14,15], antioxidant [16, 17], anti-hepatotoxic [18], nephroprotective [19], thrombosis suppressing [20], and hypoglycemic activities [21]. Curcuminoids have been identified as a potent anti-breast cancer agent available from natural food ingredients including turmeric. This effect maybe contributed through targeting the estrogen receptors [22]. Advance understanding of bioactive metabolites through chemical synthesis has further enhanced the potential of these natural products including curcumin as the anticancer agent. For example, 4-hydroxy-3-methoxybenzylidene)-N-methyl-4-piperidone (PAC), which is the analogue of curcumin were reported with enhanced antitumor effect against breast cancer via targeting the estrogen receptor [23]. On the other hand, modification of cyclohexanone derivative of curcumin was reported to enhance cytotoxicity against estrogen receptor-negative breast cancer cells [24]. Although it is well known natural remedies for pain still have bioavailability problems such as absorption, distribution, metabolism etc. [25, 26]. Due to its significant anti-cancer properties on the various cancers such as gastrointestinal, genitourinary, gynecological, hematological, pulmonary, breast, and bone diseases, curcumin becomes a promising lead compound to develop a novel drugs [27, 28].
Results and discussion
Chemistry
Curcuminoids are the derivatives of curcumin. About 24 curcuminoids have been synthesized and investigated their cytotoxic properties against various cancer lines and thus established the structure–activity relationship for the future drugs development. In our experiments, we have synthesized three series of mono-carbonyl analogues of curcuminoids with cyclohexanone (1–10), acetone (11–17) and cyclopentanone (18–24). Three series were synthesized by Claisen–Schmidt condensation reaction by coupling the appropriate aromatic aldehydes with cyclohexanone, acetone and cyclopentanone by acid or base catalysed as previously stated by Wei [29]. In this project, β-diketone moiety of curcumin was modified with mono ketone and investigated their cytotoxic properties against Hele cell lines (human cervical cancer), K562 (Leukemia) cell lines, MCF-7 (an estrogen-dependent) and MDA-MB-231 (an estrogen-independent) cancer cell lines [30]. Additionally, we are going to report first time the data of (2E, 6E)-2,6-bis(2-methoxybenzylidene)cyclohexanone (1). Recently, we have reported the in vivo anti-tumour activity of 2,6-bis(4-hydroxy-3-methoxybenzylidene)cyclohexanone (5) on 4T1 breast cancer cells [31]. Previously, we have published another curcumin derivative DK1 and naturally occurring chalcone flavokawain B and its derivatives on various breast cancer cell lines [32,33,34].
The compound was 1 purified as yellow liquid. The UV spectrum of compound 1 showed the absorption wavelength, λmax at 339 nm corresponding to the α,β conjugated carbonyl group (C=O) compound. The IR absorption bands at 1636 cm−1 corresponding to carbonyl (C=O) and 2942–3001 cm−1 referred to aromatic C–H stretching functional groups. The 1H NMR spectrum (600 MHz, CDCl3) of compound 1 appeared at δ 1.75 as multiplet (2H) was assigned to the methylene proton (CH2) at C4. A methylene protons at 2.84 as a multiplet (4H) integrated was corresponding to the C3 and C5 atoms. A singlet appeared at 3.86 integrated by 6H was assigned to the methoxy protons (OCH3) at C2′ and C2″ position. A multiplet appeared at 6.92 was assigned to the aromatic protons at C3′ and C3″ methine protons. Two protons (2H) integrated at 6.96 shown a multiplet were assigned to the C5′ and C5″ protons. Another multiplet appeared at 7.33–730 (4H) was assigned to the C4′, C4″, C6′ and C6″ as aromatic methine protons. A broad singlet appeared at 7.98 integrated by 2H was due to the olefinic protons (–C=C–H). The board band decoupled spectra 13C NMR showed the presence seven quaternary carbons, three methylene and ten methine carbons atoms. The compound showed EI-MS molecular mass was at m/z 334. The molecular formula of compound 1 was supported by HREI-MS calculated C22H22O3 334.1575, found for 334.1580, which supported the proposed structure of compound 1 (Fig. 1). Previously, the radical scavenger and enzyme inducer activity of compound 1 obtained from Aldrich was reported by Dinkova-Kostavo et al. [35]. Interestingly, the data of all the compounds were characterized precisely on 600 MHz Bruker and 500 MHz and assignments were made carefully. The data of known compounds were compared with the previously published by Wei, Hosoya and Du [29, 36, 37].

Chemical structures of curcuminoids (1–24) and curcumin
Structure–activity relationship
All the curcuminoids have been screened against HeLa, K562, MCF-7 and MDA-MB-231 cancer cell lines and results are depicted in Table 1. Among the cyclohexanone series 1–10, compound 4 was the most potent cytotoxic against four cancer lines especially breast can cell lines exhibited (IC50 = 11.04 ± 2.80, 6.50 ± 01.80, 8.70 ± 3.10 and 2.30 ± 1.60 µg/mL), respectively. Compound 5 possess the partial structure of curcumin showed (IC50 = 6.03 ± 1.70 and 3.03 ± 1.00 µg/mL) against MCF-7 and MDA-MB-231 breast cancer cell lines almost three to four times more active than curcumin (Table 1). Others curcuminoids 1, 3, 6, 7, 8, 10 also showing good cytotoxicity against breast cancer lines MCF-7 and MDA-MB-231 and moderated against HeLa and K562 cell lines. Curcuminoids with acetone series 11–17 exhibited more potential cytotoxic effects on four type cancers cell lines, which is comparable with curcumin IC50 values in Table 1. Among acetone series, the compound 14 was found to be the most cytotoxic in breast cancer lines MCF-7 and MDA-MB-231 and moderate against HeLa and K562 cell lines. Compound 11 also exhibited (IC50 = 11.31 ± 1.33, 4.50 ± 1.20 and 2.07 ± 1.75 µg/mL) against HeLa, MCF-7 and MDA-MB-231, respectively. Other curcuminoids 15, 16, and 17 possessing Cl, Br and F substituted showing moderate cytotoxicity against four cancer lines (Table 1). Compound 17 with trimethoxy substituted also exhibiting potential cytotoxicity with (IC50 = 2.50 ± 1.10 and 3.10 ± 1.06 µg/mL) against breast cancer lines MCF-7 and MDA-MB-231, which is compatible with the previously published by Fuchs [38]. Curcuminoids 18–24 with cyclopentanone series did not show any significant cytotoxicity against all types of cancer lines except compound 22, showing better cytotoxic effects against Hela and MCF-7 and MDA-MB-231 cancer then curcumin. The lower cytotoxicity of compounds 18–24 possibly due to the ring strain, which could be sterically not well-fitted with the estrogen receptors. Cytotoxic results of curcuminoids with acetone series 1–10 and methoxy substituted exhibiting selectively more potential than cyclohexanone (11–17) and cyclopentanone (18–24) series. The results are summarized in Table 1.
Most of curcuminoids are potent as compared to the curcumin with (IC50 = 22.50 ± 5.50 and 26.50 ± 1.40 µg/mL) against MCF-7 and MDA-MB-231 (Table 1). Several reports on curcuminoids with mono-carbonyl (acetone series) have been even better pharmacological properties than curcumin [22, 38]. Due to enolization and chelating (hydrogen bonding with the diketone), curcumin exhibited slightly lower cytotoxic effect than the modified derivatives. This could be due to the weak binding with the receptors, thus cause the weak pharmacokinetic profiles [39]. All curcuminoids possessed bis-enone conjugated system, which is quite reasonable site to binding with the Michael receptor selectivity with target nucleophile [30, 40,41,42]. The curcuminoids with mono-carbonyl 1–10 could be potential analogues for the drug discovery against cancer. In this respect, curcumin derivatives bearing a mono-carbonyl and methoxy groups especially cyclohexanone (1–10) and acetone 11–17 series could be a remarkable approach for the improvement of bioavailability problems related to curcumin [43, 44].
X-ray structure description
Crystal data of compound 4 was given in Table 2. One crystal structure was determined by using X-ray diffraction method. Figure 2 showed the molecular structure of compound 4. Compound 4 crystalized in orthorhombic crystal system, space group Pna21.

Molecular structures of compound 4 showing the atomic numbering scheme
Experimental
Chemistry
General
Melting points were determined on Electrothermal IA 9100 capillary melting point apparatus and are uncorrected. UV spectra were recorded on UV–Vis spectrophotometer model type of Genesys 10 s and expressed in nm. Thermo Scientific. Glass cuvettes were used. All the samples were dissolve in chloroform or methanol. FT-IR spectroscopic studies were carried out on FTIR spectrophotometer 1000 model Perkin Elmer at room temperature 25 °C. KBr pellets were dried in oven and scanned for calibration purpose. 1H NMR spectra of compounds were recorded on a Bruker Ascend TM 600 MHz machine, while the spectra of compounds 12, 16, 17 were recorded on 500 MHz NMR spectrometers. The chemical shifts (δ) are presented with references to CDCl3 (δ: 7.25) and TMS (δ: 0.00) as the internal reference. Electron-spray ionization mass spectra in positive mode (ESI–MS) were recorded on a Bruker Esquire 3000 spectrometer. Column chromatography purifications were carried out on Silica Gel 60 (Merck, 70–230 mesh, ASTM) and flash silica gel (230–400 mesh, ASTM, Merck). The purity of all compounds were checked by thin-layer chromatography (TLC) and 1H-NMR spectra. All reagents used were of analytical grade. All the chemicals were purchased from Aldrich, U.S.A. Other reagents were purchased from Sinopharm Chemical Reagent Co. Ltd., China.
Synthetic procedures
Method A (acid-catalyzed)
A typical Claisen-Schmidt condensation reaction procedure was used to prepare all curuminoids. Appropriate mono ketone (cyclohexanone, acetone and cyclopentanone) 10 mol (1 equiv) was dissolved in absolute ethanol (15–20 mL). Substituted benzaldehydes 20 mol, (2 equiv) was added slowly. About 1–2 mL concentrated HCl was added drop wise over 5–10 min in a stirred mixture of ketone. The reaction mixture was stirred overnight (12–24 h). The product was monitored by comparing the Co-TLC with the starting material. The products were extracted with ethyl acetate by dissolving the compounds in distilled water (100 mL). Curcuminoids were purified by silica gel column chromatography (ethyl acetate/hexane) and re-crystallized with hot solution of ethyl acetate and ethanol.
Method B (base-catalyzed)
The general procedure Claisen–Schmidt condensation reaction was used to synthesize curcuminoids by using this method involved in addition of certain amount of mono ketone (cyclohexanone, acetone and cyclopentanone) to a solution of substituted aldehydes in MeOH or C2H5OH by adding KOH or NaOH. The reaction mixture is stirred at room temperature and monitored by TLC. The products are extracted and purified as described as in acid catalysed [43, 44].
(2E,6E)-2,6-bis(2-Methoxybenzylidene)cyclohexanone (1)
Yellow liquid; yield (86%); UV–Vis (CHCl3) λmax: 302, 339 nm; IR (KBr,) v 3023 (Ar C–H stretch), 1636 (C=O), 1527 (Ar C=C<) cm−1; 1H NMR (CDCl3, 600 MHz) δ 1.75 (m, 2H, 4-H), 2.84 (m, 4H, 3, 5-H), 3.86 (s, 6H, OCH3, C-2′ & C-2″), 6.92 (m, 2H, 3′, 3″-H), 6.96 (m, 2H, 5′, 5″-H), 7.32 (m, 2H, 4′, 4″-H), 7.33–7.30 (m, 4H, 4′, 4″, 6′, 6″-H), 7.98 (brs, 2H, –C=C–H). 13C NMR (CDCl3, 150 MHz) δ 23.5 (C-4), 28.6 (C-3, C-5), 55.5 (OCH3), 110.6 (C-3′, C-3″), 119.9 (C-5′, C-5″), 125.2 (C-4′, C-4″, C-6′, C-6″), 130.3 (C-1′, C-1″), 132.5 (C-2, C-6), 136.6 (–C=C–H), 158.4 (C-2′, C-2″), 190.6 (C=O); EI-MS m/z 334.0 (10), 303.1 (20), 240.3 (14), 161.2 (19), 107.4 (23), 77.0 (64); HREI-MS for C22H22O3 M+, calcd.: m/z 334.1575, found: m/z 334.1589.
(2E,6E)-2,6-bis(4-Methoxybenzylidene)cyclohexanone (2)
Yellow crystals; yield (74%); m.p. 152–153 °C (lit. [29] 148–149 °C); UV–Vis (CHCl3) λmax: 362 nm; IR (KBr) v 3010 (Ar C–H stretch), 1660 (C=O), 1508–1594 (Ar C=C) cm−1; 1H NMR (CDCl3, 600 MHz) δ 1.80 (m, 2H, 4-H), 2.92 (m, 4H, 3, 5-H), 3.84 (s, 6H, OCH3, C-4′, 4″), 6.93 (d, 4H, 3′, 3″, 5′, 5″-H, J = 6.78 Hz), 7.45 (d, 4H, 2′, 2″, 6′, 6″-H, J = 6.78 Hz), 7.76 (brs, 2H, –C=C–H); EI-MS m/z 334.0 (100), 303.45 (36), 240.1 (23), 161.2 (10), 107.0 (28); HREI-MS for C22H22O3 M+, calcd.: m/z 334.1568, found: m/z 334.1573.
(2E,6E)-2,6-bis(2,3-Dimethoxybenzylidene)cyclohexanone (3)
Yellow crystals; yield (92%); m.p. 105–106 °C (lit. [36] 107–109 °C); UV–Vis (CHCl3) λmax: 324 nm; IR (KBr) v 3023 (Ar C–H stretch), 1622 (C=O), 1536–1536 (Ar C=C) cm−1; 1H NMR (CDCl3, 600 MHz) δ 1.75 (m, 2H, 4-H), 2.80 (m, 4H, 3, 5-H), 3.82 (s, 6H, OCH3, C3′, 3″), 3.88 (s, 6H, OCH3, C-2′, 2″), 6.93 (m, 4H, 4′, 4″, 6′, 6″-H), 7.06 (brt, 2H, 5′, 5″-H, J = 7.98 Hz), 7.94 (brs, 2H, –C=C–H); 13C NMR, (150 MHz, CDCI3) δ 23.3 (C-4), 28.78 (C-3, C-5), 55.9 (OCH3), 61.2 (OCH3), 112.8 (C-5′, C-5″), 122.2 (C-4′, C-4″), 123.5 (C-6′, C-6″), 130.5 (C-1′, C-1″), 132.5 (C-2, C-6), 137.5 (C=C–H), 152.9 (C-2′, C-2″, C-3′, C-3″), 190.4 (C=O); EI-MS m/z 394 (5), 363.0 (100), 331.2 (68), 161.23 (86), 227.33 (24), 136.18 (29); HREI-MS for C24H26O5 M+, calcd.: m/z 394.1783, found: m/z 394.1778.
(2E,6E)-2,6-bis(4-Hydroxy-3-methoxybenzylidene)cyclohexanone (5)
Synthesis, purification and experimental data of compound 5 was recently published by us [31].
(2E,6E)-2,6-bis(2-Chlorobenzylidene)cyclohexanone (6)
Yellow crystals; yield (68%); m.p. 109–110 °C (lit. [36] 94–95 °C); UV–Vis (CHCl3) λmax: 320 nm; IR (KBr) v 3073 (Ar C–H stretch), 1663 (C=O), 1574–1433 (Ar C=C) cm−1; 1H NMR (CDCl3, 600 MHz) δ 1.76 (m, 2H, 4-H), 2.78 (m, 4H, 3, 5-H), 7.33 (m, 2H, 3′, 3″-H), 7.28 (m, 4H, 4′, 4″, 5′, 5″-H), 7.44 (m, 2H, 6′, 6″-H), 7.91 (brs, 2H, –C=C–H); EI-MS m/z 343.0 (5), 307 (100), 272 (8), 166 (4), 138 (6), 112 (17); HREI-MS for C20H16Cl2O M+, calcd.: m/z 342.0578, found: m/z 342.0572.
(2E,6E)-2,6-bis(4-Chlorobenzylidene)cyclohexanone (7)
Yellow crystals; yield (86%); m.p. 149–153 °C (lit. [29] 147–149 °C); UV–Vis (CHCl3) λmax: 335 nm; IR (KBr) v 3063 (Ar C–H stretch), 1604 (C=O), 1576–1487 (Ar C=C) cm−1; 1H NMR (CDCI3, 500 MHz) δ 1.80 (m, 2H, 4-H), 2.89 (m, 4H, 3, 5-H), 7.34 (m, 2H, 2′, 2″-H), 7.34 (m, 2H, 3′, 3″-H), 7.34 (m, 2H, 5′, 5″-H), 7.34 (m, 2H, 6′, 6″-H), 7.73 (brs, 2H, –C=C–H); EI-MS m/z 343 (76), 307 (87), 272 (71), 244 (31), 166 (14), 138 (22), 112 (9); HREI-MS for C20H16Cl2O M+, calcd.: m/z 342.0678, found: m/z 342.0672.
(2E,6E)-2,6-bis(3,4-Dimethoxybenzylidene)cyclohexanone (10)
Yellow crystals; yield (74%); m.p. 146–149 °C (lit. [37] 148–150 °C); UV–Vis (CHCl3) λmax: 373 nm; IR (KBr) v 3036 (Ar C–H stretch), 1614 (C=O), 1489–1462 (Ar C=C) cm−1; 1H NMR (CDCl3, 600 MHz) δ 1.83 (m, 2H, 4-H), 2.95 (m, 4H, 3, 5-H), 3.90 (s, 6H, OCH3, C-3′, 3″), 3.92 (s, 6H, OCH3, C-4′, 4″), 6.91 (d, 2H, 5′, 5″-H, J = 8.34 Hz), 7.02 (d, 2H, 2′, 2″-H, J = 1.92 Hz), 7.12 (dd, 2H, 6′, 6″-H, J = 8.34, 1.92 Hz), 7.76 (brs, 2H, –C=C–H); EI-MS m/z 394 (3), 363 (100), 331 (9), 161 (4), 227 (23), 136 (3), 77 (31); HREI-MS for C24H26O5 M+, calcd.: m/z 394.1784, found: m/z 394.1787.
(1E,4E)-1,5-bis(2-Methoxyphenyl)-penta-1,4-dien-3-one (11)
Yellow crystals; yield (66%); m.p. 111–114 °C (lit. [45] 118–120 °C); UV–Vis (CHCl3)λmax: 312, 360 nm; IR (KBr) v 3023 (Ar C–H stretch), 1614 (C=O), 1489–1462 (Ar C=C) cm−1; 1H NMR (CDCl3, 600 MHz) δ 3.90 (s, 6H, OCH3, C-2′, 2″), 6.93 (d, 2H, 3′, 3″-H, J = 8.34 Hz), 6.99 (t, 2H, 4′, 4″-H, J = 8.46, 7.4 Hz), 7.18 (d, 2H, 2, 4-H, J = 16.08 Hz), 7.36 (td, 2H, 5′, 5″-H, J = 7.4, 1.6 Hz), 7.62 (dd, 2H, 6′, 6″-H, J = 7.6, 1.6 Hz), 8.08 (d, 2H, 1, 5-H, J = 16.08 Hz); EI-MS m/z 294 (100), 263 (8), 234 (15), 186 (50), 161 (36), 133 (33), 77 (16); HREI-MS for C19H18O3 M+, calcd.: m/z 294.1255, found: m/z 294.1251.
(1E,4E)-1,5-bis(4-Methoxyphenyl)-penta-1,4-dien-3-one (12)
Yellow crystals; yield (79%); m.p. 121–122 °C (lit. [46] 119–120 °C); UV–Vis (CHCl3) λmax: 354 nm; IR (KBr) v 3033 (Ar C–H stretch), 1624 (C=O), 1590–1488 (Ar C=C) cm−1. 1H NMR (CDCl3, 500 MHz) δ 3.87 (s, 6H, OCH3, C-4′, 4″), 6.94 (d, 4H, 3′, 3″, 5′, 5″-H, J = 8.75 Hz), 6.99 (d, 2H, 2, 4-H, J = 16.0 Hz), 7.60 (d, 4H, 2′, 2″, 6′, 6″-H, J = 8.75 Hz), 7.74 (d, 2H, 1, 5-H, J = 16.0 Hz); EI-MS m/z 294.14 (100), 263 (15), 234 (20), 186 (54), 161 (38), 133 (36), 77 (21); HREI-MS for C19H18O3 M+, calcd.: m/z 294.1264, found: m/z 294.1257.
(1E,4E)-1,5-bis(2,3-Dimethoxyphenyl)-penta-1,4-dien-3-one (13)
Yellow solid; yield (68%); m.p. 103–104 °C (lit. [36] 106–108 °C); UV–Vis (CHCl3) λmax: 330 nm; IR (KBr) v 3011–2943 (Ar C–H stretch), 1619 (C=O), 1577–1479 (Ar C=C) cm−1; 1H NMR (CDCl3, 600 MHz) δ 3.87 (s, 12H, OCH3, C-2′, 2″, 3′, 3″), 6.97 (dd, 2H, 4′, 4″-H, J = 8.16, 1.44 Hz), 7.10 (t, 2H, 5′, 5″-H, J = 8.04, 8.00 Hz), 7.16 (d, 2H, 2, 4-H, J = 16.1 Hz), 7.26 (dd, 2H, 6′, 6″-H, J = 8.00, 1.44 Hz), 8.04 (d, 2H, 1, 5-H, J = 16.1 Hz); EI-MS m/z 354 (5), 323 (3), 230 (4), 186 (9), 132 (13), 191 (4), 163 (7), 77 (52); HREI-MS for C21H22O5 M+, calcd.: m/z 354.1467, found: m/z 394.1462.
(1E,4E)-1,5-bis(4-Chlorophenyl)-penta-1,4-dien-3-one (16)
Yellow solid; yield (72%); m.p. 193–195 °C (lit. [36] 192–193 °C); UV–Vis (CHCl3) λmax: 333 nm; IR (KBr) v 3065 (Ar C–H stretch), 1608 (C=O), 1584–1489 (Ar C=C str.) cm−1; 1H-NMR (CDCl3, 500 MHz) δ 7.04 (d, 2H, 2, 4-H, J = 15.9 Hz), 7.40 (dd, 4H, 3′, 3″, 5′, 5″-H, J = 8.60 Hz), 7.56 (d, 4H, 2′, 2″, 6′, 6″-H, J = 8.60 Hz), 7.70 (d, H, 1, 5-H, J = 15.9 Hz); 13C NMR (150 MHz, CDCl3) δ 126.0 (C-2, 4), 128.7 (C-3′, 3″), 128.7 (C-5′, 5″), 129.3 (C-2′, 2″), 129.3 (C-6′, 6″), 133.3 (C-1′, 1″), 136.5 (C-4′, 4″), 142.1 (C-1, 5), 188.3 (C=O); EI-MS m/z 302 (60), 267 (32), 232 (5), 203 (20), 165 (35), 137 (49), 77 (5); HREI-MS for C17H12Cl2O M+, calcd.: m/z 302.0265, found: m/z 302.0259.
(1E,4E)-1,5-bis(2,4,6-Trimethoxyphenyl)-penta-1,4-dien-3-one (17)
Yellow solid; yield (68%); m.p. 213–215 °C (lit. [36] 209–211 °C); UV–Vis (CHCl3) λmax: 381 nm. IR (KBr) 3002 (Ar C–H str.), 1629 (C=O), 1561–1466 (Ar C=C) cm−1; 1H NMR (CDCl3, 600 MHz) δ 3.74 (s, 6H, 2 × OCH3, C-4′, 4″), 3.85 (s, 12H, 4 × OCH3, C-2′, 2″, 6′, 6″), 6.13 (brs, 4H, 3′, 3″, 5′, 5″-H), 7.46 (d, 2H, 2, 4-H, J = 16.30 Hz), 8.12 (d, 2H, 1, 5-H, J = 16.30 Hz); EI-MS m/z 414 (5), 131 (6), 105 (10); HREI-MS for C23H26O7 M+, calcd.: m/z 414.1671, found: m/z 414.1679.
(2E,5E)-2,5-bis(4-Methoxybenzylidene)cyclopentanone (19)
Yellow solid; yield (66%); m.p. 150–155.5 °C (lit. [29] 158–161 °C); UV–Vis (CHCl3) λmax: 391 nm; IR (KBr) v 2964 (Ar C–H stretch), 1696 (C=O), 1597–1509 (Ar C=C) cm−1; 1H NMR (CDCI3, 600 MHz) δ 3.09 (brs, 4H, 3, 4-H), 3.86 (s, 6H, 2 × OCH3, C-4′, 4″), 6.98 (brd, 2H, 5′, 5″-H, J = 8.34 Hz), 6.98 (brd, 2H, 3′, 3″-H, J = 8.34 Hz), 7.57 (brt, 2H, 2′, 2″-H, J = 8.52 Hz), 7.57 (brt, 2H, 6′, 6″-H, J = 8.52 Hz), 7.58 (brs, 2H, –C=C–H); EI-MS m/z 320 (11), 213 (8), 183 (5), 131 (12), 77 (16); HREI-MS for C21H20O3 M+, calcd.: m/z 320.1412, found: m/z 320.140.
(2E,5E)-2,5-bis(2,3-Dimethoxybenzylidene)cyclopentanone (20)
Yellow solid; yield (54%); m.p. 156–158 °C (lit. [36] 155–157 °C); UV–Vis (CHCl3) λmax: 346 nm; IR (KBr) v 3032 (Ar C-H stretch), 1694 (C=C), 1622 (C=O), 1584–1489 (Ar C=C) cm−1; 1H NMR (CDCI3, 600 MHz) δ 3.02 (brs, 4H, 3, 4-H), 3.87 (s, 6H, OCH3, C-2′, 2″), 3.88 (s, 6H, OCH3, C-3′, 3″), 6.96 (m, 2H, 4′, 4″-H), 7.10 (t, 2H, 5′, 5″-H, J = 7.9 Hz), 7.16 (dd, 2H, 6′, 6″-H, J = 7.9 Hz), 7.93 (brs, 2H, –C=C–H); EI-MS m/z 380 (3), 349 (4), 163 (10), 137 (10), 98 (18); HREI-MS for C23H24O5 M+, calcd.: m/z 380.1618, found: m/z 380.1623.
(2E,5E)-2,5-bis(4-Hydroxy-3-methoxybenzylidene)cyclopentanone (22)
Yellow solid; yield (58%); m.p. 212–214 °C (lit. [47] 214 °C); UV–Vis (CHCl3) λmax: 388 nm; IR (KBr) v 3043 (Ar C–H stretch), 1690 (C=C), 1620 (C=O), 1588–1485 (Ar C=C) cm−1; 1H NMR (CDCl3, 500 MHz) δ 3.03 (s, 4H, 3, 4-H), 3.88 (s, 6H, OCH3, C-3′, 3″), 6.92 (d, 2H, 5′, 5″-H, J = 8.30 Hz), 7.04 (brs, 2H, 2′, 2″-H), 7.14 (dd, 2H, 5′, 5″-H, J = 8.30, 1.65 Hz), 7.46 (brs, 2H, –C=C–H); EI-MS m/z 352; HREI-MS for C21H20O5 M+, calcd.: m/z 352.1310, found: m/z 352.1305.
(2E,5E)-2,5-bis(3,4-Dimethoxybenzylidene)cyclopentanone (23)
Yellow solid; yield (54%); m.p. 191–193 °C (lit. [37] 188–190 °C); UV–Vis (CHCl3) λmax: 368 nm; IR (KBr) v 3006 (Ar C–H stretch), 1693 (C=O), 1592–1515 (Ar C=C) cm−1; 1H NMR (CDCl3, 600 MHz) δ 3.12 (brs, 4H, 3, 4-H), 3.94, 3.93 (s, 12H, 4 × OCH3, C-3′, 3″, 4′, 4″), 6.96 (d, 2H, 5′, 5″-H, J = 8.34 Hz), 7.14 (s, 2H, 2′, 2″-H), 7.24 (dd, 2H, 6′, 6″-H, J = 8.34 Hz), 7.55 (brs, 2H, –C=C–H); 13C NMR (150 MHz, CDCl3) δ 26.3 (C-3, 4), 56.0 (C–O), 111.2 (C-2′, 2″), 113.5 (C-5′, 5″), 124.6 (C-6′, 6″), 129.0 (C-1′, 1″), 133.7 (–C=C–H), 148.9 (C-2, 5), 150.3 (C-3′, 3″), 150.3 (C-4′, 4″), 196.0 (C=O); EI-MS m/z 380.1 (5), 191.0 (10), 132.2 (18), 77.2 (55); HREI-MS for C23H24O5 M+, calcd.: m/z 380.1624, found: m/z 380.1619.
Anticancer activity
Sample preparation
Stock samples at 1 mg/mL of dimethyl sulfoxide (DMSO) (Sigma-Aldrich, USA) were prepared and keep at 4 °C.
MTT cell viability assay
Breast cancer MCF-7 and MDA-MB-231 cells, chronic myelogenous leukemia K562 cells, and cervical cancer HeLa cells lines were purchased from American Type Culture Collection (ATCC, USA) and cultured at 37 °C, 5% CO2 and 90% humidity using RPMI-1640 medium (Sigma-Aldrich, USA) supplemented with 10% Foetal Bovine Serum (FBS) (Thermo Fisher Scientific, USA). For MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) cell viability assay [48], MCF-7, MDA-MB-231, K562 and HeLa cells were seeded overnight in 96-well plates at 8 × 104 cells/well at 37 °C of CO2 [49]. Then, 100 µL of media was discarded for all well-plates and compounds were serially diluted into the seeded cells at the concentration ranging between 30–0.47 µg/mL with cells treated with 3% DMSO (Sigma-Aldrich, USA) as the negative control. All samples were tested for triplicates. After 72 h of incubation, all well was added with 20 µL of MTT solution (5 mg/mL) and further incubated for 3 h. At that point, 170 µL of solution were discarded and 100 µL of DMSO (Sigma-Aldrich, USA) was added to all wells. Finally, absorbance was recorded by ELISA plate reader (Biotek-Instruments, USA) at the wavelength of 570 nm. Percentage of cell viability was calculated using following formula [38, 39]. The assay was performed in triplicate to calculate the half maximal inhibitory concentration (IC50) values. Doxorubicin was used as a positive control.
Cell viability (%) = [OD sample at 570 nm/OD negative control at 570 nm] × 100%
IC50 value (concentration of compounds inhibited 50% of cell viability) was determined from the graph of cell viability vs absorbance.
X-ray crystallographic analysis
X-ray analysis for all these samples were performed using Bruker APEX II DUO CCD diffractometer, employing MoKα radiation (λ = 0.71073 Å) with φ and ω scans, at room temperature. Data reduction and absorption correction were performed using SAINT and SADABS programs [50,51,52,53]. The structures of compound 4 was solved by direct methods and refined by full-matrix least-squares techniques on F2 using SHELXTL software package. Crystallographic data of the reported structures have been deposited at the Cambridge Crystallographic Data Centre with CCDC deposition numbers of 1548735. Copy of available material can be obtained free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (Fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk).
Conclusions
In conclusion, we have examined three series of curcumin analogues against four types (HeLa, K562, MCF-7 and MDA-MB-231) cancer cell lines. Curcuminoids with diferuloyl (4-hydroxy-3-methoxycinnamoyl) moiety with mono carbonyl exhibiting potential cytotoxic properties. The compound 14 was exhibited (IC50 = 3.02 ± 1.20 and 1.52 ± 0.60 µg/mL) against MCF-7 and MDA-MB-231 breast cancer cell lines. Structure activity relationship revealed that the role of methoxy groups are important. Curcumin derivatives, 4, 5, 9, 14, 11 and 17 exhibited significant cytotoxic activity (Table 1). Curcuminoids with acetone series such as 2,5-dimethoxy substituted with mono ketones were found to be more selective and potential cytotoxic agents, which could be the best templet for future drug discovery against selective cancer especially breast cancer lines.
Abbreviations
- HeLa:
-
Henrietta Lacks
- MCF-7:
-
Michigan Cancer Foundation-7
- HCl:
-
hydrochloric acid
- TMS:
-
tetramethylsilane
- CDCI3 :
-
chloroform
- TLC:
-
thin layer chromatography
- MeOH:
-
methanol
- EtOH:
-
ethanol
- KOH:
-
potassium hydroxide
- NaOH:
-
sodium hydroxide
- NMR:
-
nuclear magnetic resonance
- IR:
-
infrared radiation
- UV-Vis:
-
ultraviolet visible
- MS:
-
mass spectrometry
- DMSO:
-
dimethyl sulfoxide
- MTT:
-
(3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide)
References
Ferlay J, Seorjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2013) GLOBOCAN 2012 v1.0, cancer incidence and mortality worldwide. IARC cancer base no. 11
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127:2893–2917
Siegel RL, Miller KD, Jemal A (2016) Cancer statistics. CA Cancer J Clin 66:7–30
Smith RA, Brooks D, Cokkinides V, Saslow D, Brawley OW (2013) Cancer screening in the United States, 2013: a review of current American Cancer Society guidelines, current issues in cancer screening, and new guidance on cervical cancer screening and lung cancer screening. CA Cancer J Clin 63:88–105
McCracken M, Olsen M, Chen MS Jr, Jemal A, Thun M, Cokkinides V, Deapen D, Ward E (2007) Cancer incidence, mortality, and associated risk factors among Asian Americans of Chinese, Filipimo, Vietnamese, Korean, and Japanese ethnicities. CA Cancer J Clin 57:190–205
Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ (2006) Cancer statistics, 2006. CA Cancer J Clin 56:106–130
Kuttan R, Bhanumathy P, Nirmala K, George MC (1985) Potential anticancer activity of turmeric. Cancer Lett 29:197–202
Ruby AJ, Kuttan G, Babu KD, Rajasekharan KN, Kuttan R (1995) Anti-tumour and antioxidant activity of natural curcuminoids. Cancer Lett 94:79–83
de Oliveira Silva E, Batista R (2017) Ferulic acid and naturally occurring compounds bearing a feruloyl moiety: a review on their structures, occurrence, and potential health benefits. Compr Rev Food Sci Food Saf 16:580–616
Shishodia S, Chaturvedi MM, Aggarwal BB (2007) Role of curcumin in cancer therapy. Curr Prob Cancer 31:243–305
Aggarwal BB, Kumar A, Bharti AC (2003) Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res 23:363–398
Ishida J, Ohtsu H, Tachibana Y, Nakanishi Y, Bastow KF, Nagai M, Wang HK, Itokawa H, Lee KH (2002) Antitumor agents. Part 214: synthesis and evaluation of curcumin analogues as cytotoxic agents. Bioorg Med Chem 10:3481–3487
Duvoix A, Blasius R, Dehalle S, Schnekenburger M, Morceau F, Henry E, Dicato M, Diederich M (2005) Chemopreventive and therapeutic effects of curcumin. Cancer Lett 223:181–190
Chainani-Wu NJ (2002) Safety and anti-inflammatory activity of curcumin. J Altern Complement Med 9:161–168
Dikshit M, Rastogi L, Shukla R, Srimal RC (1995) Prevention of ischaemia induced biochemical changes by curcumin and quinidine in the cat heart. Indian J Med Res 101:31–35
Sharma OP (1976) Antioxidant activity of curcumin and related compounds. Biochem Pharmacol 25:1811–1812
Venkatesan N (1998) Curcumin attenuation of acute adriamycin myocardial toxicity in rats. Br J Pharmacol 124:425–427
Kiso Y, Suzuki Y, Watanabe N, Oshima Y, Hikino H (1983) Antihepatotoxic principles of Curcuma longa. Planta Med 49:185–187
Venkatesan N, Punithavathi D, Arumugam V (2000) Curcumin prevents adriamycin nephrotoxicity in rats. Br J Pharmacol 129:231–234
Srivastava R, Dikshit M, Srimal RC, Dhawan BN (1985) Anti-thrombotic effect of curcumin. Thromb Res 40:413–417
Srinivasan M (1972) Effect of curcumin on blood sugar as seen in a diabetic subject. Indian J Med Sci 26:269–270
Al-Howail HA, Hakami HA, Al-Otaibi B, Al-Mazrou A, Daghestani MH, Al-Jammaz I, Al-Khalaf HH, Aboussekhra A (2016) PAC down-regulates estrogen receptor alpha and suppresses epithelial-to-mesenchymal transition in breast cancer cells. BMC Cancer 16:1–11
Hallman K, Aleck K, Dwyer B, Lloyd V, Quigley M, Sitto N, Siebert AE, Dinda S (2017) The effects of turmeric (curcumin) on tumor suppressor protein (p53) and estrogen receptor (ERα) in breast cancer cells. Breast Cancer Targets Ther 9:153–161
Somers-Edgar TJ, Taurin S, Larsen L, Chandramouli A, Nelson MA, Rosengren RJ (2011) Mechanisms for the activity of heterocyclic cyclohexanone curcumin derivatives in estrogen receptor negative human breast cancer cell lines. Invest New Drugs 29:87–97
Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB (2007) Bioavailability of curcumin: problems and promises. Mol Pharm 4:807–818
Anand P, Sundaram C, Jhurani S, Kunnumakkara AB, Aggarwal BB (2008) Curcumin and cancer: an “old-age” disease with an “age-old” solution. Cancer Lett 267:133–164
Shehzad A, Lee J, Lee YS (2013) Curcumin in various cancers. BioFactors 1:56–68
Zhao C, Liu Z, Liang G (2013) Promising curcumin-based drug design: mono-carbonyl analogues of curcumin (MACs). Curr Pharm Des 19:2114–2135
Wei X, Du ZY, Zheng X, Cui XX, Conney AH, Zhang K (2012) Synthesis and evaluation of curcumin-related compounds for anticancer activity. Eur J Med Chem 53:235–245
Nakhjiri M, Safavi M, Alipour E, Emami S, Atash AF, Jafari-Zavareh M, Ardestani SK, Khoshneviszadeh M, Foroumadi A, Shafiee A (2012) Asymmetrical 2,6-bis(benzylidene)cyclohexanones: synthesis, cytotoxic activity and QSAR study. Eur J Med Chem 50:113–123
Abd Razak N, Akhtar MN, Abu N, Lim KL, Ky H, Ho WY, Tan SW, Zareen S, Tajuddin SN, Alitheen NB, Yeap SK (2017) The in vivo anti-tumor effect of curcumin derivative (2E,6E)-2,6-bis(4-hydroxy-3-methoxybenzylidene)cyclohexanone (BHMC) on 4TI breast cancer cells. RSC Adv 7:36185–36192
Ali NM, Yeap SK, Abu N, Lim KL, Ky H, Mat Pauzi AZ, Ho WY, Tan SW, Kiat AOH, Zareen S, Alitheen NB, Akhtar MN (2017) Synthetic curcumin derivative DK1 possessed G2/M arrest and induced apoptosis through accumulation of intracellular ROS in MCF-7 breast cancer cells. Cancer Cell Int 17:1–12
Abu N, Akhtar MN, Yeap SK, Lim KL, Ho WY, Abdullah MP, Ho CL, Omar AR, Ismail J, Alitheen NB (2016) Flavokawain B induced cytotoxicity in two breast cancer cell lines, MCF-7 and MDA-MB231 and inhibited the metastatic potential of MDA-MB231 via regulation of several tyrosine kinases in vitro. BMC Complement Altern Med 16:1–14
Yeap SK, Akhtar MN, Lim KL, Abu N, Ho WY, Zareen S, Roohani K, Ky H, Tan SW, Lajis N, Alitheen NB (2015) Synthesis of an anthraquinone derivative (DHAQC) and its effect on induction of G2/M arrest and apoptosis in breast cancer MCF-7 cell line. Drug Des Dev Ther 9:983–992
Dinkova-Kostova AT, Abeygunawardana C, Talalay P (1998) Chemoprotective properties of phenylpropenoids, Bis(benzylidene)cycloalkanones, and related michael reaction acceptors: correlation of potencies as phase 2 enzyme inducers and radical scavengers. J Med Chem 41:5287–5296
Hosoya T, Nakata A, Yamasaki F, Abas F, Shaari K, Lajis NH, Morita H (2012) Curcumin-like diarylpentanoid analogues as melanogenesis inhibitors. J Nat Med 66:166–176
Du ZY, Liu RR, Shao WY, Mao XP, Ma L, Gu LQ, Huang ZS, Chan ASC (2006) α-Glucosidase inhibition of natural curcuminoids and curcumin analogs. Eur J Med Chem 41:213–218
Fuchs JR, Pandit B, Bhasin D, Etter JP, Regan N, Abdel Hamid D, Li C, Lin J, Li PK (2009) Structure–activity relationship studies of curcumin analogues. Bioorg Med Chem Lett 19:2065–2069
Zhang Q, Fu Y, Wang HW, Gong T, Qin Y, Zhang ZR (2008) Synthesis and cytotoxic activity of novel curcumin analogues. Chin Chem Lett 19:281–285
Sardjiman SS, Reksohadiprodjo MS, Hakim L, van der Goot H, Timmermanz H (1997) 1,5-Diphenyl-1,4-pentadiene-3-ones and cyclic analogues as antioxidative agents. Synthesis and structure–activity relationship. Eur J Med Chem 32:625–630
Yin S, Zheng X, Yao X, Wang Y, Liao D (2013) Synthesis and anticancer activity of mono-carbonyl analogues of curcumin. J Cancer Ther 4:113–123
Ahn BZ, Sok DE (1996) Michael acceptors as a tool for anticancer drug design. Curr Pharmaceut Des 2:247–262
Landais I, Hiddingh S, McCarroll M, Yang C, Sun A, Turker MS, Snyder JP, Hoatlin ME (2009) Monoketone analogs of curcumin, a new class of Fanconi anemia pathway inhibitors. Mol Cancer 8:1–13
Katsori AM, Chatzopoulou M, Dimas K, Kontogiorgis C, Patsilinakos A, Trangas T, Hadjipavlou-Litina D (2011) Curcumin analogues as possible anti-proliferative & anti-inflammatory agents. Eur J Med Chem 46:2722–2735
Liang G, Yang S, Jiang L, Zhao Y, Shao L, Xiao J, Ye F, Li Y, Li X (2008) Synthesis and anti-bacterial properties of mono-carbonyl analogues of curcumin. Chem Pharm Bull 56:162–167
Al-Kadhimi AA, Al-Hamdany AJ, Jasim SS (2012) Synthesis and antibacterial evaluation of bis-pyrrolidinyl ketones. Res J Pharm Biol Chem Sci 3:908–921
Liang G, Shao L, Wang Y, Zhao C, Chu Y, Xiao J, Zhao Y, Li X, Yang S (2009) Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorg Med Chem 17:2623–2631
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
Ali NM, Akhtar MN, Ky H, Lim KL, Abu N, Zareen S, Ho WY, Alan-Ong HK, Tan SW, Alitheen NB, Ismail JB, Yeap SK, Kamarul T (2016) Flavokawain derivative FLS induced G2/M arrest and apoptosis on breast cancer MCF-7 cell line. Drug Des Dev Ther 10:1897–1907
Bruker AXS Inc. (2009) APEX2, SAINT and SADABS. Bruker AXS Inc., Madison
Sheldrick GM (2008) A short history of SHELX. Acta Cryst 64:112–122
Spek AL (2009) Structure validation in chemical crystallography. Acta Cryst 65:148–155
Macrae CF, Edgington PR, McCabe P, Pidcock E, Shields GP, Taylor R, Towler M, van de Streek J (2006) Mercury: visualization and analysis of crystal structures. J Appl Cryst 39:453–457
Authors’ contributions
SNHZ, MNA and SZ carried the literature and designed synthetic schemes (synthesis and purification), SKY, NBA and YH contributed to study of cancer cell lines of curcuminoids, CKQ and WSL contributed to X-ray analysis of compound, SAAS record the NMR of all compounds. All authors read and approved the final manuscript.
Acknowledgements
We are thankful to the Universiti Malaysia Pahang (http://www.ump.edu.my) and Ministry of Education Malaysia for award of the FRGS l Grant RDU 150109, 150349 and 150356. The authors thankful to USM for Fundamental Research Grant Scheme (FRGS) (203/PFIZIK/6711411) and RUPRGS Grant (1001/PFIZIK/846076). For the analysis of HREI-MS, greatly thankful to HEJ Research Institute of Chemistry, Universiti of Karachi, Pakistan.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zamrus, S.N.H., Akhtar, M.N., Yeap, S.K. et al. Design, synthesis and cytotoxic effects of curcuminoids on HeLa, K562, MCF-7 and MDA-MB-231 cancer cell lines. Chemistry Central Journal 12, 31 (2018). https://doi.org/10.1186/s13065-018-0398-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-018-0398-1