
Abstract
Background
Isoxazoles exhibit interesting biological activities, and the 1,3-dipolar cycloaddition(13DC) reactions play an important role in both mechanistic and synthetic organic chemistry. Pyrazoles and annulated pyrazoles exhibit some diverse biological activities. They are used as antipyretic, analgesic drugs, tranquilizing, and herbicidal agents. Pyrazoles are also used extensively as useful synthons in organic synthesis. Pyrazolo[3,4-d]pyridazines showed good antimicrobial, anti-inflammatory and analgesic activities. Several oximes are found to be hyperglycemic, anti-neoplastic, anti-inflammatory, anti-leishmanial and VEGFR-2 kinase inhibitors.
Results
The present work describes an efficient synthesis protocol and molecular orbital calculations of isoxazoline and pyrrolo[3,4-d]isoxazole-4,6-dione derivatives from the reaction of hydroximoyl chloride with acrylonitrile, acrylamide, and N-arylmalemides. In addition, pyrazoles and pyrazolo[3,4-d]pyridazines are obtained via the reaction of 3-(dimethylamino)-1-(2,4-dimethyl-1,3-thiazol-5-yl)prop-2-ene-1-one with hydrazonoyl halides. Pyrazolo[1,5-a]pyrimidines were derived from condensation of either Sodium Salt of 3-Hydroxy-1-(2,4-dimethylthiazol-5-yl)prop-2-en-1-one (10) or 3-(dimethylamino)-1-(2,4-dimethyl)(1,3-thiazol-5-yl)prop-2-en-1-one (11) with aiminopyrozoles. A comparative study of the biological activity of the synthesized compounds with ampicillin and tetracycline is compiled in Table 3. Generally, all synthesized compounds showed an adequate inhibitory efficiency of growth of gram-positive and gram-negative bacteria. Structures of the newly synthesized compounds were elucidated by elemental analysis, spectral data and a computational study.
Conclusions
In summary, new and efficient synthetic routes of isoxazoline, pyrrolo[3,4-d]isoxazole-4,6-dione derivatives, pyrazoles, pyrazolo[3,4-d]pyridazines and pyrazolo[1,5-a]pyrimidines have been achieved and the biological activity has been investigated.

New and efficient synthetic routes of isoxazoline, pyrrolo[3,4-d]isoxazole-4,6-dione derivatives, pyrazolo[3,4-d]pyridazines and pyrazolo [1,5-a] pyrimidines
Similar content being viewed by others


Background
Isoxazoles exhibit interesting biological activities [1, 2], and oxazoles are widely recognized for their therapeutic purposes, especially as tranquillizing agents and CNS regulates. They are known to have bacteriostatic, bactericidal and fungicidal activities [3]. The 1,3-dipolar cycloaddition(13DC) reactions play an important role in both mechanistic and synthetic organic chemistry. Pyrazoles and annulated pyrazoles exhibit some diverse biological activities. They are used as antipyretic [4], analgesic drugs [5–7], tranquilizing [8] and herbicidal [9] agents. Pyrazoles are also used extensively as useful synthons in organic synthesis [10–17]. Recently, the synthesis of biologically active compounds based on pyrazolo[3,4-d]pyridazines systems are of outstanding importance for medicinal and biological utilities [18, 19], Generally, pyrazolo[3,4-d]pyridazines showed good antimicrobial, anti-inflammatory and analgesic activities [20]. Herein, we report a facile synthesis procedure for some new derivatives of the newly developed isoxazoline, pyrrolo[3,4-d]isoxazole-4,6-dione derivatives, pyrazoles, pyrazolo[3,4-d]pyridazines and pyrazolo[1,5-a] pyrimidines.
Results and discussion
Chemistry
The reaction of 1-(2,4-dimethyl(1,3-thiazol-5-yl))-2-bromoethan-1-one (1) [21] with dimethylsulfide in refluxing ethanol has afforded 1-(2,4-dimethylthiazol-5-yl)-2-oxodimethylsulfonium bromide (2) [21], furthermore, the nitrosation of (2) in dioxane-water solution acidified with hydrochloric acid gave 2-chloro-2-(hydroximino)-1-(2,4-dimethyl-1,3-thiazol-5-yl)ethanone (3). The chemical structure of (3) was confirmed based on elemental analysis, spectral data, and chemical transformations. The 1H NMR spectrum showed signals at δ = 2.47 (s, 3H, CH3), 2.71 (s, 3H, CH3) and 13.18 (s, 1H, NOH). The IR spectrum revealed absorption bands at 3370 cm−1 (OH) and 1655 cm−1 (CO conjugated) (Scheme 1).

Synthesis of 2-chloro-2-(hydroximino)-1-(2,4-dimethylthiazol-5-yl)ethanone (3)
Moreover, the treatment of 2-chloro-2-(hydroximino)-1-(2,4-dimethylthiazol-5-yl)ethanone (3) with acrylonitrile in boiling toluene afforded an insoluble product, according to TLC, of which structures (4) and (5) seemed to be possible (Scheme 2). The 1H NMR spectrum of the product showed signals at δ = 2.57 (s, 3H, CH3) 2.80, (s, 3H, CH3), 2.90–2.94 (d, 2H, CH2, J = 10 Hz, isoxazoline C-4) and 3.85 (t, 1H, J = 10 Hz, isoxazoline C-5). Its IR spectrum revealed bands at 1665 cm−1 (CO). However, no absorption bands appeared at 2200 cm−1 corresponding to the CN group in support of the 5-cyano structure [22]. The product was readily hydrolyzed by dilute sulfuric acid to give the corresponding 3-[(2,4-dimethyl-1,3-thiazol-5-yl)carbonyl]-4,5-dihydroisoxazole-5-carboxamide (6) (IR spectral bands at 3350, 3170 cm−1 (NH2) and 1680 cm−1 (CO)). In addition, refluxing of 2-chloro-2-(hydroximino)-1-(2,4-dimethylthiazol-5-yl)ethanone (3) with acrylamide in a boiling toluene furnished an identical product with compound (6) in all aspects (m.p., mixed m.p., spectra). Hence, the proposed structure (4) was excluded and the product was assigned to a formulated structure of 3-[(2,4-dimethyl-1,3-thiazol-5-yl)carbonyl]-4,5-dihydro-isoxazole-5-carbonitrile (5). Also, the compound (3) was reacted with the appropriate N-arylmalemides (7a–c) in boiling toluene and produced 3-[(2,4-dimethyl-1,3-thiazol-5-yl)carbonyl]-5-substituted-3(aH)-pyrrolo[3,4-d]isoxazole-4,6 (5H,6aH)dione (8a-c), respectively (Scheme 2).

Synthesis of isoxazoline, pyrrolo[3,4-d]isoxazole 5 and (8a–c)
The structures of compounds (8) were confirmed by elemental analysis and spectral data. The IR spectra of (8a–c) revealed bands at 1730 and 1637 cm−1 assigned for CO and -CO-NAr-CO- groups [23]. The 1H NMR spectrum of (8a) showed signals at δ = 2.66 (s, 3H, CH3), 2.92 (s, 3H, CH3), 5.23–5.24 (d, 1H, J = 7.4 Hz, isoxazoline C-4), 5.81–5.88 (d, 1H, J = 7.4 Hz, isoxazoline C-5), and 7.22–7.33 (m, 5H, ArH’s).
Quantum chemical calculations
Computational methods
All calculations have been carried out using the Gaussian 09 suite of programs [24]. The geometries of the reactants, transition states and products have been fully optimized at the DFT/B3LYP/6-311 ++G** level of the theory [25–28]. Frequency calculations were performed at the same level of the theory in order to characterize the stationary points and to evaluate the zero-point energy (ZPE), free energies (G) and enthalpies (H) at 298.15 K. TSs had only one imaginary frequency.
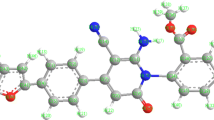
The interaction between acrylonitrile and 2,4-dimethylthiazol (13DC) can give two isomeric structure 5 (head-to-head) or 4 (head-to-tail) as shown in Scheme 3. There are some theoretical studies of the 1,3-dipolar cycloadditions of carbon materials [29–31]. Density functional theory (DFT) is employed to investigate the 13DC reaction. We report a computational study of regioselectivity of 2,4-dimethylthiazol (3) cycloadditions to acrylonitrile dipolarophiles. Our main objective in obtaining these results is to calculate the energy barrier for the 13DC reaction. B3LYP method confirms that the 5 geometry is preferred by 3.789 kJ mole−1, see Table 1. Our results are in complete agreement with experimental which indicated that the 5 conformer is the product from the above reaction. The calculated geometries of the stationary points corresponding to this 13DC reaction (reactants, transition states, and products) are presented in Fig. 1. The total and relative energies are shown in Tables 1 and 2. The TSs structures from B3LYP calculations for 13DC are very similar with minor changes in the bond distances and energies, see Tables 1 and 2. Four bond distances are important in 13 DC reaction. Two existing double bonds elongate (C=C and C=N) and two new bonds form (C–C and C–O) during this cycloaddition reaction. The C–C bond lengths of the acrylonitrile are only ~0.02 longer in the transition states than in the reactant. Similarly, the C–N and N–O bond lengths of structure 3 are only changed by 0.013 Å longer in the transition states than in the reactant. It is clear to note that, each transition state involves significant bending of structure 3 angle from its planar ground-state geometry to a product-like bending angle. The distorted structure for 3 structure with bending angle is named 3D as presented in Fig. 1. The bending angle changes from the ground state to the transition state range from 180° to ~120°. The distortion energy for structure 3 is 199.247 kJ mole−1. The corresponding activation barrier, enthalpy, free energy and reaction energies are given in Table 2. As mentioned before, the studied 13DC reaction favors structure 5 product with the lower activation energy (72.328 kJ mole−1) and high negative values of enthalpy and free energy.

Regioisomeric pathways for the studied cycloaddition reaction

Optimized geometries obtained by B3LYP/6-311 ++G** of all spices in the studied 13DC reaction. The bond lengths are given in angstroms
The frontier molecular orbital (FMO) obtained by B3LYP/6-311 ++G** level of the theory of the studied molecules are plotted in Fig. 2. The energies and shape of the FMO (HOMO and LUMO) for both 1,3-dipole 3 and dipolarophile determine the chemical reactivity in cycloaddition reactions. Hence, the interaction between the FMO is important to rationalize of the cycloaddition processes. The computations demonstrate that the distortion structure 3 to give 3D structure decrease HOMO–LUMO separation energy, which is capable to react with acrylonitrile. When the separation energy between the interaction orbital small, the better they interact. Comparing the energies of the FMO, HOMO–LUMO of the dipolarophile and 3, we can suggest that 13DC reaction as HOMO for 3 controlled. The interaction of the dipole HOMO with the dipolarophile LUMO is greater, due to small separation interaction energy between them. The computations show that the structure 3 has a small HOMO–LUMO gap energy (4.358 eV) compared to 3D structure (3.319 eV). As shown in Fig. 2, the interaction energy separation between HOMO of 3D (1,3 dipole) and LUMO of dipolarophile is 4.480 eV compared to the value of HOMO for 3 and LUMO for dipolarophile which it is 5.074 eV. It is shown from the calculations that the reaction of 3 structure (1,3 dipolar) with acrylonitrile (dipolarophiles) is occurred during charge transfer from HOMO of structure 3 to LUMO of acrylonitrile. With respect to the shape of HOMO and LUMO, only if the interacting lobes are in phase the reaction is thermally feasible. The lobes of HOMO for 3D and LUMO of dipolarophile are in the same phase, which it is thermally feasible.

FMO interactions in 13DC and the HOMO, LUMO, gap energy (HOMO–LUMO) and interaction separation energies (HOMO–LUMO) (eV) are calculated at B3LYP/6-311 ++G**. Values given in red color are for distortion structure 3D
Besides, the compound 1-(2,4-dimethyl-1,3-thiazol-5-yl)ethanone (9) was reacted with ethyl formate in dried ether containing sodium methoxide and afforded the sodium salt of 3-hydroxy-1-(2,4-dimethylthiazol-5-yl)prop-2-en-1-one (10). The structure of compound (10) was elucidated by its chemical transformations. Furthermore, treatment of compound (10) with 3-amino-4-phenyl-1H-pyrazole or 3-amino-5-phenyl-1H-pyrazole in glacial acetic acid containing piperidenum acetate afforded compounds 5-(2,4-dimethyl-1,3-thiazol-5-yl)-3(or 4)-phenylpyrazolo[1,5-a]pyrimidines (12a,b), respectively (Scheme 4). The structures of compounds (12a,b) were elucidated by elemental analysis, spectral data, and an alternate synthetic route. The 1H NMR spectrum of (12a) showed signals at δ = 2.59 (s, 3H, CH3), 2.83 (s, 3H, CH3), 6.53 (s, 1H, pyrazole H-4), 7.13 (d, 1H, J = 4 Hz, pyrimidine H-5), 7.54–7.92 (m, 5H, ArH’s) and 8.74 (d, 1H, 8 Hz, pyrimidine H-6). On the other hand, refluxing of 5-acetyl-2,4-dimethylthiazole (9) with dimethylformamide-dimethylacetal in boiling dry xylene gave the compound 3-(dimethylamino)-1-(2,4-dimethyl-1,3-thiazol-5-yl))prop-2-ene-1-one (11) in a good yield. Chemical elucidation of compound (11) was confirmed by elemental analysis, spectral data, and chemical transformation. The 1H NMR spectrum showed signals at δ = 2.49 (s, 3H, CH3), 2.78 (s, 3H, CH3), 2.98 (s, 3H, CH3), 3.15 (s, 3H, CH3), 5.49–5.54 (d, 1H, J = 12 Hz, CH=CH–N) and 6.90–7.28 (d, 1H, J = 12 Hz, CH=CH–N). Further reaction of the compound (11) with 3-amino-4-phenylpyrazole or 3-amino-5-phenyl-1H-pyrazole in a mixture of acetic acid and ammonium acetate gave identical products in all aspects (m.p., mixed m.p., spectra) to (12a,b) (Scheme 4).

Synthesis of pyrazolo[1,5-a]pyrimidines (12a,b)
Finally the treatment of C-ethoxycarbonyl-N-phenylhydrazonoyl chloride [32–35] (13) with compound (11) in refluxing toluene containing triethylamine catalyst yielded a new isolable product, which formulated as either ethyl 3-[(2,4-dimethyl-1,3-thiazol-5-yl)carbonyl]-1-phenyl-pyrazole-4-carboxylate (20a) or ethyl 3-[(2,4-dimethyl-1,3-thiazol-5-yl)carbonyl]-1-phenyl-pyrazole-5-carboxylate (21a) (Scheme 5). The structures of compounds (20) were elucidated by their spectral, elemental analysis and chemical transformation. The 1H NMR spectrum of compound (20a) showed a characteristic signals at δ = 1.22 (t, 3H, CH3, J = 7 Hz), 2.44 (s, 3H, CH3), 2.69 (s, 3H, CH3), 4.33 (q, 2H, CH2, J = 7 Hz), 7.44–7.89 (m, 5H, ArH’s) and 8.19 (s, 1H, pyrazole C-5). Formation of compounds (20) can be verified via chemical reaction of nitrilum imide (17), formed in situ from hydrazonoyl halides and triethylamine, with compound (11) which afforded the cycloadduct intermediate (18) or (19). After elimination of dimethylamine, the pyrazoles were obtained as final products (20) or (21). Similarly, the appropriate hydrazonoyl halides (14–16) reacted with a compound (11) to afford the corresponding pyrazoles (20b–d).

Synthesis of pyrazolo[3,4-d]pyridazines (22a–c)
Boiling of the appropriate pyrazoles (20a–d) with hydrazine hydrate in ethanol yielded Pyrazolo[3,4-d]pyridazines (22a–c) (Scheme 5). The chemical structures of compounds (22a–c) were elucidated via elemental analysis, spectral data and alternative synthesis. The 1H NMR spectrum of compound (22b) depicted signals at δ = 2.46 (s, 3H, CH3), 2.77 (s, 3H, CH3), 3.01 (s, 3H, CH3), 7.32–8.11 (m, 5H, ArH’s), 8.76 (s, 1H, pyrazole C-5). Alternatively, a new route for the synthesis of the compound (22a), the compound (20d) was refluxed with hydrazine hydrate in ethanol to give an identical product in all aspects (m.p., mixed m.p., and spectra) with compound (22a).
Conclusions
In summary, new and efficient synthetic routes of isoxazoline, pyrrolo[3,4-d]isoxazole-4,6-dione derivatives, pyrazoles, pyrazolo[3,4-d]pyridazines and pyrazolo [1,5-a] pyrimidines have been achieved, and computational investigations are in complete agreement with experimental. Moreover, the selected newly synthesized products were evaluated for their antimicrobial activity against gram positive and gram negative bacteria as well as some fungal-plants. The results revealed all synthesized compounds showed an adequate inhabitory efficiency of growth of gram positive and gram negative bacteria.
Experimental section
General methods
All melting points were determined on an electrothermal apparatus and are uncorrected. IR spectra were recorded (KBr discs) on a Shimadzu FT-IR 8201 PC spectrophotometer. 1H NMR and 13C NMR spectra were recorded in CDCl3 and (CD3)2SO solutions on a Varian Gemini 300 MHz spectrometer, and chemical shifts are expressed as δ using TMS as an internal reference. Mass spectra were recorded on a GC–MS QP1000. Elemental analyses were carried out at the Microanalytical Center of Cairo University. The hydrazonoyl halides [32–35] and hydroximoyl chloride [36] were prepared as previously described.
Synthesis of 2-chloro-2-(hydroximino)-1-(2,4-dimethyl)thiazol-5-yl)ethanone (3)
Hydrochloric acid (12 M, 100 ml) was added while stirring to a mixture of 1-(2,4-dimethylthiazol-5-yl)-2-oxodimethyl-sulfonium bromide (2) (11.8 g, 0.04 mol), sodium nitrite (3.5 g, 0.05 mol) in 1,4-dioxane (50 ml) and water (50 ml) at 25 °C. Stirring was continued for 3 h to produce a pale yellow solid, which was separated by filtration and recrystallized from ethanol to give (3). Yellow solid; Yield (62 %); m.p. 142 °C. IR (KBr) νmax: 3370 (OH), 3055, 2966 (CH), 1655 (CO conjugated) cm−1; 1H NMR (DMSO-d 6): 2.47 (s, 3H, CH3), 2.71 (s, 3H, CH3) and 13.18 (s, 1H, NOH); MS m/z (%): 218 (M+, 100). Anal. Calcd for C7H7ClN2O2S (218.66): C, 38.45; H, 3.23; N, 12.81; S, 14.66; Found: C, 38.43; H, 3.22; N, 12.79; S, 14.64 %.
Synthesis of 3-[(2,4-dimethyl-1,3-thiazol-5-yl)carbonyl]-4,5-dihydroisoxazole-5-carbonitrile (5), 3-[(2,4-dmethy-1,3-thiazol-5-yl)carbonyl]-4,5-dihydroisoxazole-5-carboxamide (6) and 3-[(2,4-dimethyl-1,3-thiazol-5-yl)carbonyl]-5-substituted 3aH-pyrrolo[3,4-d]isoxazole-4,6(5H,6aH)-dione (8a-c)
General method
Equimolar amounts of the appropriate (3), acrylonitrile, acrylamide or the appropriate N-arylmalemides (7a–c) (0.005 mol each) in toluene (30 ml) were heated under reflux for 18 h. The solvent was evaporated under vacuum and the residual oil was triturated with petroleum ether (40–60 °C). The solid products were collected and recrystallized from ethanol to give (5, 6) and (8a–c), respectively.
Alternative method for synthesis of 3-[(2,4-dimethyl-1,3-thiazol-5-yl)carbonyl]-4,5-dihydroisoxazole-5-carboxamide (6)
A mixture of 3-[(2,4-dimethyl-1,3-thiazol-5-yl)carbonyl]-4,5-dihydroisoxazole-5-carbonitrile 5 (0.5 g) and dilute sulfuric acid (5 ml) were stirred at room temperature for 1 h, and then poured onto crushed ice (20 g). The resulting solid was collected and recrystallized from ethanol to give a product identical in all aspects (m.p., mixed m.p., and spectra) with (6).
3-[(2,4-Dimethyl-1,3-thiazol-5-yl)carbonyl]-4,5-dihydroisoxazole-5-carbonitrile (5)
Yellow solid; Yield (73 %); m.p. 101 °C. IR (KBr) νmax: 2916, 2846 (CH), 1665 (CO) cm−1; 1H NMR (DMSO-d 6): 2.57 (s, 3H, CH3) 2.80, (s, 3H, CH3), 2.90–2.94 (d, 2H, CH2, J = 10 Hz, isoxazoline C-4) and 3.85 (t, 1H, J = 10 Hz, isoxazoline C-5); MS m/z (%):234 (M+, 70).Anal. Calcd for C10 H9N3O2S (235.26): C, 51.05; H, 3.86; N, 17.86; S, 13.63; Found: C, 50.80; H, 3.84; N, 17.89; S, 13.67 %.
3-[(2,4-Dimethy-1,3-thiazol-5-yl)carbonyl]-4,5-dihydroisoxazole-5-carboxamide (6)
Red solid; yield (66 %); m.p. 168 °C. IR (KBr) νmax: 3350, 3170 (NH2), 2920, 2856 (CH), 1680 (CO) cm−1; 1H NMR (DMSO-d 6): 2.46 (s, 3H, CH3), 2.79 (s, 3H, CH3), 3.54–3.60 (dd, 2H, J = 10.98 Hz), 5.20–5.29 (t, 1H, J = 8.72 Hz), 5.95 (s, br., 1H), 6.45 (s, br., 1H); MS m/z (%): 253 (M+, 67).Anal. Calcd for C10 H11 N3 O3 S (253.27): C, 47.42; H, 4.38; N, 16.59; S, 12.66; found: C, 47.40; H, 4.36; N, 16.61; S, 12.68 %.
3-[(2,4-Dimethyl-1,3-thiazol-5-yl)carbonyl]-5-phenyl-3aH-pyrrolo[3,4-d]isoxazole-4,6(5H,6aH)-dione (8a)
Yellow solid; yield (76 %); m.p. 150 °C. IR (KBr) νmax: 2926, 2855 (CH), 1717, 1639 (CO’s), 1638 (C=N) cm−1; 1H NMR (DMSO-d6): 2.66 (s, 3H, CH3), 2.92 (s, 3H, CH3), 5.23–5.24 (d, 1H, J = 7.4 Hz, isoxazoline C-4), 5.81–5.88 (d, 1H, J = 7.4 Hz, isoxazoline C-5), 7.22–7.33 (m, 5H, ArH’s); MS m/z (%): 355 (M+, 77).Anal. Calcd for C17 H13 N3 O4 S (355.36): C, 57.46; H, 3.69; N, 11.82; S, 9.02; found: C, 57.44; H, 3.71; N, 11.84; S, 8.99 %.
3-[(2,4-Dimethyl-1,3-thiazol-5-yl)carbonyl]-5-(4-methylphenyl)-3aH-pyrrolo[3,4-d]isoxazole-4,6(5H,6aH)-dione (8b)
Yellow solid; yield (80 %); m.p. 156 °C. IR (KBr) νmax: 2923, 2853 (CH), 1719, 1633 (CO’s), 1637 (C=N) cm−1; 1H NMR (DMSO-d6): 2.32 (s, 3H, 4-CH3C6H4), 2.54 (s, 3H, CH3), 2.89 (s, 3H, CH3), 5.18–5.22 (d, 1H, J = 9.78 Hz, isoxazoline C-4), 5.75–5.79 (d, 1H, J = 9.70 Hz, isoxazoline C-5) and 7.18–7.29 (m, 4H, ArH’s); MS m/z (%): 369 (M+, 90).Anal. Calcd for C18 H15 N3 O4 S (369.39): C, 58.53; H, 4.09; N, 11.38; S, 8.68; found: C, 58.55; H, 4.12; N, 11.40; S, 8.70 %.
3-[(2,4-Dimethyl-1,3-thiazol-5-yl)carbonyl]-5-(4-methoxyphenyl)-3aH-pyrrolo[3,4-d]isoxazole-4,6(5H,6aH)-dione (8c)
Yellow solid; yield (77 %); m.p. 165 °C. IR (KBr) νmax: 2926, 2918, 2849 (CH), 1716, 1634 (CO’s), 1640 (C=N) cm−1; 1H NMR (DMSO-d6): 2.52 (s, 3H, CH3), 2.89 (s, 3H, CH3), 3.83 (s, 3H, 4-OCH3C6H4), 5.15–5.17 (d, 1H, J = 7.34 Hz, isoxazoline C-4), 5.73–5.77 (d, 1H, J = 9.40 Hz, isoxazoline C-5) and 6.95–7.29 (m, 4H, ArH’s); MS m/z (%): 385 (M+, 88).Anal. Calcd for C18 H15 N3 O5 S (385.39): C, 56.10; H, 3.92; N, 10.90; S, 8.32; Found: C, 56.12; H, 3.94; N, 10.88; S, 8.34 %.
Synthesis of sodium salt of 3-hydroxy-1-(2,4-dimethylthiazol-5-yl)prop-2-en-1-one (10), [37]
A mixture of 1-(2,4-dimethylthiazol-5-yl)ethanone (9) (1.55 g, 10 mmol) and ethylformate (0.74 g, 10 mmol) in dry ether (20 ml) was added portion wise while stirring to solution sodium methoxide (0.54 g, 10 mmol) in dry ether (10 ml) at 0–5 °C. The resulting solid was collected, dried, and was used without purification.
Synthesis of 3-(dimethylamino)-1-(2,4-dimethyl)(1,3-thiazol-5-yl))prop-2-en-1-one (11)
A mixture of 1-(2,4-dimethyl-1,3-thiazol-5-yl)ethanone (9) (1.55 g, 0.01 mol) and dimethylformamide-dimethylacetal (1.47 g, 0.01 mol) were refluxed in dry xylene (10 ml) for 4 h. The hot solution was evaporated to its half volume and then cooled. The resulting solid was collected and recrystallized from ethanol to give (11).
3-(Dimethylamino)-1-(2,4-dimethyl)(1,3-thiazol-5-yl))prop-2-ene-1-one (11)
Yellow solid; yield (69 %); m.p. 103 °C. IR (KBr) νmax: 2904 (CH) and 1655 (CO conjugated) cm−1; 1H NMR (DMSO-d 6): 2.49 (s, 3H, CH3), 2.78 (s, 3H, CH3), 2.98 (s, 3H, = NCH3), 3.15 (s, 3H, = NCH3), 5.49–5.54 (d, 1H, J = 12 Hz, CH=CH–N) and 6.90–7.28 (d, 1H, J = 12 Hz, CH=CH–N); MS m/z (%): 210 (M+, 86).Anal. Calcd for C10H14N2OS (210.29): C, 57.11; H, 6.71; N, 13.32; S, 15.25; found: C, 57.13; H, 6.69; N, 13.34; S, 15.27 %.
Synthesis of 5-(2,4-dimethyl-1,3-thiazol-5-yl)-2-phenylpyrazolo[1,5-a]pyrimidine (12a) and 5-(2,4-dimethyl-1,3-thiazol-5-yl)-3-phenylpyrazolo[1,5-a]pyrimidine (12b)
Method A
A mixture of the sodium salt (10) (1.26 g, 5 mmol) and the appropriate amount of 3-amino-4-phenylpyrazole or 3-amino-5-phenylpyrazole (5 mmol) in a solution of piperidenum acetate [piperidine (2.5 ml)], water (5 ml), and acetic acid (2.5 ml) was heated under reflux for about 10 min. Then acetic acid (1.5 ml) was added while boiling, and the resulting solid was collected and recrystallized from the appropriate solvents to give (12a) and (12b), respectively.
Method B
An equimolar amount of 3-(dimethylamino)-1-(2,4-dimethyl)(1,3-thiazol-5-yl))prop-2-ene-1-one (11). (1.05 g, 5 mmol), the appropriate amount of 3-amino-4-phenylpyrazole or 3-amino-5-phenylpyrazole (5 mmol) and ammonium acetate (5 mmol) in acetic acid (10 ml) was heated under reflux for 4 h. The resulting solid was collected and recrystallized from the appropriate solvent to give products identical in all aspects (m.p., mixed m.p., and spectra) with (12a) and (12b).
5-(2,4-Dimethyl-1,3-thiazol-5-yl)-2-phenylpyrazolo[1,5-a]pyrimidine (12a)
Yellow solid; yield (79 %); m.p. 107 °C. IR (KBr) νmax: 3317 (NH) 3076, 2998 (CH, aromatic and aliphatic), 1628 (CN), 1343 (CH3) cm−1; 1H NMR (DMSO-d 6): 2.59 (s, 3H, CH3), 2.83 (s, 3H, CH3), 6.53 (s, 1H, pyrazole H-4), 7.13 (d, 1H, J = 4 Hz, pyrimidine H-5), 7.54–7.92 (m, 5H, ArH’s) and 8.74 (d, 1H, 8 Hz, pyrimidine H-6); MS m/z (%): 306 (M+, 48).Anal. Calcd for C17H14N4S (306.38): C, 66.64; H, 4.61; N, 18.29; S, 10.47; found: C, 66.66; H, 4.63; N, 18.31; S, 10.45 %.
5-(2,4-Dimethyl-1,3-thiazol-5-yl)-3-phenylpyrazolo[1,5-a]pyrimidine (12b)
Red solid; Yield (69 %); m.p. 109 °C. IR (KBr) νmax: 3315 (NH), 3057, 2996 (CH, aromatic and aliphatic), 1624 (CN), 1343 (CH3) cm−1; 1H NMR (DMSO-d 6): 2.56 (s, 3H, CH3), 2.84 (s, 3H, CH3), 7.14 (d, 1H, J = 4 Hz, pyrimidine H-5), 7.56–7.78 (m, 5H, ArH’s), 8.74 (d, 1H, J = 8 Hz, pyrimidine H-6 and 9.05 (s, 1H, pyrazole H-3); MS m/z (%): 306 (M+, 44).Anal. Calcd for C17H14N4S (306.38): C, 66.64; H, 4.61; N, 18.29; S, 10.47; found: C, 66.63; H, 4.63; N, 18.27; S, 10.46 %.
Synthesis of 1-phenyl-4-(2,4-dimethyl)thiazol-5-yl-3-substituted pyrazoles (20a–d), [38]
Equimolar amounts of each of (11) and the appropriate hydrazonoyl halides (13–16) (0.005 mol) were refluxed in dry toluene containing triethylamine for 3 h. The hot solution was filtered off and the filtrate was evaporated and triturated with petroleum ether (40–60 °C). The resulting solid was collected and crystallized from ethanol to give (20a–d), respectively.
Ethyl 4-[(2,4-dimethyl-1,3-thiazol-5-yl)carbonyl]-1-phenyl-1H-pyrazole-3-carboxylate (20a)
Yellow solid; yield (73 %); m.p. 140 °C. IR (KBr) νmax: 3450 (NH), 3088, 2996 (CH), 1674 (CO) and 1597 (C=C) cm−1; 1H NMR (DMSO-d 6): 1.22 (t, 3H, CH3, J = 7 Hz), 2.44 (s, 3H, CH3), 2.69 (s, 3H, CH3), 4.33 (q, 2H, CH2, J = 7 Hz), 7.44–7.89 (m, 5H, ArH’s) and 8.19 (s, 1H, pyrazole C-5); MS m/z (%): 355 (M+, 56).Anal. Calcd for C18H17N3O3S (355.41): C, 60.83; H, 4.82; N, 11.82; S, 9.02; found: C, 60.85; H, 4.80; N, 11.84; S, 9.15 %.
1-{4-[(2,4-Dimethyl-1,3-thiazol-5-yl)carbonyl]-1-phenyl-1H-pyrazol-3-yl}ethanone (20b)
Yellow solid; yield (70 %); m.p. 119 °C. IR (KBr) νmax: 3039, 2985 (CH), 1648 (CO conjugated) and 1599 (C=C) cm−1; 1H NMR (DMSO-d 6): 2.57 (s, 3H, CH3), 2.69 (s, 3H, CH3), 2.78 (s, 3H, CH3), 7.15–7.75 (m, 5H, ArH’s) and 8.21 (s, 1H, pyrazole C-5); MS m/z (%): 325 (M+, 60).Anal. Calcd for C17H15N3O2S (325.38): C, 62.75; H, 4.65; N, 12.91; S, 9.85; found: C, 62.77; H, 4.66; N, 12.89; S, 9.87 %.
(2,4-Dimethyl-1,3-thiazol-5-yl)[1-phenyl-3-(phenylcarbonyl)-1H-pyrazol-4-yl]methanone (20c)
Yellow solid; Yield (78 %); m.p. 157 °C. IR (KBr) νmax: 3058, 2919 (CH), 1645 (CO conjugated) and 1598 (C=C) cm−1; 1H NMR (DMSO-d 6): 2.44 (s, 3H, CH3), 2.7 (s, 3H, CH3), 7.21–8.11 (m, 10H, ArH’s) and 8.31 (s, 1H, pyrazole C-5); MS m/z (%): 387 (M+, 80). Anal. Calcd for C22 H17N3O2S (387.45): C, 68.20; H, 4.42; N, 10.85; S, 8.28; found: C, 68.22; H, 4.40; N, 10.87; S, 8.30 %.
4-[(2,4-Dimethyl-1,3-thiazol-5-yl)carbonyl]-N,1-diphenyl-1H-pyrazole-3-carboxamide (20d)
Pale yellow solid; yield (79 %); m.p. 190 °C. IR (KBr) νmax: 3438 (NH), 3065, 2993 (CH), 1677 (CO) and 1595 (C=C) cm−1; 1H NMR (DMSO-d 6): 2.48 (s, 3H, CH3), 2.79 (s, 3H, CH3), 7.13–7.87 (m, 10H, ArH’s), 8.35 (s, 1H, pyrazole C-5) and 10.71 (s, 1H, NH); MS m/z (%): 402 (M+, 73).Anal. Calcd for C22H18N4O2S (402.46): C, 65.65; H, 4.51; N, 13.92; S, 7.97; found: C, 65.67; H, 4.49; N, 13.90; S, 7.99 %.
Synthesis of pyrazolo[3,4-d]pyridazines (22a–c), [38]
An appropriate amount of substituted pyrazole (20a-d). (0.5 g) and hydrazine hydrate (1 ml) in ethanol (15 ml) was refluxed for 1 h. The resulting solid was collected and recrystallized from ethanol to give the pyrazolo[3,4-d]pyridazines (22a-c).
4-(2,4-Dimethyl-1,3-thiazol-5-yl)-2-phenyl-2H-pyrazolo[3,4-d]pyridazin-7-ol (22a)
Yellow solid; yield (78 %); m.p. 243 °C. IR (KBr) νmax: 3450 (NH), 3088, 2996 (CH), 1674 (CO) and 1597 (C=C) cm−1; 1H NMR (DMSO-d6): 2.45 (s, 3H, CH3), 2.72 (s, 3H, CH3), 7.22–8.12 (m, 5H, ArH’s), 8.54 (s, 1H, pyrazole C-5) and 10.11 (s, 1H, NH); MS m/z (%): 323 (M+, 50).Anal. Calcd for C16H13N5OS (323.37): C, 59.43; H, 4.05; N, 21.66; S, 9.92; found: C, 59.45; H, 4.15; N, 21.64; S, 9.90 %.
4-(2,4-Dimethyl-1,3-thiazol-5-yl)-7-methyl-2-phenyl-2H-pyrazolo[3,4-d]pyridazine (22b)
Yellow solid; yield (75 %); m.p. 190 °C. IR (KBr) νmax: 3045, 2991 (CH) and 1587 (C=C) cm−1; 1H NMR (DMSO-d 6): 2.46 (s, 3H, CH3), 2.77 (s, 3H, CH3), 3.01 (s, 3H, CH3), 7.32–8.11 (m, 5 H, ArH’s) and 8.76 (s, 1H, pyrazole C-5); MS m/z (%): 321 (M+, 55). Anal. Calcd for C17H15N5S (321.39): C, 63.53; H, 4.70; N, 21.79; S, 9.98; found: C, 63.55; H, 4.72; N, 21.81; S, 10.00 %.
4-(2,4-Dimethyl-1,3-thiazol-5-yl)-2,7-diphenyl-2H-pyrazolo[3,4-d]pyridazine (22c)
Yellow solid; yield (83 %); m.p. 204 °C. IR (KBr) νmax: 3055, 2958 (CH) and 1594 (C=C) cm−1; 1H NMR (DMSO-d6): 2.44 (s, 3H, CH3), 2.89 (s, 3H, CH3), 7.23–8.75 (m, 10 H, ArH’s) and 8.81(s, 1H, pyrazole C-5); MS m/z (%): 383 (M+, 63). Anal. Calcd for C22 H17N5S (383.46): C, 68.91; H, 4.47; N, 18.26; S, 8.36; found: C, 68.89; H, 4.49; N, 18.28; S, 8.38 %.
Antimicrobial activity
The synthesized compounds were tested for their antimicrobial activity against gram positive and gram negative bacteria as well as some fungal-plants. The sensitivity of the selected microorganisms towards the compounds under investigation was determined in vitro culture dissolved in chloroform, Appling the filter paper and hole plate method [39]. The sterile filter paper disc was saturated with 10 μL of 0.5 mg ml−1 w/v solution of the compound under investigation in DMF. A comparative study of the biological activity of these compounds with Ampicillin and tetracycline is compiled in Table 3. Generally, all synthesized compounds showed an adequate inhabitory efficiency of growth of gram positive and gram negative bacteria.
Abbreviations
- m.p.:
-
melting point
- CNS:
-
the central nervous system
- 13DC:
-
1,3-dipolar cycloaddition
- ZPE:
-
the zero-point energy
- G:
-
free energies
- H:
-
enthalpies
- MW:
-
molecular weight
- TLC:
-
thin layer chromatography
References
Kamal A, Bharathi EV, Reddy JS, Ramaiah MJ, Dastagiri D, Reddy MK, Viswanath A, Reddy TL, Shaik TB, Pushpavalli SN, Bhadra MP (2011) Synthesis and biological evaluation of 3,5-diaryl isoxazoline/isoxazole linked 2,3-dihydroquinazolinone hybrids as anticancer agents. Eur J Med Chem 46(2):691–703
Kankala S1, Kankala RK, Gundepaka P, Thota N, Nerella S, Gangula MR, Guguloth H, Kagga M, Vadde R, Vasam CS (2013) Regioselective synthesis of isoxazole-mercaptobenzimidazole hybrids and their in vivo analgesic and anti-inflammatory activity studies. Bioorg Med Chem Lett 23(5):1306–1309
Preston PN (1980) The chemistry of heterocyclic compounds. In: Benzimidazoles and congeneric tricyclic compounds, vol 40(2). Wiley, New York, p 10
Kirkpatrick WE, Okabe T, Hillyard IW, Robins RK, Dren AT, Novinson T (1977) 3-Halo-5,7-dimethylpyrazolo[1,5-a]pyrimidines, a nonbenzodiazepinoid class of antianxiety agents devoid of potentiation of central nervous system depressant effects of ethanol or barbiturates. J Med Chem 20(3):386–393
Elnagdi MH (1974) Reactions with β-cyanoethylhydrazine—I: a route for the preparation of pyrazolo[1.5-a]pyrimidines and pyrrolo[1.2-b]pyrrazoles. Tetrahedron 30(16):2791–2796
Ryan AJ, Welling PG, Wright SE (1969) Further studies on the metabolism of tartrazine and related compounds in the intact rat. Food Cosmet Toxicol 7(4):287–295
Ebnöther A, Jucker E, Lindeman A (1959) Über neuartige, basisch substituierte Pyrazolon-Derivate. Untersuchungen über synthetische Arzneimittel. 3. Mitteilung. Helv Chim Acta 42:1201–1214
Mallick SK, Martin AR, Lingard RG (1971) Synthesis and antimicrobial evaluation of some 5-(5-nitrofurylidene)rhodanines, 5-(5-nitrofurylidene)thiazolidine-2,4-diones, and their vinylogs. J Med Chem 14(6):528–532
Yang JF, Cao H, Liu H, Li BQ, Ma YM (2011) Synthesis and bioactivity of novel bis-heterocyclic compounds containing pyrazole and oxadiazoline. J Chin Chem Soc 58:369–375
Mallikarjuna Rao R, Sreeramulu J, Ravindranath LK, Nagaraja Reddy G, Hanumanthurayudu K, Nageswara Reddy G, Jayaraju A, Madhusudhan P (2012) Synthesis and biological screening of some pyridine and pyrrole derivatives of pyrazolo[3, 4-c]pyrazoles. J Chem Pharm Res 4(1):272–278
Mohareb RM, El-Sayed NNE, Abdelaziz MA (2012) Uses of cyanoacetylhydrazine in heterocyclic synthesis: novel synthesis of pyrazole derivatives with anti-tumor activities. Molecules 17(7):8449–8463
Kumar KA, Jayaroopa P (2013) Pyrazoles: synthetic strategies and their pharmaceutical applications-an overview. Int J PharmTech Res 5(4):1473–1486
Piste PB (2014) Facile synthesis and antimicrobial screening of pyrazole derivatives world. J Pharm Res 3(5):735–742
Kamal A, Shaik AB, Polepalli S, Reddy VS, Kumar GB, Gupta S, Krishna KV, Nagabhushana A, Mishra RK, Jain N (2014) Pyrazole-oxadiazole conjugates: synthesis, antiproliferative activity and inhibition of tubulin polymerization. Org Biomol Chem 12(40):7993–8007
Bhalla A, Bari SS, Bhalla J (2015) Synthesis of novel pyrazolylmethylene–pyrimidine heterocycles: potential synthons for hybrid Β-lactams. Can Chem Trans 3(1):72–84
Abdelhamid AO, Zohdi HF, Sallam MMM, Ahmed NA (2000) Reactions with hydrazonoyl halides. 31. Synthesis of some new pyrrolidino[3,4-c]pyrazolines, pyrazoles, and pyrazolo[3,4-d]pyridazines. Molecules 5(7):967–973
Vera K (1993) Preparation of some thio derivatives of pyridinecarbothioamides. Collect Czech Chem Commun 58(5):1195–1197
Matyus P (1998) 3(2H)-Pyridazinones: some recent aspects of synthetic and medicinal chemistry. J Heterocycl Chem 35(5):1075–1089
Önal A, Akçamur Y, Altural B (1996) Synthesis of some pyrazolo–pyridazine compounds. Turk J Chem 20(2):159–163
Tewari AK, Mishra A (2001) Synthesis and anti-inflammatory activities of N4, N5-disubstituted-3-methyl- H-pyrazolo[3,4-c]pyridazines. Bioorg Med Chem 9(3):715–718
Abdelhamid AO, El-Ghandour AH, Hussein AH, Zaki YH (2004) Reactions of hydrazonoyl halides 44[1]: synthesis of some new 1,3,4-thiadiazolines, 1,3,4-selenadiazolines and triazolino[4,3-a]pyrimidines. J Sulfur Chem 25(5):329–342
Bellamy LJ (1975) The infrared spectra of complex molecules, 3rd ed.Wiley, New York-London, p 150
Searle NE (1948) Synthesis of nu-aryl-maleimides. US Pat 2 444:536. (Chem Abstr 1948;42:7340)
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M et al (2009) Gaussian 09 Suite of Programs. Gaussian Inc, Wallingford
Becke AD (1996) Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J Chem Phys 104(3):1040–1046
Becke AD (1997) Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals. J Chem Phys 107(20):8554–8560
Raghavachari K, Trucks GW, Pople JA, Head-Gordon M (1989) A fifth-order perturbation comparison of electron correlation theories. Chem Phys Lett 157(6):479–483
Becke AD (1993) Density functional thermochemistry. III. The role of exact exchange. J Chem Phys 98(7):5648–5652
Shawali AS, Hilal RH, El-Sheikh S (2001) Regioselectivity in the reactions of bis-hydrazonoyl halides with pyrimidine-2-thiones. Monatshefte für Chemie Chem Mon 132(6):715–720
Schoenebeck F, Ess DH, Jones GO, Houk KN (2009) Reactivity and regioselectivity in 1,3-dipolar cycloadditions of azides to strained alkynes and alkenes: a computational study. J Amr Chem Soc 131(23):8121–8133
Lan Y, Zou L, Cao Y, Houk KN (2011) Computational methods to calculate accurate activation and reaction energies of 1,3-dipolar cycloadditions of 24 1,3-dipoles. J Phys Chem A 115(47):13906–13920
Asiri AM, Zayed MEM, Ng SW (2010) Ethyl (Z)-2-chloro-2-(2-phenylhydrazin-1-ylidene)acetate. Acta Cryst E66:o2374
Eweiss NF, Osman A (1980) Synthesis of heterocycles-2. New routes to acetylthiadiazolines and arylazothiazoles. J Heterocycl Chem 17(8):1713–1717
Shawali AS, Abdelhamid AO (1976) Reaction of dimethylphenacylsulfonium bromide with N-nitrosoacetarylamides and reactions of the products with nucleophiles. Bull Chem Soc Jpn 49(1):321–324
Shawali AS, Osman A (1971) Synthesis and reactions of phenyl carbamoyl-aryl hydrazidic chlorides. Tetrahedron 27(12):2517–2528
Abdelhamid AO, Elghandour AH, Ahmed SA, Zaki YH (2006) Synthesis and reactions of 2-chloro-2-(hydroximino)-1-(4-methyl-2-phenylthiazol-5-yl)ethanone. J Heterocycl Chem 43(2):249–254
Mariella RP (1963) 3-Cyano-6-methyle-2(1)-pyridone. Org Synth 4:210
Zaki YH, Ahmed SA, Hussein AM, Abdelhamid AO (2006) Reactions of hydrazonoyl halides 471: synthesis of some new 2,3-dihydro-1,3,4-thiadiazoles, triazolo[4,3-a]pyrimidines, and pyrazolo[3,4-d]pyridazines with expected biological activity. Phosphorus Sulfur Silicon 181(4):825–837
Solomons WE, Doorenbos NJ (1974) Synthesis and antimicrobial properties of 17β-amino-4-aza-5α-androstane and derivatives. J Pharm Sci 63(1):19–23
Authors’ contributions
YHZ carried the literature study, designing part, designing of synthetic schemes, and docking studies. SAK carried out the computational chemistry section. YHZ and ARS contributed in the synthesis as well as purification of compounds. YHZ (corresponding author) did the final sequence alignment in the manuscript and drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank Prof AO Abdelhamid for the valuable discussions.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zaki, Y.H., Sayed, A.R. & Elroby, S.A. Regioselectivity of 1,3-dipolar cycloadditions and antimicrobial activity of isoxazoline, pyrrolo[3,4-d]isoxazole-4,6-diones, pyrazolo[3,4-d]pyridazines and pyrazolo[1,5-a]pyrimidines. Chemistry Central Journal 10, 17 (2016). https://doi.org/10.1186/s13065-016-0163-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-016-0163-2