
Abstract
Background
1-Octanol solubility is important in a variety of applications involving pharmacology and environmental chemistry. Current models are linear in nature and often require foreknowledge of either melting point or aqueous solubility. Here we extend the range of applicability of 1-octanol solubility models by creating a random forest model that can predict 1-octanol solubilities directly from structure.
Results
We created a random forest model using CDK descriptors that has an out-of-bag (OOB) R2 value of 0.66 and an OOB mean squared error of 0.34. The model has been deployed for general use as a Shiny application.
Conclusion
The 1-octanol solubility model provides reasonably accurate predictions of the 1-octanol solubility of organic solutes directly from structure. The model was developed under Open Notebook Science conditions which makes it open, reproducible, and as useful as possible.

:
Similar content being viewed by others


Background
The solubility of organic compounds in 1-octanol is important because of its direct relationship to the partition coefficient logP used in pharmacology and environmental chemistry. Current models that can be used to predict 1-octanol solubility include group contribution methods [1] and often include melting point as a descriptor [2–4]. The most recent model by Admire and Yalkowsky [4] gives a very useful rule of thumb to predict molar 1-octanol solubility from just the melting point
where the compound melting point mp is in °C for compounds that are solid at room temperature and is taken to be 25 for liquids. Abraham and Acree [5] refined Admire and Yalkowsky’s model by appending the melting point term to their linear free energy relationship (LFER) model
where E is the solute excess molar refractivity in units of (cm3/mol)/10, S is the solute dipolarity/polarizability, A and B are the overall or summation hydrogen bond acidity and basicity, and V is the McGowan characteristic volume in units of (cm3/mol)/100. The A·B term was added to deal with the solute–solute interactions. The coefficients were found using linear regression against the solubilities of solutes with known Abraham descriptors with the following result:
In the present study, we improve upon previous models by creating a nonlinear random forest model using solubility data from the Open Notebook Science Challenge [6], an open data, crowdsourcing research project that collects and measures the solubilities of organic compounds in organic solvents created by Jean-Claude Bradley and Cameron Neylon. The challenge is, in turn, part of Jean-Claude Bradley’s UsefulChem program, an open drug discovery project that uses open notebook science [7].
Procedure
The 1-octanol solubility data in this paper were extracted from the Open Notebook Science Challenge solubility database [8]. We removed all items that were marked “DONOTUSE.” For compounds with multiple solubility values that included values listed in the Abraham and Acree paper, we kept only the solubility values that were listed in the Abraham and Acree paper. If no Abraham and Acree paper value was available, then we kept the Raevsky, Perlovich, and Schaper value instead. In the rare case that two Abraham and Acree (or Raevsky, Perlovich, and Schaper) paper values were listed for a single chemspider ID (CSID), we kept the higher of the two values.
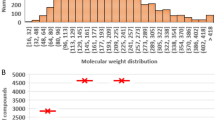
The collection and curation process left us with 261 data points to model, see Additional file 1. The structures in our dataset are not very diverse and can be characterized, in general, as relatively small organic compounds with 1-octanol solubility values between 0.01 and 1.00 M, see Figs. 1, 2, and 3.

Mass distribution of the compounds in our study. 94 % of compounds have a molecular weight between 100 and 400 Da

Solubility distribution of the compounds in our study. 76 % of compounds have solubility values between 0.01 and 1.00 M

Nearest neighbor Tanimoto similarity
Two features about the chemical space are immediately apparent. Firstly, the dataset has 50 carboxylic acids which is a common feature for both Abraham and Acree datasets and the Open Notebook Science Challenge dataset where the primary focus is on measuring solubilities for the same compound in several non-aqueous solvents. While common in non-aqueous solubility studies, sometimes one does have to consider dimerization for carboxylic acids [9]. Secondly, there are only 50 compounds that have a single Lipinski’s Rules failure (all the rest having zero failures), suggesting the dataset could be characterized as drug-like.
Principal component analysis (using the prcomp function with scale = T) and cluster analysis was performed on the dataset of 259 compounds with 86 CDK descriptors using R. The optimal number of clusters was determined to be 2 by using silhouette analysis (using the pam function) on a series ranging from 2 to 20 clusters. The silhouettes had an average width of 0.74 for 2 clusters; almost double the next closest value [10]. The clusters are shown in Fig. 4 below with the x and y axes corresponding to the first and second principal components respectively. The first two principal components explain 36 % of the variance. The first cluster (red) is typified by compounds without hydrogen bond acceptors and with ALogP >1.56 and with TopoPSA <26.48; 128 out of 157 compounds match this criteria. The blue cluster is more chemically diverse than the red cluster but even so 75 of the 102 compounds have ALogP <1.56 and TopoPSA >26.48 and at least one hydrogen bond acceptor.

Chemical space of compounds naturally separate into two distinct clusters
Results and discussion
Modeling
A Random Forest Model is a compilation of uncorrelated decision trees used to choose the best case among many. Our model used 86 variables in its calculation. In general, the less correlated that the variables are, the better the results that will occur from a random forest model. A higher strength of each individual tree also improves the accuracy of the final model—“The strength of each individual tree in the forest. A tree with a low error rate is a strong classifier. Increasing the strength of the individual trees decreases the forest error rate.” [11]. Using a random forest model allows us to get out-of-bag (OOB) estimates which are akin to cross-validation and are useful for estimating the performance of models created using small datasets.
Using Rajarshi Guha’s CDK Descriptor Calculator (v 1.4.6) [12], we calculated the CDK [13–15] descriptors for all the compounds in our refined data file, selecting the option to add explicit hydrogens. Once descriptors were calculated, we deleted all columns that had a zero standard deviation. Additional feature selection was performed by removing columns that were highly correlated (0.9 and above). Two compounds were removed as they had several “NA” values across multiple descriptors. This left us with a dataset of 259 1-octanol solubility values with 86 CDK descriptors.
The dataset was then split randomly into training and test sets (75:25). Using the random forest model package (v 4.6-10) in R (v 3.1.2), we created a random forest model using our training set data. This model had an OOB R2 value of 0.63 and an OOB MSE of 0.38. This model was then used to predict the 1-octanol solubilities of the compounds in the test-set resulting in and R2 value of 0.54 and a MSE of 0.44, see Fig. 5. The performance statistics obtained when using the model to predict test-set solubilities are comparable to the OOB values. The fact that they are slightly smaller may be an artifact of the relatively small sizes of the training and test sets and the fact that we decided to doing a single taining-set/test-set split rather than use cross-validation.

Predicted vs. measured solubility values for the randomly selected test-set coloured by AE
One of the goals of our research was to provide for the community a useful web application that can be used to predict 1-octanol solubilities directly from structure. To accomplish this, we created a random forest model using the entire dataset. This model has an OOB R2 value of 0.66 and an OOB MSE of 0.34.
The following descriptors were identified as important: ALogP, XLogP, TopoPSA, nAtomP, MDEC.23, khs.aaCH, and nHBAcc, see Fig. 6, which correspond to two models for LogP, the predicted topological polar surface area, the number of atoms in the longest pi chain, the MDE topological descriptor, a Kier and Hall smarts descriptor, and the number of hydrogen bond acceptors respectively. It is not surprising that both ALogP and XLogP would be important in predicting 1-octanol solubility, though one would have assumed that one of these descriptors would have been removed during feature selection as being highly correlated with the other. Analyzing the correlation between these two descriptors, we see that they are correlated at 0.83 and they both survived as are cutoff was at 0.90. This further confirms the problems with current Open LogP descriptors implemented in the CDK [16].

Random forest model variable importance
We tried several other models using the same training set/test set split as above with no improvement in performance. A linear model (lm) using all 86 CDK descriptors had an R2 value of 0.24 and MSE of 0.88; A tuned (using tenfold cross validation) support vector machine (epsilon = 0.3, cost = 4.3) had an R2 value of 0.35 and MSE of 0.38; and an optimized (using the train command in the caret package) artificial neural network model (nnet) had an R2 value of 0.36 and MSE of 0.74. Thus the random forest model seems the best model for the current dataset.
Previously published models only report the training set statistics, so in order to directly compare our model with previous models we used our full random forest model to predict the solubilities of the entire dataset, see Fig. 7. For the training set, the model has an R2 value of 0.94 and a MSE of 0.06. Abraham and Acree’s recommended Eq. (3), if all necessary descriptors are available, for estimations of log Soct has a training set R2 value of 0.83 [5] which is lower than our value. Our model also does not require a measured melting point. This makes our model, even with the modest OOB R2 value of 0.66, superior to all others previously published.

Training set chemical space where red indicates poor model performance
In general, we expect the performance of our model to be better for compounds similar to those in the training set, apart from obvious outliers. However, there was no statistically significant performance differential between the interior and the periphery of the chemical space as has been found previously for other properties we have modeled using similar techniques [17]. We used the free-to-use DMax Chemistry Assistant Software [18] to help discover regions of the chemical space where our random forest model performs poorly (and conversely, well). Interestingly, the only statistically noteworthy (p ~ 0.1) finding is that the model performance is dependent upon the solubility values themselves; with the model performing well for compounds with solubility values over 0.01 M and performing poorly for compounds with solubility values less than 0.01 M. This suggests that the solubility data is comparatively not as reliable for compounds with solubility values less than 0.01 M and that using the model to predict solubilities of compounds that have low solubilities should be done with caution. No other statistically significant or noteworthy differences in model performance were found based on both physical properties and structure/scaffold.
The data collection, curation, and modeling were all performed under Open Notebook Science (ONS) conditions. Additional modeling details, including our R code, can be found on the Open Notebook page [19]. We have deployed our model as a Shiny application [20].
Conclusions
We have developed a random forest model for 1-octanol solubility that has an OOB R2 value of 0.66 and an average absolute error of 0.34 that performs better than any other currently published model. Our model makes 1-octanol solubility predictions directly from structure without having to know the solute’s melting point or aqueous solubility. This makes our model the leading open model for predicting 1-octanol solubilities for a variety of applications.
Abbreviations
- LFER:
-
linear free energy relationship
- CSID:
-
chemspider ID
- CDK:
-
chemistry development kit
- OOB:
-
out-of-bag
- ONS:
-
open notebook science
- MSE:
-
mean squared error
- AE:
-
absolute error
References
Li A, Pinsuwan S, Yalkowsky SH (1995) Estimation of solubility of organic compounds in 1-octanol. Ind Eng Chem Res 34(3):915–920
Sepassi K, Yalkowsky SH (2006) Solubility prediction in octanol: a technical note. AAPS PharmSciTech 7(1):E184–E191
Raevsky OA, Perlovich GL, Schaper K-J (2007) Physicochemical properties/descriptors governing the solubility and partitioning of chemicals in water-solvent-gas systems. Part 2. Solubility in 1-octanol. SAR QSAR Environ Res 18(5–6):543–578
Admire B, Yalkowsky SH (2013) Predicting the octanol solubility of organic compounds. J Pharm Sci 102(7):2112–2119
Abraham MH, Acree WE Jr (2014) The solubility of liquid and solid compounds in dry octan-1-ol. Chemosphere 103:26–34. doi:10.1016/j.chemosphere.2013.10.095
Bradley J-C, Neylon C, Guha R, Williams A, Hooker B, Lang ASID, Friesen B, Bohinski T, Bulger D, Federici M, Hale J, Mancinelli J, Mirza K, Moritz M, Rein D, Tchakounte C, Truong H (2010) Open notebook science challenge: solubilities of organic compounds in organic solvents. Nat Precedings. doi:10.1038/npre.2010.4243.3
Open Notebook Science. Wikipedia. Wikimedia Foundation. http://en.wikipedia.org/wiki/Open_notebook_science. Accessed 1 Sept 2015
Bradley J-C, Guha R, Hooker B, Koch SJ, Lang ASID, Neylon C, Williams AJ (2015) Open Notebook Science Challenge Solubility Dataset. Figshare. doi:10.6084/m9.figshare.1514952
Bradley J-C, Abraham MH, Acree WE Jr, Lang ASID, Beck SN, Bulger DA, Clark EA, Condron LN, Costa ST, Curtin EM, Kurtu SB, Mangir MI, McBride MJ (2015) Determination of Abraham model solute descriptors for the monomeric and dimeric forms of trans-cinnamic acid using measured solubilities from the Open Notebook Science Challenge. Chem Cent J 9:11
Partitioning Around Medoids (PAM) Object. R Documentation. https://stat.ethz.ch/R-manual/R-devel/library/cluster/html/pam.object.html. Accessed 1 Sept 2015
Breiman L, Cutler A. Random Forests. https://www.stat.berkeley.edu/~breiman/RandomForests/cc_home.htm. Accessed 1 Sept 2015
Guha R. CDK Descriptor UI. https://github.com/rajarshi/cdkdescui. Accessed 1 Sept 2015
The Chemistry Development Kit http://sourceforge.net/projects/cdk. Accessed 1 Sept 2015
Steinbeck C, Han Y, Kuhn S, Horlacher O, Luttmann E, Willighagen E (2003) The Chemistry Development Kit (CDK): an open-source Java library for chemo-and bioinformatics. J Chem Inf Comput Sci 43(2):493–500
Steinbeck C, Hoppe C, Kuhn S, Floris M, Guha R, Willighagen EL (2006) Recent developments of the chemistry development kit (CDK)-an open-source java library for chemo-and bioinformatics. Curr Pharm Des 12(17):2111–2120
Guha R (2013) CDK and logP Values. So much to do, so little time. http://blog.rguha.net/?p=896. Accessed 1 Sept 2015
Bradley J-C, Abraham MH, Acree WE Jr, Lang ASID (2015) Predicting Abraham model solvent coefficients. Chem Cent J 9:12
DeGrave K (2010) DMax Chemistry Assitant. http://dtai-old.cs.kuleuven.be/ml/systems/dmax. Accessed 1 Sept 2015
Buonaiuto MA, Lang ASID. Modeling the Solubility of Organic Compounds in 1-Octanol. ORU Open Notebook Science. Open Lab Notebook Page: http://oruopennotebookscience.wikispaces.com/CDK001. Accessed 1 Sept 2015
Buonaiuto MA, Lang ASID (2015) Open 1-octanol solubility model. Shiny. http://michaeloru.shinyapps.io/Shiny. Accessed 1 Sept 2015
Authors’ contribution
MAB performed all the modeling and aided in manuscript preparation, ASIDL collected and curated all the 1-octanol solubility data from the Open Data available in the Open Notebook Science Challenge dataset and aided in manuscript preparation including addressing referee’s comments. Both authors read and approved the final manuscript.
Acknowledgements
The authors would like to acknowledge the contributors and judges of the Open Notebooks Science Challenge under the leadership of Jean-Claude Bradley without whom this work would not have been possible.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Buonaiuto, M.A., Lang, A.S.I.D. Prediction of 1-octanol solubilities using data from the Open Notebook Science Challenge. Chemistry Central Journal 9, 50 (2015). https://doi.org/10.1186/s13065-015-0131-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-015-0131-2