
Abstract
Background
Calculating Abraham descriptors from solubility values requires that the solute have the same form when dissolved in all solvents. However, carboxylic acids can form dimers when dissolved in non-polar solvents. For such compounds Abraham descriptors can be calculated for both the monomeric and dimeric forms by treating the polar and non-polar systems separately. We illustrate the method of how this can be done by calculating the Abraham descriptors for both the monomeric and dimeric forms of trans-cinnamic acid, the first time that descriptors for a carboxylic acid dimer have been obtained.
Results
Abraham descriptors were calculated for the monomeric form of trans-cinnamic acid using experimental solubility measurements in polar solvents from the Open Notebook Science Challenge together with a number of water-solvent partition coefficients from the literature. Similarly, experimental solubility measurements in non-polar solvents were used to determine Abraham descriptors for the trans-cinnamic acid dimer.
Conclusion
Abraham descriptors were calculated for both the monomeric and dimeric forms of trans-cinnamic acid. This allows for the prediction of further solubilities of trans-cinnamic acid in both polar and non-polar solvents with an error of about 0.10 log units.

Molar concentration of trans-cinnamic acid in various polar and non-polar solvents.
Similar content being viewed by others

Background
The Abraham solvation parameter model describes solute transfer between two condensed phases, or between a condensed phase and a gas phase. Specific chemical and biological processes that have been described by the basic model include water-to-organic solvent and gas-to-organic solvent partition coefficients [1], blood-to-body tissue/fluid and gas-to-body tissue/fluid partition coefficients [2], skin permeability coefficients [3], median lethal concentrations of organic compounds for toxicity towards specific aquatic organisms [3], nasal pungency thresholds [3], Draize eye irritation scores [3], and the minimum alveolar concentration for inhalation anthesia towards rats [3]. Expressed in terms of partition coefficients the Abraham general solvation equations can be formulated as:
where Ps is a water-solvent partition coefficient of a solute, Ks is a gas-solvent partition coefficient, E, S, A, B, V, and L are the solute descriptors and c, e, s, a, b, v and l are coefficients that describe the particular water-solvent or gas-solvent process. The solute descriptors each describe an important solute property: E represents the excess molar refractivity in units of (cm3 per mol)/10, S represents the dipolarity/polarity of the solute, A and B represent the hydrogen bond acidity and basicity respectively, V is the solute’s McGowan characteristic volume in units of (cm3 per mol)/100 and L is the logarithm of the gas-hexadecane partition coefficient at 298 K. [4,5]
The solute descriptor V is the easiest to obtain as it can be calculated directly from structure. It is equal to the McGowan characteristic volume (cm3 per mol)/100 [6]. V encodes sized-related solvent-solute dispersion interactions, including a measure of the solvent cavity term that will accommodate the dissolved solute.
The solute descriptor E, the excess molar refractivity, can be calculated from a refractive index at 293 K for a compound that is liquid at 293 K [4]. For other solutes E can be predicted, either directly using Absolv, part of ACD Labs proprietary ACD/ADME Suite [7], or through the predicted molar refractivity, freely available for individual compounds through ChemSpider [8], or some other source, such as the Open Source Chemistry Development Kit [9]. Another useful method for estimating E is through summation of structural fragments from compounds with known values of E.
Equation (1) can be applied to saturated molar concentrations, Cs, of a compound in various organic solvents through Equation (3),
where Cw is the aqueous solubility of the compound. If the aqueous solubility is unavailable it can either be left unknown and determined by regression or predicted using ACD Labs ACD/ADME Suite or through the freely available VCC Labs ALOGPS webservice [10].
The solute descriptors S, A, and B can also be predicted [7,11-13] or in limited cases determined experimentally [14,15]. However, accurate results, in general much more accurate than predicted values, are easily obtained by using regression with measured solubilities and/or partition coefficient values [1].
Finally, we note that the applicability of the Abraham model to the solubility of crystalline organic solutes assumes three conditions. Firstly, the solute has the same form when dissolved in any solvent, including water. That is, we assume no solvate, hydrate, or complex formation. Secondly, the secondary medium coefficient must be at or near unity. This condition generally restricts the model to solutes that are not too soluble. Thirdly, if the solute ionizes in water, the aqueous solubility, Cw, is taken to be that of the neutral form. The second restriction may not be as important as initially believed. The Abraham solvation parameter model has shown remarkable success in correlating the solubility of several very soluble crystalline solutes. For example, Equations (1) and (2) described the molar solubility of 1,4-dichloro-2-nitrobenzene in 24 organic solvents to within overall standard deviations of 0.128 and 0.119 log units, respectively [16]. Standard deviations for aspirin dissolved in 13 alcohols, 4 ethers, and ethyl ethanoate were 0.123 and 0.138 log units [17]. 1,4-Dichloro-2-nitrobenzene and aspirin exhibited solubilities exceeding 1 molar in several of the organic solvents studied.
The Open Notebook Science Challenge [18] contains a valuable collection of Open Data (CC0 1.0 License: See the creative commons website for more information about this license) solubility data that could be used to determine Abraham descriptors for a large number of compounds. We illustrate the utility of the Open Notebook Science Challenge data by determining the Abraham descriptors for both the monomeric and dimeric forms of trans-cinnamic acid. The current study represents the first time that we have calculated the solute descriptors for carboxylic acid dimers. Solute descriptors are required input parameters in order to predict solute solubilities, partition coefficients, and other chemical/biological properties for which Abraham model correlations have been developed.
Methods
The measured solubility values presented here are from the Open Notebook Science Challenge [18], an Open Science project to collect and measure the solubility of organic compounds in organic solvents, ran by Jean-Claude Bradley, and sponsored by the Royal Society of Chemistry, Sigma Aldrich, Submeta, and Nature. The method and materials used to determine the solubility values varied by experiment and researcher and can be found in the Open Notebook [19].
In addition to the measured solubility values outlined above, we collected solubility values from the literature [20-24] and partition coefficients from Bio-Loom [25]. All values (mole fraction, mass fraction and mass ratio) were converted to molarity for ease of comparison.
The combined collection numbered 69 trans-cinnamic acid/solvent values (molar concentrations) at temperatures ranging from 19.5 C to 28 C. The solubility values were all converted to values at 25 C using the Buchowski equation with the assumption of miscibility at solute melting point [26]. Multiple measurements for the same solvent were averaged (with a mean deviation of 0.067 M) giving a total of 30 solute/solvent values for trans-cinnamic acid, see Table 1 below.
The case of cinnamic acid is interesting as it conflicts with our conditions of applicability, above. As with carboxylic acids in general, cinnamic acid dimerizes in the less polar solvents, especially in the less polar aprotic solvents. Experimental dimerization constants, Kdimer, based on Equation (4) often differ somewhat for the same compound in the same solvent, but whatever the actual value it is evident that at the saturated solubility concentrations, benzoic acid, and by analogy cinnamic acid, will be dimerized in non-polar aprotic solvents. For example Kdimer for benzoic acid in cyclohexane is 11300, in tetrachloromethane is 5010 and in benzene is 590 [27].
We can use this difficulty to advantage by choosing polar solvents for the determination of descriptors for cinnamic acid monomer and by choosing non-polar solvents for the determination of descriptors for cinnamic acid dimer. A few solvents were excluded altogether as they currently do not have Abraham solvent parameters: pentachloroethane, tetrachloroethane, tetrachloroethylene, and trichloroethylene.
Calculating the Abraham descriptors for cinnamic acid monomer
As input we used solubility data in Table 1 for the polar solvents where cinnamic acid is expected to exist largely in monomeric form, together with a number of direct partition coefficients [25]. Although the latter are partitions from water to non-polar solvents, they still refer to cinnamic acid monomer because the experimental determination has either been carried out at low solute concentration or has been extrapolated to low solute concentration. The direct log Ps values that we use [25] are in Table 2.
The value for E was determined from structure, by comparing cinnamic acid fragment-wise with compounds that have known values for E; ethyl benzoate (E = 0.689), ethyl cinnamate (E = 1.102), and benzoic acid (E = 0.730). The E solute descriptors for ethyl benzoate and benzoic acid differ by 0.041, with benzoic acid having the larger E value. Maintaining the same difference between the E solute descriptors for ethyl cinnamate and trans-cinnamic acid then gives E = 1.14 for trans-cinnamic acid (rounded to the hundredths place) [11]. The solute volume descriptor, calculated from the McGowan characteristic volume, is given by V = 1.1705. We can transform all the Ps values into values of the gas-solvent partition coefficient Ks through Equation (5), where Kw is the dimensionless gas-water partition coefficient
We then have a total of 21 values of log Ps, 5 being the number of partition coefficient measurements and 16 being the number of values derived from solubility ratios, using Equation (5), with log Cw taken as −2.40 [18]. These can be converted into 21 values of log Ks. We also have two equations for log Kw, one in terms of V (Equation 1) and one in terms of L (Equation 2), and an equation for GLC retention data [28] thus leading to a total of 45 equations. The unknowns are S, A, L and log Kw. The set of 45 equations were solved by regression to yield the values of the four unknowns that gave the best fit of experimental and calculated properties, exactly as described before [29,30].
Calculating the Abraham descriptors for cinnamic acid dimer
The input data is now restricted to solubilities in the less polar solvents where cinnamic acid is expected to exist predominantly in dimeric form. We do not know the solubility of cinnamic acid dimer in water, and so log Cw is another unknown quantity to be obtained by regression. We have solubilities in nine non-polar solvents, nine corresponding values of log Ps and two equations for log Kw giving a total of 20 equations. The value of V for cinnamic acid dimer was obtained in the usual way for a compound of molecular formula C18H16O4 as V = 2.2098. There are a number of aromatic liquid carboxylic acids, with known values of the refractive index at 293 K. These values for the pure liquids will refer to the dimeric form of the carboxylic acid, and can be used to calculate E in the usual way [4] for the dimer. The value for E for the dimeric form can also be obtained by addition of fragments, as we have done for cinnamic acid monomer. We find that the two E-values are related through
For cinnamic acid, with Emonomer = 1.14 the value of Edimer is 1.68. The unknowns are then S, A, B, L, log Kw and log Cw so that it is easily possible to obtain a solution for the 20 simultaneous equations by regression.
Results and discussion
The obtained descriptors for cinnamic acid monomer and cinnamic acid dimer are in Table 3, together with values for benzoic acid (monomer) as a comparison. The statistical fits are very good, and the 20 or 45 log Ps and log Ks values are fitted with a standard deviation (SD) of about 0.1 log units. As expected, the A-descriptor for cinnamic acid dimer (0.24) is much less than that for twice the monomer (1.22) because the two OH protons are internally bonded and are less available for bonding to an external hydrogen bond base. The other descriptors for cinnamic acid dimer are also as expected. A comparison of descriptors for cinnamic acid and benzoic acid monomers shows quite close agreement. The B-descriptor (hydrogen bond basicity) of cinnamic acid is a little more than that of benzoic acid due to the extra C = C group, and this also slightly increases the S-descriptor and the L-descriptor.
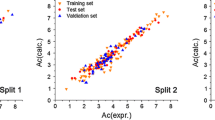
The SD values for the two sets of total equations are quite good but we decided to obtain the statistics for just the solubility data. Details are in Table 4 for the calculations of the cinnamic acid monomer. We include data on the log Ps values, but the statistics are exactly the same as for the solubilities. For the 16 solubilities, the average error (AE) between observed and fitted values is 0.006, the absolute average error (AAE) is 0.055 and the SD is 0.078 log units. Thus from the descriptors in Table 3 and the coefficients for the relevant equations, further solubilities of monomeric cinnamic acid in a large numer of polar solvents can be predicted to about 0.10 log units. The corresponding data for the cinnamic acid dimer are in Table 5. For the nine solubilities AE = 0.003, AAE = 0.053 and SD = 0.084 log units, so that solubilities in non-polar solvents can be predicted, again to within about 0.10 log units. It is interesting that the fitted and observed solubility in trifluoroethanol agree to 0.039 log units. An illustration of the results from Tables 4 and 5 can be seen in Figure 1, where the blue circles correspond to non-polar solvents and the red circles correspond to polar solvents.

Observed and fitted solubilities for trans-cinnamic acid. Red is for dimer in non-polar solvents.
Although we refer to solvents that support formation of the dimer as ‘non-polar’ solvents, the main distinguishing factor between solvents that support the dimer and those that support the monomer is the hydrogen bond basicity of the solvent. If the solvent is a hydrogen bond base, it will form solvent-solute hydrogen bonds with the OH group and will break up the dimer into the monomeric form. Trifluoroethanol as a solvent is an extremely weak hydrogen bond base. Marcus [31] gives values of the Kamlet-Taft solvent hydrogen bond basicity, β, as methanol (0.66), diethyl ether (0.47), propanone (0.43) propyl acetate (0.40), acetonitrile (0.40), nitrobenzene (0.30), trichloromethane (0.10), benzene (0.10), cyclohexane (0.00) and trifluoroethanol (0.00). It seems that for saturated solutions of cinnamic acid in solvents with β > 0.35 the monomer is mainly present but when the solvent β < 0.35 the dimer is mainly present.
Conclusion
We have determined Abraham solute descriptors for trans-cinnamic acid using solubility values measured using Open Notebook Science supplemented with values reported in the literature and with values of partition coefficients from the literature. For compounds that are not dimerized it is quite easy to perform these calculations using just solubility data. We have determined Abraham solute descriptors for the dimer of trans-cinnamic acid using just solubilities from the Open Notebook Science Challenge supplemented with values reported in the literature. This is the first time that descriptors have been assigned to carboxylic acid dimers. The Open Notebook Science Challenge details solubilities for a number of compounds that are easier to work with than cinnamic acid, because they do not form dimers. Those wishing to calculate Abraham solute descriptors for other compounds in a similar fashion can use the solubility data in the Open Notebook Science Challenge database to do so.
References
Abraham MH, Smith RE, Luchtefeld R, Boorem AJ, Luo R, Acree Jr WE. Prediction of solubility of drugs and other compounds in organic solvents. J Pharm Sci. 2010;99(3):1500–15.
Acree WE Jr, Grubbs LM, Abraham MH. Prediction of partition coefficients and permeability of drug molecules in biological systems with Abraham model solute descriptors derived from measured solubilities and water-to-organic solvent partition coefficients. In Toxicity and drug. 2012, INTECH Publishers, Chapter 5, p. 91-128.
WE Acree, Jr, LM Grubbs, MH Abraham. Prediction of toxicity, sensory responses and biological responses with the Abraham model, toxicity and drug testing, Prof. Bill Acree (Ed.). InTech. ISBN: 978-953-51-0004-1. 2012. doi:10.5772/29972. Available fromx: http://www.intechopen.com/books/toxicity-and-drugtesting/prediction-of-toxicity-sensory-responses-and-biological-responses-with-the-abraham-model.
Abraham MH. Scales of hydrogen bonding: their construction and application to physicochemical and biochemical processes. Chem Soc Rev. 1993;22:73–83.
Abraham MH, Ibrahim A, Zissimos AM. The determination of sets of solute descriptors from chromatographic measurements. J Chromatogr A. 2004;1037:29–47.
Abraham MH, McGowan JC. The use of characteristic volumes to measure cavity terms in reversed phase liquid chromatograph. Chromatographia. 1987;23(4):243–6. doi:10.1007/BF02311772.
ACD/Absolv. The Absolv prediction module calculates Abraham solvation parameters and is the result of collaboration between ACD/Labs and Prof. MH Abraham. 2014. [http://www.acdlabs.com/products/percepta/predictors/absolv/.
Pence HE, Williams AJ. ChemSpider: an online chemical information resource. J Chem Educ. 2010;87(11):1123–4 [http://www.chemspider.com/]
Steinbeck C, Han Y, Kuhn S, Horlacher O, Luttmann E, Willighagen E. The Chemistry Development Kit (CDK): an open-source Java library for chemo-and bioinformatics. J Chem Inf Comput Sci. 2003;43(2):493–500.
Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, et al. Virtual computational chemistry laboratory–design and description. J Comput Aided Mol Des. 2005;19(6):453–63.
Platts JA, Butina D, Abraham MH, Hersey A. Estimation of molecular linear free energy relation descriptors using a group contribution approach. J Chem Inf Comput Sci. 1999;39(5):835–45. doi:10.1021/ci980339t.
Jover J, Bosque R, Sales J. Determination of Abraham solute parameters from molecular structure. J Chem Inf Comput Sci. 2004;44:1098–106.
Sprunger LM, Proctor A, Acree Jr WE, Abraham MH. Computation methodology for determining Abraham solute descriptors from limited experimental data by combining Abraham model and Goss-modified Abraham model correlations. Phys Chem Liq. 2008;46:5.
Abraham MH, Abraham RJ, Byren J, Griffith L. NMR method for the determination of solute hydrogen bond acidity. J Org Chem. 2006;71(9):3389–94. doi:10.1021/jo052631n.
Poole CF, Atapattu SN, Poole SK, Bell AK. Determination of solute descriptors by chromatographic methods. Analytica Chimica Acta. 2009; 652 1-2. doi:10.1016/j.aca.2009.04.038.
Brumfield M, Wadawadigi A, Kuprasertkul N, Mehta S, Stephens TW, Barrera M et al. Determination of Abraham model solute descriptors for three dichloronitrobenzenes from measured solubilities in organic solvents. Phys Chem Liq. 2014; accepted for publication. doi:10.1080/00319104.2014.972555.
Charlton AK, Daniels CR, Acree Jr WE, Abraham MH. Solubility of crystalline nonelectrolyte solutes in organic solvents: mathematical correlation of acetylsalicylic acid solubilities with the Abraham general solvation model. J Solution Chem. 2003;32:1087–102.
Bradley JC. 2014. Open notebook science challenge. [http://onschallenge.wikispaces.com/]
Bradley JC. 2014. Open notebook science challenge - list of experiments. [http://onschallenge.wikispaces.com/list+of+experiments]
Seidell A. Solubilities of inorganic and organic compounds: a compilation of quantitative solubility data from the periodical literature. 1911. D. Van Nostrand Company. Print.
Desai PG, Patel AM. Effect of polarity on the solubilities of some organic acids. J Indian Chem Soc. 1935;12:131–6.
Erdmann OL. Untersuchungen Uber Den Indigo. J Fur Praktische Chemie. 1841;22.1:257–99. Print.
Yalkowsky SH, He Y, Jain P. Handbook of aqueous solubility data. Boca Raton, FL: CRC Press; 2010. Print.
Wang J, Hou T, Xu X. Aqueous solubility prediction based on weighted atom type counts and solvent accessible surface areas. J Chem Inform Model. 2009;49(3):571–81.
Bioloom. 2014. [http://www.biobyte.com/bb/prod/bioloom.html]
Bradley JC, Lang ASID. 2014. Cinnamic acid data temperature conversion. Open notebook science. [https://docs.google.com/spreadsheet/ccc?key=0Au_5J1f583GgdHhkY2VHNWIzanMzYzBLN1h3WVlzcWc]
Allen G, Watkinson JG, Webb KH. An infra-red study of the association of benzoic acid in the vapour phase and in dilute solution in non-polar solvents. Spectrochim Acta. 1966;22:807–14.
Schupp OE, Lewis JS. Compilation of gas chromatographic data. Philadelphia: American Society for Testing and Materials; 1967. Print.
Wilson A, Tian A, Chou V, Quay AN, Acree Jr WE, Abraham MH. Experimental and predicted solubilities of 3,4-dichlorobenzoic acid in select organic solvents and in binary aqueous ethanol mixtures. Phys Chem Liq. 2013;50:324–35.
Abraham MH, Acree Jr WE. Descriptors for artemisinin and its derivatives; estimation of physicochemical and biochemical data. Eur Chem Bull. 2013;2:1027–37.
Marcus Y. The properties of organic liquids that are relevant to their use as solvating solvents. Chem Soc Rev. 1993;22:409–16.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
J-CB was the principal investigator and supervised every solubility measurement. MHA, WEA Jr., and ASL modelled the data and prepared the manuscript for publication, SB, DB, EC, LC, SC, EC, SK, MM, and MM measured and recorded the solubility of trans-cinnamic acid in various solvents using various techniques as part of the Open Notebook Science Challenge. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bradley, JC., Abraham, M.H., Acree, W.E. et al. Determination of Abraham model solute descriptors for the monomeric and dimeric forms of trans-cinnamic acid using measured solubilities from the Open Notebook Science Challenge. Chemistry Central Journal 9, 11 (2015). https://doi.org/10.1186/s13065-015-0080-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-015-0080-9