
Abstract
Background
Despite therapeutic hypothermia (TH) and neonatal intensive care, 45–50% of children affected by moderate-to-severe neonatal hypoxic-ischemic encephalopathy (HIE) die or suffer from long-term neurodevelopmental impairment. Additional neuroprotective therapies are sought, besides TH, to further improve the outcome of affected infants.
Allopurinol — a xanthine oxidase inhibitor — reduced the production of oxygen radicals and subsequent brain damage in pre-clinical and preliminary human studies of cerebral ischemia and reperfusion, if administered before or early after the insult.
This ALBINO trial aims to evaluate the efficacy and safety of allopurinol administered immediately after birth to (near-)term infants with early signs of HIE.
Methods/design
The ALBINO trial is an investigator-initiated, randomized, placebo-controlled, double-blinded, multi-national parallel group comparison for superiority investigating the effect of allopurinol in (near-)term infants with neonatal HIE.
Primary endpoint is long-term outcome determined as survival with neurodevelopmental impairment versus death versus non-impaired survival at 2 years.
Results
The primary analysis with three mutually exclusive responses (healthy, death, composite outcome for impairment) will be on the intention-to-treat (ITT) population by a generalized logits model according to Bishop, Fienberg, Holland (Bishop YF, Discrete Multivariate Analysis: Therory and Practice, 1975) and .”will be stratified for the two treatment groups.
Discussion
The statistical analysis for the ALBINO study was defined in detail in the study protocol and implemented in this statistical analysis plan published prior to any data analysis. This is in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines.
Trial registration
ClinicalTrials.gov NCT03162653. Registered on 22 May 2017.
Similar content being viewed by others


Introduction
During labor and childbirth various events (such as placental abruption, uterine rupture, umbilical cord complications, etc.) may result in impaired oxygenation and/or perfusion of the newborn brain which may result in brain injury termed “hypoxic-ischemic encephalopathy” (HIE) (reviewed in [14]). HIE is associated with long-term motor, cognitive, and neurosensory disability, seizure disorders, and death and is one of the fundamental problems in perinatal medicine affecting about 5000–20,000 infants/year in Europe (or 1–4/1000 live births in Western societies) and approximately 1 million infants/year worldwide.
In recent years, therapeutic hypothermia became the only established therapy to improve outcomes after perinatal HIE. Despite hypothermia and modern supportive neonatal intensive care, 45–50% of children with moderate or severe HIE (i.e., 2500–10,000 infants per year in Europe) still die or suffer from long-term neurodevelopmental impairments [4]. Therefore, additional neuroprotective interventions, besides hypothermia, are warranted to further improve outcomes.
Allopurinol is a xanthine oxidase inhibitor and reduces the degradation of purines (especially adenosine) and the production of oxygen radicals and, subsequently, reduced brain damage in experimental and early human studies of ischemia and reperfusion.
This paper describes the statistical analysis plan to evaluate the efficacy and safety of allopurinol administered immediately after birth to near-term infants with perinatal asphyxia and early potential signs of HIE to attenuate long-term neurodevelopmental impairment.
The study protocol version 5 of the ALBINO study was published previously [7]. The statistical analysis was predefined in detail in the study protocol. Substantial changes were made concerning the analysis of the primary endpoint in version 6 and the definition of an interim analysis in version 7 of the study protocol. Any deviation from the originally planned statistical analysis was described within protocol amendments and accepted by ethics committees and national regulatory authorities. The statistical analysis plan (SAP) conforms with the guidelines for the content of statistical analysis plans in clinical trials [5]], please refer to checklist in supplementary material. This SAP includes the interim and the final analysis and describes the analysis principles, definition of outcomes, and methods for their analyses.
Background information
Rationale
Perinatal hypoxic/ischemic events can cause immediate (necrosis) and delayed death (apoptosis) of (especially neuronal) cells, the latter responsible for a substantial amount of HIE-associated permanent brain damage. Whereas no intervention is known to prevent necrosis, the delayed cell death by apoptosis can be reduced by therapeutic interventions:
Apoptosis is in part caused by secondary energy failure which can be reduced by hypothermic treatment [6, 4, 13].
Apoptosis is also caused by xanthine oxidase-mediated production of cytotoxic oxygen radicals during reperfusion, and there is evidence that allopurinol, a xanthine-oxidase inhibitor, reduces delayed cell death in animal models of perinatal asphyxia and ischemia/reperfusion [9, 15, 3]. Allopurinol, a xanthine-oxidase inhibitor, blocks purine degradation. It also seems to result in the accumulation of adenosine during hypoxia, since allopurinol treatment increases brain tissue levels of adenosine after hypoxic-ischemic injury [8]. Adenosine is a potent inhibitory neuromodulator providing additional neuroprotection in HIE. In higher concentrations, allopurinol acts as an iron chelator and direct scavenger of free radicals [12]. Allopurinol pretreatment preserves cerebral energy metabolism as shown by 31P NMR during perinatal hypoxia-ischemia in immature rats [17], and thus prevents cerebral damage [9].
The evidence for a potential neuroprotective effect of allopurinol and the preclinical and early clinical studies on allopurinol for HIE have been reviewed [1]. The suggested neuroprotective effect is the basis of the ALBINO study, which has been described in detail in the publication of the study protocol [7].
Objectives
The primary objective of the ALBINO trial is to determine whether in newborns with perinatal asphyxia and early clinical signs of HIE, early postnatal allopurinol compared to placebo administered in addition to standard of care (including therapeutic hypothermia if indicated) reduces the incidence of death or severe neurodevelopmental impairment (defined as cerebral palsy, or cognitive or language impairment) at 24 months of age.
The secondary objectives are to evaluate the effect of allopurinol in addition to hypothermia (if indicated) on biomarkers such as:
-
Brain injury assessed by magnetic resonance imaging
-
Brain injury assessed by (amplitude integrated) electroencephalogram
Trial design
This is an investigator-initiated, randomized, placebo-controlled, (double-)blinded, multi-national, parallel-group comparison for superiority (phase III study) of allopurinol compared to placebo in preventing death or neurodevelopmental impairment at 24 months postnatal age in infants with perinatal asphyxia and early signs of evolving HIE. Essential components of its study protocol have been published [7] and, subsequently, a protocol amendment introducing an interim analysis after 300 included infants reached 24 months postnatal age was approved by ethics committees and authorities in 2023.
Eligibility
Inclusion criteria
Term and near-term infants with a history of disturbed labor who meet at least one criterion of severe perinatal acidosis (or ongoing resuscitation) (as a surrogate for “asphyxia”) are considered eligible for this study.
Criteria for severe perinatal acidosis are defined as
-
Umbilical (or arterial or reliable venous) blood gas within 30 min after birth with pH < 7.0
-
Umbilical (or arterial or reliable venous) blood gas within 30 min after birth with base deficit ≥ 16 mmol/l (i.e., a base excess ≤ − 16 mmol/l)
-
Need for an ongoing cardiac massage at/beyond 5 min postnatally
-
Need for adrenalin administration during resuscitation
-
APGAR score ≤ 5 at 10 min
Additionally, at least two out of the following four criteria of evolving HIE must be met:
-
Altered state of consciousness (reduced or absent response to stimulation or hyperexcitability)
-
Severe muscular hypotonia or hypertonia,
-
Absent or insufficient spontaneous respiration (e.g., gasping only) with the need for respiratory support at 10 min postnatally
-
Abnormal primitive reflexes (absent suck or gag or corneal or Moro reflex) or abnormal movements (e.g., potential clinical correlates of seizure activity)
Exclusion criteria
-
Gestational age below 36 weeks
-
Birth weight below 2500 g
-
Postnatal age >30 min at the end of the screening phase
-
Severe congenital malformation or syndrome requiring neonatal surgery or affecting long-term outcome
-
Patient considered “moribund”/“non-viable” (e.g., lack of spontaneous cardiac activity and ongoing chest compression at 30 min)
-
Decision for “comfort care only” before study drug administration
-
Parents declined study participation as a response to measures of community engagement
-
Both parents are insufficiently fluent in the study site’s national language(s) or English or do not seem to have the intellectual capacity to understand the study procedures and to give consent as judged by the personnel who had been in contact with the mother/father before delivery.
-
Both parents/guardians less than 18 years of age, in case of single parent/guardian this one less than 18 years of age
Interventions
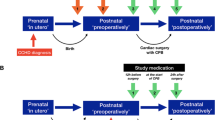
Allopurinol, as a powder for injection (PFI), is administered in two doses. The first dose (20 mg/kg in 2 ml/kg sterile water for injection) is given as soon as intravenous access is established. The start of infusion of study medication should be within 30 min (no later than 45 min) after birth and the second dose (10mg/kg in 1ml/kg sterile water for injection) 12 ± 0.5 h after the (beginning of the infusion of the) first dose. The second dose will only be administered to infants treated with therapeutic hypothermia. Infants who recover quickly and do not qualify for and hence do not undergo hypothermia do not receive a second dose. Administration is by infusion over 10 min using a syringe pump through secure venous access.
Mannitol infusion, as a powder for injection (PFI), is given as a placebo treatment. Dosing and application are the same as for allopurinol, that is 20 mg/kg in 2 ml/kg sterile water given as soon as intravenous access is established (within 45 min after birth) followed by a second dose (10 mg/kg in 1 ml/kg sterile water) — administered over 10 min using a syringe pump through secure venous access, where the second dose is only given in infants that were treated with therapeutic hypothermia.
Definition of primary and secondary outcomes
Primary outcome
The primary endpoint is defined as three mutually exclusive outcomes: survival with neurodevelopmental impairment (NDI, defined as cerebral palsy or severe cognitive and/or language delay at 2 years) or death or survival without NDI at 2 years postnatal age.
Secondary outcomes
-
Death or survival with NDI versus survival without NDI (primary endpoint will be reconstituted as dichotomized composite secondary outcome — survival without NDI versus Death or NDI)
-
Posterior probability that treatment is better than placebo concerning the rate of healthy survivors (survival without NDI), estimated using a Bayesian approach.
-
Incidence of Death
-
Incidence of cerebral palsy (CP)
-
Gross motor function classification system (GMFCS)-Score
-
Motor-Composite-Score of the Bayley III
-
Cognitive-Composite Score of the Bayley III
-
Language-Composite Score of the Bayley III
Further relevant endpoints
-
Anthropometric measures, neurological status, milestones, seizure activity, as well as visual and hearing impairment at 2-year follow-up (in detail: cognitive and language score of PARCA-R-questionnaire, progress concerning weight, head circumference and length, incidence of severe visual and hearing impairment, neurological status, milestones at follow-up concerning right and left hand as well as leg control and speech, incidence of persisting seizure activity and need for anticonvulsive therapy)
-
Results of central reading of magnetic resonance Imaging (MRI) (in detail: Weeke scores [16] and ADC-map-measurement)
-
Results of central reading of electroencephalogram (EEG) epochs 0–12, 12–24 h, 24–48 h, 48–72 h, and 72–96 (in detail: most abnormal background pattern, dominant background pattern, seizure activity, time from birth until onset of any appreciable sleep-wake cycling, time from birth until onset of fully developed sleep-wake cycling, time from birth until onset of first normalization of aEEG trace)
Level of significance
The intention-to-treat (ITT) population is the basis for the confirmatory analysis of the primary endpoint with a significance level of 0.001 in the interim analysis and 0.05 in the final analysis (according to Peto/Haybittle [10]). The analysis of the primary endpoint is based on the PP population and all analyses of secondary and further endpoints are descriptive and will be regarded as remarkable if p < 0.05.
Sample size and power
An incidence of death or severe NDI of 27% in the Allopurinol group compared to 37% in the control group is expected. A total of 682 infants (341 per treatment group) will be required in whom the primary outcome can be ascertained. Assuming a drop-out rate of 10% for loss to follow-up, a total of 760 infants need to be enrolled with formal written consent. Assuming that 10% of parents will refuse continuing participation after the initial dose of the study drug (following short oral or deferred consent procedures, depending on the country) 846 infants have to be randomized immediately after birth.
Intervention allocation and blinding
Clinicians, caregivers, and trial outcome assessors are masked.
Randomization lists have been prepared by the CPCS and were sent to ACE Pharmaceuticals for blinded labeling and packaging of the study medication. Randomization has been done in blocks of four in a 1:1 ratio, with an equal number of patients in each treatment arm. Each shipment of study medication to study centers comprised complete blocks of 4, thereby achieving stratification by center and allocation concealment.
Justification:
Although a variable block size would have been desirable for best allocation concealment, a fixed block size of 4 was selected for the prevention of an uneven distribution of verum/placebo in this study with a low anticipated recruitment rate per center (on average < 10–15) — as well as for practical reasons of study medication distribution to numerous study sites.
Stratification for therapeutic hypothermia — although desirable — was impossible, because the clinical indications for therapeutic hypothermia evolve with time and may not be apparent at the 1st dose of study medication.
Data collection schedule
Data are collected in electronic case record forms (eCRFs) into the study’s secuTrial® electronic database by the staff of each participating center.
The eCRFs to be completed are as follows:
-
Screening
-
Randomization and first dose of study medication
-
Full written informed consent
-
Baseline — infant data
-
Baseline — maternal data
-
Hypothermia treatment and 2nd dose of study medication
-
Thompson score at 1–6 h and 84–106 h or discharge (whichever comes first)
-
Blood gas analysis
-
Cell injury markers and documentation of hypereosinophilia
-
Medication before or during aEEG and mchEEG measurement
-
Neonatal outcome until day 14
-
Discharge
-
Follow-up (Overall, Bayley, Parent QuestionnairesFootnote 1)
-
End of study
-
Adverse events
The following documentation will be done in the respective eCRFs after central assessment by the respective staff (Table 1):
-
MRI central reading
-
aEEG central reading
-
Cerebral ultrasound (0–24 h, 48–72 h, 96–120 h)
-
Peroxidase products (blood)
-
Peroxidase products (urine)
-
S100B and inflammasome-mediated cytokines
Regular safety reporting to the DMC
Safety reporting to the data monitoring committee (DMC) is done after 10, 30, 50, 100, 200, 300, 400, and 600 patients have reached 44 weeks postmenstrual age (PMA).
The following safety parameters are reported and were predefined in the study protocol and described in detail in the DMC charter:
-
Patient characteristics (birth weight, gestational age at birth, gender, umbilical artery pH and lactate)
-
Compliance with the protocol (1st dose of study medication (age at start of administration and administered dose), 2nd dose of study medication (administered dose and interval after 1st dose)
-
Safety parameters (mortality, HIE severity, blood gas analyses at 0.5–6 h/6 to 12 h/12 to 24 h after birth, cell injury markers and plasma osmolality at 24 h ± 6 h after birth, results of portal vein ultrasound in the subgroup of patients with administration of medication through umbilical venous catheter, potential clinical/laboratory signs of allopurinol hypersensitivity reactions, organ failure until day 14 after birth or discharge home (whichever comes first), clinical seizures, health status, support on discharge, MRI — Weeke scores)
-
Listings and aggregate summary tabulation of adverse reactions and adverse events
Interim analysis and stopping rules
No interim analysis was intended at the beginning of the study due to the fact that the primary endpoint will be determined at 2 years follow-up, and recruitment should have been already terminated before 50% of recruited infants have reached 2 years of age according to the original recruitment plan.
A protocol amendment (protocol version 7) approved in 2023 defined an interim analysis after 300 patients had reached follow-up within 2 years. Interim Analysis will be done according to Peto/Haybittle [10]) on a two-sided level of significance of 0.001, leaving a two-sided level of significance of 0.05 for the final analysis.
Depending on the result of the interim analysis, the study will be stopped or continued according to predefined criteria:
-
1)
In the event that the null hypothesis of equal proportions of primary endpoint in the two groups is rejected (based on a two-sided p-value <0.001), the trial statistician will recommend immediate stop of recruitment.
-
2)
Also, in the event that the point estimate for the rate of the primary outcome “survival without severe neurodevelopmental impairment” is exactly equal for the experimental group compared to the placebo group, the trial statistician will recommend an immediate stop of recruitment for the futility of the trial.
-
3)
In all other cases, the results will be reported directly to the members of the independent DMC with data being identified as Group A or Group B first. The DMC may request unblinding.
The DMC will decide about whether or not to advise the steering committee to discontinue the study. “Proposed” decisions depending on the results of the interim analysis of the primary outcome are listed in Table 2 (for guidance), but safety data will additionally be considered and biomarker data may be taken into account.
No adjustment of the significance level due to interim analysis will be done.
In addition to the analysis of the primary endpoint, the DMC will be provided with the data usually included in DMC reports (refer to section “DMC reporting”).
If the DMC recommends to continue the study, the detailed results of the interim analysis will not be communicated to any person directly involved in the conduct of the trial until the final analysis will be done.
Trial reporting
The trial will be reported according to the CONSORT principles [11]. The final analysis will be done after a 2-year follow-up of the last patient.
Protocol non-compliances
Regular remote and on-site monitoring according to a predefined monitoring manual ensures high quality of the data. The following protocol non-compliances will be listed in the final report:
Major
-
Any protocol deviation that may influence the results of the study
-
This may include the following deviations from the protocol that will lead to exclusion from the per-protocol population:
-
Participants randomized in error, i.e., not fulfilling all inclusion or fulfilling an exclusion criterion
-
Time to administration of 1st dose exceeded 45min postnatally
-
Deviation of actually administered dose by more than 10% from the intended dose
-
Any open-label allopurinol
Minor
-
Protocol deviations that will likely not influence study results (e.g., incorrect timing of a brain ultrasound/blood sample)
Treatment non-compliances
-
Timing and dose of study medication, are subject to source data verification by monitoring.
-
Participants in whom the above-listed major protocol deviations occurred will be excluded from the per-protocol population
Analysis populations
Post-randomization exclusions
No post-randomization exclusions will be done except for the case that there would be a patient for whom fraudulent data are detected.
Population definitions
Intention-to-treat population
The intention-to-treat population (ITT) will be all patients included in the study. Patients for whom informed consent was withdrawn will be included in the analysis with all their data that were collected before the withdrawal of consent.
Interim analysis population
The interim analysis will include all randomized patients with the date of birth before the pre-defined reference date minus 24 months.
Safety population
The safety population consists of all patients included in the study.
Descriptive analyses
Numerical items will be summarized as number, number missing, mean, standard deviation, minimum, q25, median, q75, and maximum, if appropriate. Categorical items will be summarized as numbers and percentages.
Representativeness of the trial population and participants throughout
Participant’s flow through each stage of the study will be presented in a CONSORT scheme.
Baseline characteristics of treatment groups
Baseline characteristics of infants and their mothers will be described for the ITT population stratified for the treatment group. The characteristics presented are:
Mother’s baseline characteristics
-
Age in years
-
Maternal ethnic background
-
Pregnancy-related items (diabetic condition, hypertensive disorder, pathological umbilical or Doppler examination)
-
Delivery-related items (mode of delivery, general anesthesia before delivery, clinical chorioamnionitis, uterine rupture, placental abruption, cord complications, other complications
Infant’s characteristics at trial entry
-
Demographic data (gender, place of birth)
-
Basic infant data (birthweight, head circumference, length at birth, APGAR score at 5 and 10 min, late umbilical cord clamping or milking of the cord)
-
Blood gas analysis (pH, lactate, base excess)
-
Delivery room and NICU data (body temperature on admission to NICU/neonatal ward, duration of bag and mask ventilation — if done, age at ET-tube placement — if done, age at the resumption of spontaneous respiration if resumed in the delivery room, age at return of sufficient spontaneous circulation if in the delivery room, cumulative dose of crystalloid volume, colloid volume, erythrocyte concentrate, adrenalin, and bicarbonate — if any, suspected meconium or blood aspiration, suspected sepsis)
Losses to follow-up
The primary endpoint will be assessed at follow-up within 2 years. We expect a certain proportion of patients that will withdraw their informed consent or be lost to follow-up. To avoid losses to follow-up several other sources for information on neurodevelopmental outcomes besides regular follow-up at the study center and the performance of the Bayley III examination will be taken into account (refer to the “Primary endpoint” section and Fig. 1).

Definition of primary endpoint. If data are not available for the first level of definition, the second level applies, and so on. NDI, neurodevelopmental impairment
Comparative analyses
According to the intention-to-treat principle infants will be analyzed according to the treatment group they were randomized to, regardless of the treatment they may have received.
Numerical items will be summarized as number, number missing, mean, standard deviation, minimum, 25% quantile, median, 75% quantile, and maximum, if appropriate. Categorical items will be summarized as numbers and percentages.
Comparative analyses will be stratified for the treatment group only. Due to the fact that there are so many centers participating in this study, the analyses will not be stratified for centers. This is in accordance with ICH E9 for multicenter trials if it is recognized from the start that the limited numbers of subjects per center will make it impracticable to include the center effects in the statistical models.
Detailed definition of outcomes
The primary outcome is composed by cognitive and language development, motor development, and survival status at the age of 2 years.
The assessment of motor development is based on the presence or absence of cerebral palsy (CP). CP is diagnosed if the child has a non-progressive motor impairment characterized by abnormal muscle tone and impaired range or control of movements, according to the criteria defined by the European network “Surveillance of CP in Europe.” A severely abnormal neurological status is classified as unilateral spastic CP, bilateral spastic CP, ataxic CP, dyskinetic CP, or no CP, but other severe abnormality.
To avoid missing data the following hierarchy applies:
-
a)
Neurological classification by the study team indicates cerebral palsy
-
b)
Assessment of motor/neurological development by other health care professionals or other sources (i.e., parents) indicates cerebral palsy
Cognitive and language development is based on the presence or absence of an abnormal cognitive and/or language development assessed by the cognitive-composite-score and the language-composite-score on the Bayley Scales of Infant and Toddler Development (3rd edition). An abnormal development is defined as a composite score of < 85 in at least one of the two scales.
To avoid missing data the following hierarchy applies:
-
a)
Bayley III: Composite cognition score and/or language cognition score < 85
-
b)
Explanation why composite cognition and/or language score has not been provided despite Bayley III having been attempted is indicating abnormal cognitive and/or abnormal language development
-
c)
Bayley II: mental development index (MDI) < 85
-
d)
Rating done by health care professional: other test, assessment, or reason for no test indicates abnormal cognitive and/or abnormal language development
-
e)
PARCA-R (documented by parents) with language and/or cognition standardized score <85
-
f)
Rating done by other source, i.e., parents: other test, assessment, or reason for no test indicates abnormal cognitive and/or abnormal language development
-
g)
Other parental questionnaire results indicate abnormal neurodevelopment
The hierarchy of the definition of the primary endpoint is displayed in Fig. 1.
Detailed derivations of all other outcomes are described in a separate document.
Primary analysis
Primary endpoint with three mutually exclusive responses (healthy, death, composite outcome for impairment) will be analyzed — stratified for the two treatment groups — by a generalized logits model according to Bishop, Fienberg, Holland [2] with SAS 9.4 procedure proc catmod within the intention-to-treat (ITT) population.
Secondary analysis
Secondary endpoints will be analyzed by the Cochrane-Mantel-Haenzel-χ2-test in case of categorial binary data and by the Wilcoxon-Mann-Whitney test in case of score data. Bayley-III-scores will be cut due to lack of sensitivity below 50 points and therefore only fit for non-parametric methods.
Bayesian approach will be done by SAS 9.4 procedure proc genmod.
Analysis of further relevant endpoints
Further relevant endpoints will be analyzed by the Cochrane-Mantel-Haenzel-χ2-test in the case of categorial binary data. Numerical endpoints will be analyzed using parametric or non-parametric methods as appropriate. The proportional odds model will be applied for the analysis of non-binary categorial endpoints.
Multivariate analyses
Multivariate analyses of the primary endpoint will be done including gender, postnatal age at administration of a first dose of study medication (< 15 min after birth vs. 16–30 min after birth vs. >30 min after birth), encephalopathy, where the degree of HIE severity will be derived from the Thompson Score assessed at 3–6 h (before hypothermia) and the initial aEEG findings (first epoch and before any brain-acting medication and/or hypothermia), need for therapeutic hypothermia (yes versus no).
Analysis will be performed by a generalized logits model according to Bishop, Fienberg, Holland [2] with SAS 9.4 procedure proc catmod, adjusted for the treatment group.
The final multivariate model will only include those risk factors with p-value <0.05. These will be checked for interactions one interaction term at a time. A term will suggest an interaction if it reveals a p-value <0.05.
Appropriate subgroup analyses will be performed if these multivariate analyses suggest an interaction between the intervention and one of the risk factors. These post-hoc subgroup analyses are meant to be exploratory (hypotheses generating).
Significance levels and adjustment of p-values for multiplicity
All analyses will be done to assess the superiority of the study medication compared to placebo treatment. Only the result of the analysis of the primary endpoint in the intention-to-treat population will be regarded to be confirmative. Consequently, no adjustment of p-values for multiplicity will be done.
Missing data and sensitivity analysis
In case of more than 10% missing values after hierarchical substitution of data concerning the primary outcome as described in the “Detailed definition of outcomes” section and Fig. 1, a worst case/best case analysis for this endpoint will be performed in the intention-to-treat population as sensitivity analyses and results will be included in the final report.
No imputation will be done for secondary or further relevant endpoints.
Statistical software employed
SAS 9.4 will be used for all analyses.
Additional exploratory analysis
Analyses not specified in the study protocol and the statistical analysis plan will be exploratory in nature and have to be defined in a separate statistical analysis plan. Any post hoc analysis requested by the DMC, a journal editor, or anyone else will be labeled explicitly as such.
Discussion
This article presents the statistical analysis plan for the ALBINO study which has been described in detail in the study protocol and has been substantially changed twice by choosing a more powerful analysis strategy for the primary outcome and by defining an interim analysis. Both substantial changes were included in protocol amendments, approved by ethics committees and national authorities, and implemented before any analysis was started.
Strengths
This is a state-of-the-art randomized trial in a challenging indication and study population. Even if closed after interim analysis, the Albino study will be one of the largest studies so far to evaluate the efficacy and safety of pharmaceutical intervention to improve long-term outcomes after perinatal asphyxia in the era of therapeutic hypothermia and the only one that faced the challenge of administration of study medication immediately after birth. This study will give valuable insight into the application of biomarkers for HIE and the effect of allopurinol.
Limitations
The limitations of the study are the struggle with several problems concerning approval by national authorities and the down-scaling of allopurinol to achieve a pediatric formulation. This caused a delayed start of the recruitment. Additionally, recruitment has been very slow due to a limited number of suitable inborn patients at the participating almost 70 trial sites and the fact that in the majority of sites short oral consent by at least one parent has to be achieved within the first 45 min after birth. Due to limited resources, this may necessitate preterm termination of the study before reaching the calculated sample size potentially preventing conclusive results.
Trial status
At present, the study has been registered at www.clinical.trials.gov (NCT03162653, on May 22, 2017) and the study protocol has been published [7]. The first patient in was on March 27, 2018, and the status of recruitment is at 460 patients recruited and 300 recruited patients have reached 2 years’ postnatal age.
Deviation from analysis described in protocol
None yet.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Change history
15 March 2024
A Correction to this paper has been published: https://doi.org/10.1186/s13063-024-08031-x
Notes
Paper version of PARCA-R questionnaire will be filled by parents and inserted into the eCRF by the staff of each center.
Abbreviations
- CFR:
-
Code of Federal Regulations
- CP:
-
Cerebral palsy
- CPCS:
-
Center for Pediatric Clinical Studies
- DMC:
-
Data monitoring committee
- eCRF:
-
Electronic case report form
- EEG:
-
Electroencephalogram
- FDA:
-
Food and Drug Administration
- GCP:
-
Good clinical practice
- GMFCS:
-
Gross motor function classification system
- HIE:
-
Hypoxic-ischemic encephalopathy
- ICH:
-
International Conference of Harmonization
- ITT:
-
Intention-to-treat
- MDI:
-
Mental development index
- MRI:
-
Magnetic resonance imaging
- NDI:
-
Neurodevelopmental impairment
- PFI:
-
Powder for injection
- PMA:
-
Postmenstrual age
- PP:
-
Per protocol
- SAP:
-
Statistical analysis plan
- TH:
-
Therapeutic hypothermia
References
Annink KV, Franz AR, Derks JB, Rüdiger M, van Bel F, Benders MJ. Allopurinol: old drug, new indication in neonates? Curr Pharm Des. 2017;23(38):5935–42. https://doi.org/10.2174/1381612823666170918123307.
Bishop YF. Discrete Multivariate Analysis: Therory and Practice. Cambridge MA: The MIT Press; 1975.
Braunersreuther V, Jaquet V. Reactive oxygen species in myocardial reperfusion injury: from physiopathology to therapeutic approaches. Current Pharmaceutical Biotechnology. 2012;13:97–114.
Edwards AD, Bocklehurst P, Gunn AJ, Halliday H, Juszczak E, Levene M, Azzopardi D. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ. 2010;340:c363.
Gamble C, Krishan A, Stocken D, Lewis S, Juszczak E, Doré C, Loder E. Guidelines for the content of statistical analysis plans. JAMA. 2017;318(23):2337–43.
Jacobs SE, Berg M, Hunt R, Tarnow-Mordi WO, Inder TE, Davis PG. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev. 2013;2013(1):CD003311.
Maiwald CA, Annink KV, Rüdiger M, Benders MJNL, van Bel F, Allegaert K, Albino Study Group. Effect of allopurinol in addition to hypothermia treatment in neonates for hypoxic-ischemic brain injury on neurocognitive outcome (ALBINO): study protocol of a blinded randomized placebo-controlled parallel group multicenter trial for superiority (phase III). BMC Pediatric. 2019;19(1):210. https://doi.org/10.1186/s12887-019-1566-8.
Marro PJ, Mishra OP, Delivoria-Papadopoulos M. Effect of allopurinol on brain adenosine levels during hypoxia in newborn piglets. Brain Res. 2006;1073–1074:444–50.
Palmer C, Vannucci RC, Towfighi J. Reduction of perinatal hypoxic-ischemic brain damage with allopurinol. Pediatric Res. 1990;27:332–6.
Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, Howard SV, Smith PG. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. I. Introduction and design. Br J Cancer. 1976;34(6):585–612. https://doi.org/10.1038/bjc.1976.220.
Schulz KF, Altman DG. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised. BMJ. 2010;210(340):c332.
Shadid M, Buonocore G, Groenendaal F, Moison R, Ferrali M, Berger HM, van Bel F. Effect of deferoxamine and allopurinol on non-protein-bound iron concentrations in plasma and cortical brain tissue of newborn lambs following hypoxia-ischemia. Neuroscience Lett. 1998;248:5–8.
Tagin MA, Woolcott CG, Vincer MJ, Whyte RK, Stinson DA. Hypothermia for neonatal hypoxic ischemic encephalopathy: an updated systematic review and meta-analysis. Arch Pediatr Adolesc Med. 2012;166:558–66.
Volpe J. Neonatal encephalopathy: An inadequate term for hypoxic-ischemic encephalopathy. Ann Neurol. 2012;72:156–66.
Warner DS, Sheng H, Batinic-Haberle I. Oxidants, antioxidants and the ischemic brain. J Exp Biol. 2004;207:3221.
Weeke LC, Groenendaal F, Mudigonda K, Blennow M, Lequin MH, Meiners LC, de Vries LS. January). A Novel magnetic resonance imaging score predicts neurodevelopmental outcome after perinatal asphyxia and therapeutic hypothermia. J Pediatrics. 2018;192:33–42.e2. https://doi.org/10.1016/j.jpeds.2017.09.043.
Williams GD, Palmer C, Heitjan DF, Smith MB. Allopurinol preserves cerebral energy metabolism during perinatal hypoxia-ischemia: A 31P NMR study in unanesthetized immature rats. Neuroscience Letters. 1992;144:103–6.
Acknowledgements
We thank Thirza van Ramshorst and Edwin Spaans for carefully reading the manuscript. We would also like to thank the members of the data monitoring committee: Michael Weindling (University of Liverpool), Sandra Juul (University of Washington), Steven Miller (Hospital for Sick Children Toronto), Paula Pokorná (General University Hospital Prague), and Josef Högel (University of Ulm), and the members of the ALBINO External Advisory Board: Seetha Shankaran (Wayne State University Detroit) and Neil Marlow (University College London). We acknowledge support from the Open Access Publication Fund of the University of Tübingen.
ALBINO study group
Coordinating Investigators: Axel R. Franz (University Hospital Tuebingen, Germany; corresponding and senior author) and Mario Rüdiger (University Hospital C.G. Carus ‑ Medizinische Fakultät der TU Dresden, Germany). Beneficiaries / National Coordinators: Axel R. Franz and Christian F. Poets (Tuebingen, Germany), Mario Rüdiger (Dresden, Germany), Manon Benders and Frank van Bel (Utrecht, the Netherlands), Karel Allegaert and Gunnar Naulaers (Leuven, Belgium), Dirk Bassler (Zurich, Switzerland), Katrin Klebermaß-Schrehof (Vienna, Austria), Maximo Vento (Valencia, Spain), Hercilia Guimarães (Porto, Portugal), Tom Stiris (Oslo, Norway), Luigi Cattarossi (Udine, Italy), Marjo Metsäranta (Helsinki, Finland), Sampsa Vanhatalo (Helsinki, Finland), Jan Mazela (Poznan, Poland), Tuuli Metsvaht (Tartu, Estonia), Cees K.W. van Veldhuizen (Zeewolde, the Netherlands).
Data Management, Biometry, Monitoring, and Study Coordination, all at the Center for Pediatric Clinical Studies, University Hospital Tuebingen: Corinna Engel, Christian A. Maiwald, Iris Bergmann, Monika Weiss, Andreas Eichhorn, Michael Raubuch, Birgit Schuler.
Industry Partner: Cees K.W. van Veldhuizen, Bas Laméris, Roselinda van der Vlught-Meijer, Thirza van Ramshorst (all ACE Pharmaceuticals BV, Zeewolde, the Netherlands).
Recruiting Hospitals and Local Principal Investigators:
Austria:
Medizinische Universitaet Wien Katrin Klebermaß-Schrehof, Medizinische Universität Graz Gerhard Pichler, Tirol Kliniken - Universitätskliniken Innsbruck Elke Griesmaier, Uniklinikum Salzburg Johannes Brandner. Martin Wald
Belgium:
University Hospitals Leuven Gunnar Naulaers, CHU St. Pierre University Hospital Brüssel Marie Tackoen and Ruth Reibel, CHR - Grand Hopital de Charleroi Chantal Lecart, UZ Brussel Filip Cools, AZ Sint‑Jan Brugges Luc Cornette, Tivoli, La Louviere Genevieve Malfilatre, CHR Citadelle, Liege Renaud Viellevoye.
Estonia:
Tartu University Hospital Tuuli Metsvaht, Tallinn Children’s Hospital Mari‑Liis Ilmoja, West Tallinn Central Hospital Pille Saik and Ruth Käär, East Tallinn Central Hospital Pille Andresson.
Finland:
Helsinki University Hospital (HUS) Marjo Metsäranta, Kristiina Saarinen, Krista Rantakari
Germany:
University Hospital Tuebingen Axel R. Franz, Klinikum der J. W. Goethe-Universität Frankfurt am Main Rolf Schloesser, Universitätsklinikum Münster Torsten Ott, Jenny Potratz Universitätsklinikum C. G. Carus - Medizinische Fakultät der TU Dresden Barbara Seipolt, Universitätsklinikum Duesseldorf Thomas Hoehn, Universitätsklinikum der Ruhr-Universität Bochum Norbert Teig, Cnopf’sche Kinderklinik/Klinik Hallerwiese Nürnberg Michael Schroth, Universitätsklinik der Paracelsius Med. Privatklinik, Klinikum Nürnberg Süd Christoph Fusch, Universitätsklinikum Leipzig Ulrich H. Thome, Universitätsklinik für Kinder- und Jugendmedizin Lübeck Philipp Jung Department of General Pediatrics and Neonatology, Justus-Liebig-University Gießen Harald Ehrhardt Birgit Kampschulte. KJF Klinik Josefinum, Thomas Michael Volkl AUF DER BULT Kinder- und Jugendkrankenhaus Hannover Anna Koluch Universitätsklinikum Erlangen Patrick Mohrhart.
Italy:
Azienda sanitaria universitaria integrata di Udine Luigi Cattarossi and Isabella Mauro, Università degli studi di Padova Eugenio Baraldi, Azienda ospedaliero universitaria Ospedali Riuniti di Ancona Virgilio Carnielli, Fondazione MBBM - Ospedale San Gerardo di Monza Giuseppe Paterlini, Ospedale Evangelico Betania (Naples) Marcello Napolitano, Ospedale Valduce Como Paola Francesca Faldini, ASST FBF-Sacco Ospedale dei Bambini “V.Buzzi” Milano Gianluca Lista, Ospedale di Treviso Gianluca Visintin, ASST-Lariana, Ospedale Sant’anna San Fermo della Battaglia Mario Barbarini and Laura Pagani, Presidio Ospedaliero S.Anna, Città della Salute e della Scienza di Torino Emmanuele Mastretta, Fondazione Policlinico Universitario A. Gemelli IRCCS – Università Cattolica del Sacro Cuore Rome Giovanni Vento, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico Milano Monica Fumagalli, Ospedale Maggiore della Carità Novara Marco Binotti.
Netherlands:
VU Medical Center Mirjam M. van Weissenbruch, Isala Klinieken Henrica L.M. van Straaten, Universitair Medisch Centrum Utrecht Manon J.N.L. Benders, Kim V. Annink, Frank van Bel, Jeroen Dudink, Jan B. Derks, Diakonessenhuis Utrecht Inge P. de Boer, Meander Medisch Centrum Amersfoort Clemens B. Meijssen, Academic Medical Center Timo R. de Haan, Medisch Spectrum Twente Linda G. van Rooij, St. Antonius Ziekenhuis Jacqueline L. van Hillegersberg and Minouche van Dongen, Elisabeth Tweesteden Ziekenhuis Jos Bruinenberg, Deventer Ziekenhuis A.C.M. Dassel, Maxima Medical Center Veldhoven Koen P. Dijkman, Spaarne Gasthuis Marlies A. van Houten, OLVG Sophie R.D. van der Schoor.
Erasmus MC-Sophia Children's Hospital Gerbrich Engelien van den Bosch Isala Kliniek Zwolle Marianne van der Heide Sint Elisabeth Ziekenhuis Tilburg Carien C.C.J.M. Smeets
Norway:
Oslo Universitetssykehus HF Tom Stiris, Haukeland Univ. Sykehus Bergen Bodil Salvesen, Vestfold Hospital Trust Tonsberg Moritz Schneider and Eirik Nestaas, Akershus University Hospital (AHUS) Lørenskog Britt Nakstad, Sykehuset Innlandet Lillehammer Dag Helge Frøisland.
Poland:
Poznan University of Medical Sciences ‑Department of Neonatology Jan Mazela and Lukas Karpinski, Instytut Centrum Zdrowia Matki Polki Ewa Gulczynska, Wroclaw Medical University Department of Neonatology Barbara Królak‑Olejnik, Neonatal and Intensive Care Department Medical University of Warsaw Renata Bokiniec
Portugal:
Centro Hospitalar Universitário São João (CHUSJ) Porto Ana I. Vilan, Centro Materno Infantil do Norte (CMIN) Porto Liliana Flores de Pinho, Hospital Pedro Hispano (HPH) Porto Claudia Ferraz, Hospital de Braga (HB) Almerinda Pereira, Hospital Fernando Fonseca (HFF) Amadora (Lisboa) Rosalina Barroso, Hospital Santa Maria - Centro Hospitalar Universitario de Lisboa Norte André Mendes da Graça, Centro Hospitalar Universitário de Lisboa Central (CHULC) Teresa Tomé and Filomena Pinto.
Spain:
Hospital Universitario y Politécnico La Fe, Valencia, Maximo Vento and Juan Martínez Rodilla, Complejo Hospitalario Universitario Santiago de Compostela Maria Luz. Couce Pico, Hospital Puerta del Mar Cádiz Simón Lubián, General University Hospital of Alicante Caridad Tapia Collados, Quironsalud Madrid University Hospital Fernando Cabañas, Hospital Sant Joan de Déu Barcelona Marta Camprubí Camprubí, Hospital Virgen de las Nieves Granada José Antonio Hurtado Suazo, Hospital Universitario La Paz Madrid Eva Valverde, Hospital Reina Sofía Córdoba Inés Tofé, Complejo Hospitalario Universitario Vigo José Ramón Fernández Lorenzo, Ana Concheiro Guisá Hospital Clínico San Carlos Madrid José Martinez Orgado, Hospital Vall de Hebrón Barcelona Héctor Boix, Hospital Regional Universitario de Málaga Mercedes Chaffanel, Hospital Virgen del Rocío Sevilla Francisco Jimenez Parrilla, Hospital Gregorio Marañón Madrid Dorotea Blanco, Hospital de Cruces Barakaldo Begoña Loureiro, Hospital 12 de Octubre Madrid Maria Teresa Moral‑Pumarega, Hospital Miguel Servet Zaragoza Segundo Rite.
Switzerland:
UniversitaetsSpital Zuerich Dirk Bassler, Julia Maletzki and Claudia Knoepfli, Mara Hesse Kinderspital Zürich (KiSpi ZH) Cornelia Hagmann, Kantonsspital Winterthur Michael Kleber, Universitäts-Kinderspital beider Basel (UKBB) Sven Schulzke, Kantonsspital Luzern Martin Stocker, Ostschweizer Kinderspital (St.Gallen) André Birkenmaier, Kantonsspital Graubünden (Chur) Thomas Riedel. Bjarte Rogdo Kantonsspital Aarau AG Philipp Meyer Spital Zöllikerberg Vera Bernet.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study is funded under the Horizon 2020 Framework Program of the European Union, call H2020-PHC-2015-two-stage, grant 667224. The European Union/European Commission had no influence on the design of the study; on collection, analysis, and interpretation of data; and on writing this manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
CE drafted the first version of the manuscript on behalf of the ALBINO study group (first authorship). All other members of the ALBINO study group revised the manuscript, making important contributions, and approved the final version of the manuscript. CE is the senior statistician of the project and AF is the clinically responsible PI.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The ALBINO trial is performed in accordance with the Declaration of Helsinki and the guidelines of Good Clinical Practice (GCP). Written informed consent must be obtained by the parents or legal guardians before full participation in the study (i.e., before administration of the second dose of study medication (if indicated) and before data entry into the database). Whether oral consent by at least one parent is obtained following short information or an approved waiver of consent is applied before administration of the first dose of study medication, depends on the approvals of the responsible national ethics committees (as detailed elsewhere). At the time of publication, the ALBINO trial is currently taking place in 10 European countries and may expand to other countries, including Poland and Portugal, once ethical approval has been obtained.
Austria: Ethics: Ethikkommission Medizinische Universität Wien, reference no. 1731/2017, approved with deferred consent; Authority: Bundesamt für Sicherheit im Gesundheitswesen, reference no. 10680185 approved conduct. Belgium: Coordinating ethical committee: Ethical Committee UZ Leuven reference no. S60224, approved with deferred consent; Authority: Federal Agency for Medicines and Health Products Brussels, reference no. FAGG/ R&D/MMN approved conduct. Estonia: Ethics: Research Ethics Committee of the University of Tartu (UT REC), reference no. 272/T-13, approved with deferred consent; Authority: State Agency of Medicines clinical trial, reference no. 17–044 approved conduct. Finland: Ethics: Naisten, lasten ja psykiatrian eettinen toimikunta, Helsingin ja Uudenmaan sairaanhoitopiiri reference no. HUS/1528/2017 approved with deferred consent; Authority: Finnish Medicines Agency (FIMEA) reference no. 44/ 2017 approved conduct. Germany: Ethics: Ethics Committee at the University Hospital Tuebingen, reference no. 703/2016AMG1, approved with short oral consent; Authority: Bundesinstitut für Arzneimittel und Medizinprodukte, reference no. 4041912 approved conduct. Italy: Ethics: COMITATO ETICO UNICO REGIONALE sede operative CENTRO di RIFERIMENTO ONCOLOGICO reference no. 6.1 21/11/2017 - ID 2167 approved with short oral consent; Authority: AIFA- Agenzia Italiana del Farmaco reference no. 97707 approved conduct. Netherlands: Ethics: The ethical committee of the University Medical Center Utrecht reference no. NL57237.041.16 approved with short oral consent; Authority: Centrale Commissie Mensgebonden Onderzoek (CCMO) reference no. NL57237.041.16 approved conduct. Norway: Ethics: REK — Regionale komiteer for medisinsk og helsefaglig forskningsetikk reference no. 2017/800 approved with deferred consent; Authority: Norwegian Medicines Agency reference no. 17/04729–11 approved conduct. Poland: Ethics: to be submitted Authority: to be submitted. Portugal: Ethics: CEIC - Comissão de Ética para a Investigação Clínica - Waiting for approval; Authority: INFARMED — Autoridade Nacional do Medicamento e Produtos de Saúde, I.P. — approved conduct. Spain: Ethics: Ethics Committee for Research with Medications at the Hospital Universitario y Politécnico de La Fe. reference no. 2016–000222-19 approved with Short Oral Consent; Authority: Spanish Agency of Medicines reference no.2016–000222-19 approved with Short Oral Consent. Switzerland: Ethics: Kantonale Ethikkommission Zürich, reference no. 2017/00961, approved with short oral consent; Authority: Swissmedic — Swiss agency for therapeutic products, reference no. 2017DR3135 approved conduct.
Consent for publication
Not applicable
Competing interests
R. van der Vlught-Meijer is an employee of ACE Pharmaceuticals, the company that holds the Dutch marketing authorization registration for Acepurin® (allopurinol 1 g/100 ml) for intravenous application for the treatment of gout. C. van Veldhuizen and B. Laméris are the former owners of ACE Pharmaceuticals. All three contributed to the development of the study protocol and therefore the statistical analysis plan. All other contributors declare that they do not have competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: "Following publication of the original article, we have been notified that updates were needed to the acknowledgements, ALBINO study group section, Recruiting Hospitals and Local Principal Investigator."
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Engel, C., Rüdiger, M., Benders, M.J.N.L. et al. Detailed statistical analysis plan for ALBINO: effect of Allopurinol in addition to hypothermia for hypoxic-ischemic Brain Injury on Neurocognitive Outcome — a blinded randomized placebo-controlled parallel group multicenter trial for superiority (phase III). Trials 25, 81 (2024). https://doi.org/10.1186/s13063-023-07828-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-023-07828-6