
Abstract
Background
Despite the fast establishment of new therapeutic agents in the management of COVID-19 and large-scale vaccination campaigns since the beginning of the SARS-CoV-2 pandemic in early 2020, severe disease courses still represent a threat, especially to patients with risk factors. This indicates the need for alternative strategies to prevent respiratory complications like acute respiratory distress syndrome (ARDS) associated with COVID-19. Aviptadil, a synthetic form of human vasoactive intestinal peptide, might be beneficial for COVID-19 patients at high risk of developing ARDS because of its ability to influence the regulation of exaggerated pro-inflammatory proteins and orchestrate the lung homeostasis. Aviptadil has recently been shown to considerably improve the prognosis of ARDS in COVID-19 when applied intravenously. An inhaled application of aviptadil has the advantages of achieving a higher concentration in the lung tissue, fast onset of activity, avoiding the hepatic first-pass metabolism, and the reduction of adverse effects. The overall objective of this project is to assess the efficacy and safety of inhaled aviptadil in patients hospitalized for COVID-19 at high risk of developing ARDS.
Methods
This multicenter, placebo-controlled, double-blinded, randomized trial with 132 adult patients hospitalized for COVID-19 and at high risk for ARDS (adapted early acute lung injury score ≥ 2 points) is conducted in five public hospitals in Europe. Key exclusion criteria are mechanical ventilation at baseline, need for intensive care at baseline, and severe hemodynamic instability. Patients are randomly allocated to either inhale 67 μg aviptadil or normal saline (three times a day for 10 days), in addition to standard care, stratified by center. The primary endpoint is time from hospitalization to clinical improvement, defined as either hospital discharge, or improvement of at least two levels on the nine-level scale for clinical status suggested by the World Health Organization.
Discussion
Treatment strategies for COVID-19 are still limited. In the context of upcoming new variants of SARS-CoV-2 and possible inefficacy of the available vaccines and antibody therapies, the investigation of alternative therapy options plays a crucial role in decreasing associated mortality and improving prognosis. Due to its unique immunomodulating properties also targeting the SARS-CoV-2 pathways, inhaled aviptadil may have the potential to prevent ARDS in COVID-19.
Trial registration
ClinicalTrials.gov, NCT04536350. Registered 02 September 2020.
Similar content being viewed by others


Administrative information
Note: the numbers in curly brackets in this protocol refer to SPIRIT checklist item numbers. The order of the items has been modified to group similar items (see http://www.equator-network.org/reporting-guidelines/spirit-2013-statement-defining-standard-protocol-items-for-clinical-trials/).
Title {1} | Inhaled Aviptadil for the possible treatment of COVID-19 in patients at high risk for ARDS: A randomized, placebo-controlled, and multicentre trial. |
Trial registration {2a and 2b}. | ClinicalTrials.gov, NCT04536350 Registered on 2nd September 2020 |
Protocol version {3} | 30.06.2022, Version 8 |
Funding {4} | This trial is financed by AdVita Lifescience GmbH, Gundelfingen, Germany. Co-Investigator Maria Boesing receives a personalized research grant within the program “Young talents in clinical research” from the Swiss Academy of Medical Sciences. |
Author details {5a} | Maria Boesing, University Clinic of Medicine, Cantonal Hospital Baselland and Faculty of Medicine, University of Basel Kristin Abig, University Clinic of Medicine, Cantonal Hospital Baselland Michael Brändle, Cantonal Hospital St. Gallen Martin Brutsche, Cantonal Hospital St. Gallen Emanuel Burri, University Clinic of Medicine, Cantonal Hospital Baselland Björn C. Frye, Department of Pneumology, Medical Center University of Freiburg Stéphanie Giezendanner, University Clinic of Medicine, Cantonal Hospital Baselland Jan C. Grutters, St. Antonius Hospital Nieuwegein and Division of Heart & Lungs, University Medical Center Utrecht Philippe Haas, AdVita Lifescience GmbH Justian Heisler, University Clinic of Medicine, Cantonal Hospital Baselland and Faculty of Medicine, University of Basel Fabienne Jaun, University Clinic of Medicine, Cantonal Hospital Baselland Anne B. Leuppi-Taegtmeyer, Department of Clinical Pharmacology and Toxicology, University Hospital Basel and Cantonal Hospital Baselland Giorgia Lüthi-Corridori, University Clinic of Medicine, Cantonal Hospital Baselland and Faculty of Medicine, University of Basel Joachim Müller-Quernheim, Department of Pneumology, Medical Center University of Freiburg Reto Nüesch, Hospital Schwyz Wolfgang Pohl, Karl Landsteiner Institute for Clinical and Experimental Pneumology, Clinic Hietzing Frank Rassouli, Cantonal Hospital St. Gallen, Jörg D. Leuppi, University Clinic of Medicine, Cantonal Hospital Baselland and Faculty of Medicine, University of Basel |
Name and contact information for the trial sponsor {5b} | Prof. Jörg D. Leuppi, MD, PhD Clinical Professor of Internal Medicine, University of Basel Head of the University Clinic of Medicine Cantonal Hospital Baselland Rheinstrasse 26 CH-4410 Liestal Phone: +41-61-925-21-80 E-Mail: joerg.leuppi@ksbl.ch |
Role of sponsor {5c} | Professor Jörg D. Leuppi and his research team composed this study protocol together with collaborating partners. The study is conducted under the supervision of Jörg D. Leuppi. Jörg D. Leuppi and his research team are responsible for all submissions to relevant local authorities in order to obtain study approval. Prof. Leuppi is involved and has ultimate authority in every step of this study, including data collection, management, and analysis, interpretation of results, as well as composition of scientific reports and their submission for publication. AdVita Lifescience GmbH provides financial means for the conduction of this trial. Associates of AdVita contribute their experience with the investigational drug and its inhaled application. |
Introduction
Background and rationale {6a}
The world has been experiencing an exceptional state, due to the SARS-CoV-2 pandemic. In the beginning of 2020, about 20% of individuals with the SARS-CoV-2 associated corona virus disease (COVID-19) suffered from a severe course, characterized by significant respiratory symptoms including the potentially lethal acute respiratory distress syndrome (ARDS) [1]. Since then, numerous studies have been initiated in order to evaluate therapeutic agents for the treatment of COVID-19. To date, an improved outcome in hospitalized patients has been shown in randomized controlled trials for anti-cytokine monoclonal antibodies (tocilizumab), Janus kinase inhibitors (baricitinib), and dexamethasone [2–4], which is reflected in current treatment recommendations by national and international health organizations and societies [5–8].
Further available therapy options for early outpatient treatment in high-risk patients include the monoclonal antibodies sotrovimab and bebtelovimab, antiviral substances remdesivir, nirmatrelvir/ritonavir, and molnupiravir, as well as inhaled corticosteroid budesonide and serotonin reuptake inhibitor fluvoxamine [9–14]. The monoclonal antibody combination casirivimab/imdevimab had been effective in reducing hospitalization risk and mortality till late 2021 but proved to have no benefit for the treatment of the emerging omicron variant [15]. At the time of setting up this study, other approaches like convalescent plasma, further antiviral antibodies and other experimental substances are being investigated [3, 16–18].
Despite the fast establishment and adaptation of recommendations for the clinical management of COVID-19, as well as large-scale vaccination campaigns, severe disease-courses still represent a threat, especially to patients with risk factors such as old age, arterial hypertension, or diabetes mellitus [19]. This indicates the need for alternative strategies to prevent and ameliorate respiratory complications associated with COVID-19, in order to effectively prevent intensive care (ICU) admissions and reduce mortality.
The results of the RECOVERY trial indicate that an excessive inflammatory reaction plays an important role in the pathophysiology of severe COVID-19 and the progression to ARDS [2]. In fact, severe cases of COVID-19 are associated with elevated serum levels of pro-inflammatory mediators [1, 20, 21]. SARS-CoV-2 specifically targets the surfactant-producing pulmonary alveolar type II (ATII) cells by binding to their angiotensin converting enzyme 2 (ACE2-) receptors and entering the cell [22]. Viral replication and infection of adjacent ATII cells then lead to a massive cytokine release and, consequently, to apoptosis and a critical decrease of surfactant production, which disrupts the alveolar gas exchange resulting in ARDS [22, 23].
Vasoactive intestinal peptide (VIP), a gut peptide hormone containing 28-residue amino acid peptides, was discovered and first synthesized in the seventies [24–26]. Next to fulfilling various effects in the nervous, digestive, cardiovascular, respiratory, and reproductive systems, VIP is physiologically highly localized in the lungs [27, 28]. There, it binds with ATII cells via the VPAC1 receptor, the same cell type to which the SARS-CoV-2 virus binds via the ACE2-receptor [22, 27]. When VIP binds to the ATII-cells, it inhibits NMDA-induced caspase-3 activity inside the cell, which in turn decreases production of the pro-inflammatory cytokines interleukin-6 and TNF-alpha [29–33]. It has been proposed as a modulator of lung inflammation and airway constriction [25, 26, 34], and its protective effects on pulmonary tissue have been shown in numerous animal models of lung injury in rats, guinea pigs, dogs, and sheep [35–37]. VIP was also shown to increase surfactant production by upregulation of choline phosphate cytidylyltransferase and C-Fos protein expression in ATII cells [38–40]. Recent data demonstrated that plasma levels of VIP are higher in patients with severe COVID-19, compared to healthy individuals and those with mild COVID-19 [41, 42], and that VIP can block SARS-CoV-2 virus replication in vitro [32].
Aviptadil, a synthetic form of VIP, might prevent COVID-19 patients from developing ARDS due to the above described anti-inflammatory properties. The presumed primary therapeutic mechanism of action of inhaled aviptadil is a combination of anti-inflammatory properties and induction of tolerogenic immune response of immune cells localized in the lungs [43, 44]. It has been shown to reduce interferon producing T cells, to dampen Th17-T-cells and to promote regulatory T-cells [43, 45, 46]. These immune-dampening effects have been previously described to occur in the alveolar compartment of sarcoidosis patients after inhalation of aviptadil, which demonstrates that its local application by inhalation is feasible and results in relevant immunological changes [43]. There is further promising evidence from a case report of a patient with pneumonitis resulting from check-point-inhibitor therapy for melanoma, in which the administration of inhaled aviptadil was well tolerated and led to dampening of alveolar inflammation, radiological and clinical improvement [47].
To date, there is no clinical evidence for the efficacy of inhaled aviptadil in COVID-19. However, it was recently observed in vitro that VIP can inhibit SARS-CoV-2 replication and reduce cellular proinflammatory cytokine production [32]. Two phase II trials have been announced recently on the US National Library of Medicine platform “ClinicalTrials.gov” (COVID-AIV (NCT04311697), AVICOVID-2 (NCT04360096)), which investigate aviptadil in patients with COVID-19 in the USA. Preliminary results of the COVID-AIV trial, which has been recruiting patients with COVID-19 and respiratory failure, indicate a promising antiviral effect of the intravenous administration of aviptadil [42]. In contrast, this current trial investigates the inhaled application of aviptadil in an earlier stage of disease, in patients at increased risk for ARDS. Inhaled aviptadil likely circumvents several potential side effects, like hypotension and tachycardia [43, 47].
Daily doses of up to 300 μg inhaled aviptadil have been shown to be safe in phase II trials for the treatment of sarcoidosis and pulmonary hypertension, as well as in a recently published phase I trial in the treatment of ARDS [43, 48–50]. Aviptadil has been given Orphan Drug Designation in the European Union and the USA for the treatment of ARDS and the inhaled application has been observed to be safe without severe side effects [43, 47, 51].
Objectives {7}
The primary objective of this trial is to investigate the efficacy and safety of inhaled aviptadil in hospitalized COVID-19 patients at high risk for developing ARDS. The study will assess whether patients with COVID-19 under high risk for developing ARDS recover faster when they receive inhaled aviptadil in addition to standard care, compared to patients receiving standard care only. A secondary objective is the investigation of the overall course of disease under inhaled aviptadil in terms of need for mechanical ventilation, time requiring oxygen supplementation, infection-related biomarkers, and subjective severity of symptoms. The safety objective is to assess any potential harm of inhaled aviptadil.
Trial design {8}
This study is a multicenter, placebo-controlled, double-blinded, randomized phase II trial with 132 adult patients. In a parallel group design, patients will be randomly allocated in a 1:1 ratio to inhale either aviptadil (67 μg three times a day) or normal saline (1 ml three times a day) for 10 days, or until hospital discharge, in addition to standard care. The study is conducted as a superiority trial.
Public and patient involvement in the design of this protocol
Due to the novelty of the disease under investigation and the prevalent time pressure in the design phase of the trial, patient and public involvement in the design of this protocol was not applied.
Methods: participants, interventions, and outcomes
Study setting {9}
Recruitment of hospitalized patients takes place at the Cantonal Hospitals of Liestal and St. Gallen and the Hospital Schwyz in Switzerland, as well as the Clinic Hietzing, Vienna in Austria and the Antonius Hospital Nieuwegein in the Netherlands between May 2021 and December 2022 (estimated).
Eligibility criteria {10}
Hospitalized patients with diagnosed COVID-19 are asked to take part in this study when the following eligibility criteria are fulfilled:
-
▪ SARS-CoV-2 infection, verified according to current in-house guidelines
-
▪ High risk for the development of ARDS:
i.e., within 24 h before inclusion at least 2 points on an adapted EALI (early acute lung injury) score, with at least one point from the original EALI score [52, 53]
-
Original EALI score:
-
◦ 2–6 l O2 supplementation to achieve a SaO2 > 90%: 1 point
-
◦ > 6 l O2 supplementation to achieve a SaO2 > 90%: 2 points
-
◦ Respiratory rate ≥ 30/min: 1 point
-
◦ Immunosuppression: 1 point
-
-
Modification adapting for risk factors for ARDS in COVID-19 affected patients [54]
-
◦ Arterial hypertension: 1 point
-
◦ Diabetes: 1 point
-
◦ Fever > 39 °C: 1 point
-
-
-
▪ Age ≥ 18 years
-
▪ Ability to comply with the inhalation maneuver
-
▪ Ability to understand the clinical trial and sign the informed consent
The presence of any of the following exclusion criteria will lead to exclusion of the patient:
-
▪ Mechanical ventilation or intensive care treatment within current hospitalization
-
▪ Nasal high-flow cannula or continuous positive airway pressure (cPAP) ventilation at time of inclusion
-
▪ Inability to conduct inhalation therapy
-
▪ Hemodynamic instability with requirement of vasopressor therapy
-
▪ Severe comorbidities interfering with the safe participation according to the treating physician
-
▪ Previous participation in this trial, or current participation in another interventional study
-
▪ Pregnancy
-
▪ Systemic immunosuppression for chronic underlying condition (corticosteroid treatment as part of “standard care” allowed)
“Drop-out” is defined as the patient’s withdrawal of consent at any time after inclusion into the study.
Who will take informed consent? {26a}
After a patient is identified as a potential participant, a Good Clinical Practice (GCP)-trained physician from the study team will inform the patient personally about the trial and ask for written consent by signature on the informed consent form. Each potential participant will be informed that participation in the trial is voluntary that he/she may withdraw from the study at any time with no need of justification and that withdrawal of consent will not affect his/her subsequent medical assistance and treatment. Consent or assent from authorized surrogates is not intended, patients who are not able to understand the trial or sign the informed consent form of their own accord are excluded from participation.
Additional consent provisions for collection and use of participant data and biological specimens {26b}
Additional collection and use of participant data and biological specimens are not intended.
Interventions
Explanation for the choice of comparators {6b}
Due to the novelty of the investigated disease COVID-19, global guidelines or a clear definition of standard care are not available at the time of implementation of the study. Additionally, standard treatment strategies may change during the study period in accordance with new research findings. At each study center, there are internal written guidelines for the clinical management of hospitalized COVID-19 patients, depending on the severity of the case. At the time of implementation of the study, elements of the standard care used in the participating study centers include oxygen therapy, systemic glucocorticoids, remdesivir, tocilizumab, nutritive measures, and prevention and management of ARDS, bacterial superinfections, and sepsis, as well as thrombembolic, neurologic, cardiac, and renal complications. Since there is no clear definition of standard care to date, the only ethically justifiable comparator is the best available care, which is adapted throughout the trial according to the current state of research.
Intervention description {11a}
Patients in the intervention group inhale 1 ml aviptadil solution (67 μg/ml) three times a day (morning, noon, evening), while participants in the control group inhale 1 ml of NaCl 0.9% three times a day. In both groups, the respective treatment is given in addition to standard care and lasts for 10 days, or until hospital discharge, whichever occurs first. The study drug is administered with the M-neb® dose+mesh nebulizer MN-300/8 in both study groups, and each application will approximately last for 10 min.
Criteria for discontinuing or modifying allocated interventions {11b}
Dose or device modifications are not intended. If clinically indicated, treating doctors can stop the treatment with the study drug. Participants are given the possibility to withdraw from the study at any time. Treating physicians can independently decide about additional treatment options and concomitant medication can be re-evaluated and changed at any time of this trial. A termination of the supplementation is reported to the coordinating study center immediately.
Strategies to improve adherence to interventions {11c}
All participants are hospitalized and medication is administered by qualified ward personnel. Ward personnel is responsible that participating patients receive the study medication in a correct manner and each administration will be documented in the patient records. Any complication or non-adherence with the administration is reported as a note to file.
Relevant concomitant care permitted or prohibited during the trial {11d}
All treatments considered necessary by treating doctors are permitted and their use is recorded in the case report form. Because inhaled aviptadil does not reach the systemic circulation and is mainly metabolized in the lung, pharmacokinetic interactions with aviptadil are not expected. In order to account for potential bias, we aim to analyze data from patients, who received potentially biasing concomitant treatment (e.g., immunosuppressants or immunomodulators) in an adjusted manner (see Statistical methods for primary and secondary outcomes {20a}).
Provisions for ancillary and post-trial care {30}
Patients are carefully monitored until discharge from the hospital and followed-up until four weeks after inclusion. Necessary aftercare is organized by treating physicians independently of this trial. No additional specific ancillary and post-trial care is planned. Despite the efforts of the research team to mitigate risks associated with the intervention, potential small harms may occur. Any potential damage or harm to participants in connection with this trial is covered by the obligatory trial insurance.
Outcomes {12}
The primary endpoint is time to clinical improvement up to day 28, defined as the time (in days) from randomization to the decrease of at least two levels on the WHO-suggested nine-level ordinal scale (see Table 1) [55] or alive discharge from hospital, whichever occurs first. This standardized primary endpoint allows comparison with other efficacy studies in the context of the treatment of COVID-19.
Key secondary endpoints are:
-
▪ Need for mechanical ventilation, non-invasive ventilation, and intensive care during hospitalization
-
▪ Occurrence of multi organ dysfunction syndrome during hospitalization
-
▪ Number of days requiring oxygen supplementation
-
▪ Change from baseline to discharge of the following biomarkers in patients’ blood samples
-
◦ C-reactive protein (CRP)
-
◦ Neutrophil-lymphocyte ratio
-
◦ Interleukin-6
-
◦ Procalcitonin
-
-
▪ Change from baseline to follow-up in patient-reported dyspnea, cough, and fatigue on a visual analog scale
-
▪ Patient-reported impact on health by 12-item Short Form Survey version 2 (SF-12v2) at follow-up
Other clinical endpoints include the length of hospital stay until discharge or death (in days) and mortality rate. CRP, interleukin-6, and procalcitonin are measured at baseline, at least every seven days, and at discharge. The safety endpoints adverse events (AEs), including those leading to discontinuation of treatment, serious adverse events (SAEs), and death are also reported.
Participant timeline {13}
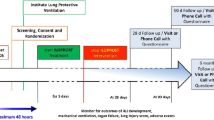
The participant timeline is exhibited in Fig. 1.

Participant timeline. Asterisk symbol (*) indicates the following: if hospital discharge occurs first, intervention is stopped at discharge. Superscript digit one (1) indicates the following: demographics, clinical status according to nine-level ordinal scale, medical history, smoking status, COVID-19 symptoms, and COVID-19 vaccination status. Superscript digit two (2) indicates the following: C-reactive protein, neutrophil-lymphocyte ratio, interleukin-6, and procalcitonin. Superscript digit three (3) indicates the following: on a visual analog scale from 0 to 10. Superscript digit four (4) indicates the following: Clinical status on nine-level ordinal scale, admission to ICU, ventilation, mortality, complications. MRC, Medical Research Council dyspnea scale; SF-12v2, 12-item Short Form Survey version 2
Sample size {14}
Based on the effect of aviptadil in previous trials described in the introductory section, we assume that patients in the experimental group will show a 1.75-fold hazard to have a clinical improvement as compared to patients in the control group (hazard ratio (HR) = 1.75) [42, 50]. Furthermore, based on previously published results for current standard care, we assume an overall probability of 80% for reaching the primary endpoint (clinical improvement) within 28 days (d = 0.8) [56, 57]. Group allocation shall be divided equally (\(\frac{1}{2}n={n}_1={n}_2\)).
Testing for two-sided equality in a Cox proportional hazards model at a significance level of α = 0.05 and aimed power of 80% (β = 0.2), we are applying the sample-size formula in Chow as follows [58]:
where b = log (HR) = log (1.75).
Estimating a drop-out rate of 3% and considering block randomization, we aim to include 132 patients into the study.
Recruitment {15}
All hospitalized patients at a study center who fulfill the eligibility criteria are asked to take part in this study. In order to reach the target sample size within the planned time frame, the study is geographically expanded to several centers within Europe.
Assignment of interventions: allocation
Sequence generation {16a}
The group-allocating randomization code is a computer-generated sequence, using block randomization with block size of four. Each block determines assignment of four patients to the two groups, while two patients are assigned to the intervention group and two patients are assigned to the control group in a random order. Randomization is stratified by study center in order to account for differences in standard of care. Block randomization also accounts for differences in standard of care according to time elapsed since the start of the pandemic.
Concealment mechanism {16b}
The entire concealment process is handled in the pharmacy that produces the study drug. Identically looking vials with doses of either 3 ml aviptadil solution (67 μg/ml, experimental group) or 3 ml NaCl 0.9% (placebo group) are filled in the pharmacy in a 1:1 ratio. Kits of 10 vials of the same content (one vial per treatment day, for a maximum of 10 days) are then packed and labeled with a continuous kit number. The actual content of each kit (aviptadil or placebo) is determined by the randomization sequence described above, while the kit number represents the concealed randomization code. For each kit, the description of the actual content (aviptadil or placebo) is packed in a separate concealed envelope, with only the kit number visible from the outside. The respectively allocated kits and concealed envelopes are shipped to the coordinating study center in Liestal. From here, they are distributed to each study center and used in the order of the kit number indicated on the label. Upon inclusion of a patient, the allocated kit number is documented in the CRF. The concealed envelopes containing the description of group allocation are stored at the coordinating study center in Liestal.
Implementation {16c}
The entire concealment process is handled in the pharmacy that produces the study drug. Study physicians at the respective study center will enroll participants into the study and hand out the kits containing the study drug in the order of the assigned kit numbers. Group assignment is pre-defined by order of the kit numbers and the content of the respective kit.
Assignment of interventions: blinding
Who will be blinded {17a}
Trial participants, investigators, treating physicians, study personnel administering the inhalation, and data analysts are blinded to group allocation. Since the study drug is filled into identically looking vials and aviptadil solution and placebo are not visually distinguishable, blinding is ensured until unblinding procedures are actively undertaken.
Procedure for unblinding if needed {17b}
If a treating physician of a participating patient needs to know for medical reasons (i.e., AE, SAE), if the patient is assigned to the interventional or control group, he/she can call a 24-h phone hotline to receive information about the patient’s group allocation. Unblinding is undertaken by a trained study team member, who opens only the envelope with the respective kit number.
Data collection and management
Plans for assessment and collection of outcomes {18a}
All required patient data at baseline, during hospitalization, at discharge and follow-up are recorded on paper-based case report forms (CRFs). Trained study personnel at each site transfer the data into a web-based electronic case report form. A copy of the CRF used in the trial is available on reasonable request. Laboratory analyses comprise only standard parameters and are performed in the accredited in-hospital laboratories at each site. Patient dyspnea is assessed at inclusion and follow-up by means of the Medical Research Council dyspnea scale (MRC), which has been studied in a variety of respiratory conditions, including COVID-19. Patient-reported outcome is assessed at follow-up by means of the instrument SF-12v2, which is a validated survey for investigating health-related quality of life in a variety of both acute and chronic conditions, including lung diseases.
Plans to promote participant retention and complete follow-up {18b}
Patients participating in the study do not receive financial reimbursement. Expenses for the study medication and diagnostics performed only during this study are covered by the study budget. The 28-day follow-up is a phone call, initiated by a study team member at the respective study center. In case of withdrawal of consent or premature termination of the study, data and samples are evaluated in encrypted form until termination of the study.
Data management {19}
Data acquisition and entry into the web-based and password-protected electronic data capture system (SecuTrial®) are performed by trained staff at the participating study centers. Personal contact information, which is needed for follow-up phone calls, is stored separately and only accessible for the staff members executing these phone calls. Any paper documents, such as informed consent forms, are stored in locked cabinets in restricted access areas at the respective participating study centers. Electronic records are stored on a password-protected server. All records are archived for a minimum of 10 years after study termination or premature termination of the clinical trial at the respective participating study center. A data management plan documents details for the data processing and is available from the authors upon reasonable request.
Confidentiality {27}
All collected patient data are treated as confidential and stored and analyzed in a coded way in accordance with data protection principles. Results will be published in anonymized fashion. Direct access to source documents is permitted for purposes of monitoring, audits, and inspections. The study data and protocol shall be accessible to regulatory authorities for at least ten years after study termination.
Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in this trial/future use {33}
The trial does not involve collecting biological specimens for genetic or molecular analysis.
Statistical methods
Statistical methods for primary and secondary outcomes {20a}
R will be used for all statistical analyses [59]. Primary and secondary endpoints will be summarized by group using descriptive statistics (mean ± standard deviation for normally distributed data, median and interquartile range for other continuous data, absolute and relative frequencies for categorical data). A log-rank test will be used to compare Kaplan-Meier curves for the primary endpoint “time to clinical improvement,” with failure to reach clinical improvement or death before day 28 being right-censored at day 28. Sensitivity analyses will be performed with “death” and “clinical improvement” as competing risks. For potential confounders that are not fully balanced by randomization, adjusted analyses for the primary outcome may be performed using Cox regression models to estimate hazard ratios with corresponding two-sided 95% confidence intervals to compare with the respective unadjusted Cox model. The proportionality assumption for the Cox proportional hazards models will be tested by assessing Schoenfeld residuals.
Treatment effects on binary secondary endpoints will be analyzed by means of chi-squared test, or Fisher’s exact test, in case expected sub-group size is smaller than five. For discrete secondary endpoints, group differences will be assessed using a Poisson or negative binomial regression, depending on the underlying distribution. Treatment effects on patient-reported severity of symptoms will be analyzed with an ordinal logistic regression and an additional longitudinal analysis with cumulative link mixed models. Effects on longitudinal changes in infection-related biomarkers will be assessed using mixed effect linear regression and analysis of covariance. Patient-reported impact on health at day 28 will be compared between the two groups by means of Student’s T-test, or Mann-Whitney-U test, depending on normality of the data.
All statistical tests will be performed using two-sided tests at the significance level 0.05. Detailed methodology for statistical analyses of the data collected in this trial is documented in a separate statistical analysis plan (SAP). The SAP is finalized before database closure and can be obtained from the authors upon reasonable request. A summary of the SAP is provided in Additional file 1.
Interim analyses {21b}
Since a relatively small number of patients is studied during a short study period (max. 28 days) and the treatment harm is considered to be very small due to the short treatment period, we do not consider stopping criteria nor interim analyses to assess the probability that the benefit exceeds the clinically important difference.
Methods for additional analyses (e.g., subgroup analyses) {20b}
See Statistical methods for primary and secondary outcomes {20a}.
Methods in analysis to handle protocol non-adherence and any statistical methods to handle missing data {20c}
The primary analysis will be done in the intention-to-treat population and safety analysis will be done on all patients who started their assigned treatment. The time to clinical improvement will be assessed after all patients will have reached day 28, with failure to reach clinical improvement or death before day 28 considered as right censored at day 28. Careful trial planning and conduct will minimize the occurrence of missing data as far as possible. In case of missing data, treating physicians are contacted with the aim to complete missing data from patients’ records. No data imputation is planned.
Plans to give access to the full protocol, participant level-data and statistical code {31c}
Further and updated trial information can be found at www.ClinicalTrials.gov, NCT04536350. Data and materials that support this protocol, such as a detailed data management plan, CRFs and informed consent form are available from the authors on reasonable request.
Oversight and monitoring
Composition of the coordinating center and trial steering committee {5d}
The coordinating study center at the Cantonal Hospital Baselland, Liestal, Switzerland, consisting of the sponsor-investigator, co-investigators, study coordinators, study physicians, study nurses, and study statistician supervises and coordinates the study. This includes trial management and coordination, statistical, and economic and data management, as well as organizational support for participating study centers. It furthermore acts as a trial steering committee.
Composition of the data monitoring committee, its role and reporting structure {21a}
The monitoring of this study is performed by the independent clinical trial unit (CTU) of the University Hospital Basel, Switzerland, in collaboration with qualified personnel of the coordinating study center. Reporting is done directly to the Investigator. The CTU is independent of the sponsor and without competing interests.
Adverse event reporting and harms {22}
During the entire duration of the trial, all AEs and SAEs are collected, fully investigated, and documented, irrespective of whether they are related or unrelated. Participating study centers are obliged to report SAEs within 24 h to the sponsor-investigator, who, in case of death, reports to the ethics committee within 7 days. AEs and SAEs are followed up until resolution or stabilization. Adverse events will be reported in any associated relevant publication arising from this trial.
Frequency and plans for auditing trial conduct {23}
The study is conducted in accordance with the currently approved protocol, GCP standards, and relevant regulations. Regular monitoring is performed following GCP and the trial monitoring plan. Data is evaluated for protocol compliance, integrity, and accuracy in relation to source documents. Authorities can audit this trial independently from the Sponsor-Investigator and the data monitoring committee. Study documentation and data are accessible to auditors and all involved parties must treat participant data as strictly confidential.
Plans for communicating important protocol amendments to relevant parties (e.g., trial participants, ethical committees) {25}
Important protocol modifications are submitted to the relevant authorities (ethics commission, local drug authorities) for approval before implementation. Changes are communicated to other relevant parties (investigators, study physicians, study nurses) via E-Mail newsletters and personal phone calls.
Dissemination plans {31a}
Our aim is to publish the results of this study in a peer-reviewed journal, without the engagement of professional writers.
Discussion
Our described study protocol presents the design for a randomized controlled trial to investigate the efficacy and safety of inhaled aviptadil in patients hospitalized for COVID-19 at high risk for ARDS. In the context of upcoming new variants of SARS-CoV-2 and a possible future endemic state, the investigation of alternative therapy options still plays a crucial role in decreasing associated mortality and improving prognosis. Due to its unique immunomodulating properties specifically targeting the SARS-CoV-2 pathways, inhaled aviptadil may have the potential to prevent ARDS in COVID-19.
This trial is conducted in different European hospitals in order to ensure generalizability and meet the recruitment target. Patients of all adult age groups and with a wide range of co-morbidities are included into the study, allowing for a broad representation of COVID-19 patients. The primary endpoint as suggested by the WHO allows a comparison with other investigated substances for the management of COVID-19. The evaluation of the secondary endpoints will give further insight into the effects of inhaled aviptadil with regard to systemic inflammation markers, as well as patient-reported outcomes.
Due to the novelty of the disease and new research results, standard care is constantly adapted, which may lead to changes in the relative effect of aviptadil over time. However, the block-randomization with a small block-size ensures that this circumstance will not bias the results. Furthermore, with COVID-19 being the research focus of many current projects, which are “competing” for participating patients, recruitment may become difficult. This challenge is addressed by increasing the number of collaborating centers and assuring, that no competing projects are recruiting at the involved sites.
To date, there are no published data about the administration of inhaled aviptadil in COVID-19. Because of the promising properties of aviptadil, there is an urgent need to close this research gap with a well-designed randomized controlled trial. If the results of this study show that inhaled aviptadil is effective and safe, they may ultimately lead to the introduction of a new substance for the management of COVID-19.
Trial status
The first patient was enrolled into this study on May 18, 2021. The study is currently ongoing with active recruitment under protocol version 8, dated June 30, 2022. Recruitment is anticipated to be completed in December 2022.
Availability of data and materials {29}
Data and materials that support this protocol, such as a detailed data management plan, CRFs, and informed consent form are available from the authors on reasonable request. Following completion of the trial, anonymized datasets and statistical code used in this study will be available from the authors on reasonable request.
Abbreviations
- AE:
-
Adverse event
- ARDS:
-
Acute respiratory distress syndrome
- ATII:
-
Alveolar type-II
- cPAP:
-
Continuous positive airway pressure
- CRF:
-
Case report form
- CRP:
-
C-reactive protein
- CTU:
-
Clinical trial unit
- DMC:
-
Data monitoring committee
- EALI:
-
Early acute lung injury
- EKNZ:
-
Ethics Committee for the Region of Northwestern and Central Switzerland
- EKOS:
-
Ethics Commission for Eastern Switzerland
- GCP:
-
Good Clinical Practice
- ICU:
-
Intensive care unit
- SAE:
-
Serious adverse event
- SAP:
-
Statistical analysis plan
- SF-12v2:
-
12-item Short Form survey, version 2
- VIP:
-
Vasoactive intestinal polypeptide
- WHO:
-
World Health Organization
References
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet (London, England). 2020;395(10223):507–13.
Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, et al. Dexamethasone in hospitalized patients with COVID-19. N Engl J Med. 2021;384(8):693–704.
Swiss National COVID-19 Science Task Force, Clinical Care Group Writing committee. Reduction of COVID-19-associated mortality by drug therapies. 2021. https://sciencetaskforce.ch/en/policy-brief/reduction-of-covid-19-associated-mortality-by-drug-therapies/. Accessed 21 July 2022.
Marconi VC, Ramanan AV, de Bono S, Kartman CE, Krishnan V, Liao R, et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir Med. 2021;9(12):1407–18.
World Health Organization: Therapeutics and COVID-19: living guideline. 2022. https://www.who.int/publications/i/item/WHO-2019-nCoV-therapeutics-2022.4. Accessed 26 July 2022.
Infectious Diseases Society of America: IDSA guidelines on the treatment and management of patients with COVID-19. 2022. https://www.idsociety.org/practice-guideline/covid-19-guideline-treatment-and-management/. Accessed 26 July 2022.
National Institutes of Health: Coronavirus disease 2019 (COVID-19) treatment guidelines. 2022. https://www.covid19treatmentguidelines.nih.gov/. Acessed 26 July 2022.
Swiss Society of Infectiology: SARS-CoV-2 /COVID-19 - antiviral and immunomodulatory treatment considerations. 2022. https://ssi.guidelines.ch/guideline/3352/de/31355. Accessed 26 July 2022.
Gupta A, Gonzalez-Rojas Y, Juarez E, Crespo Casal M, Moya J, Falci DR, et al. Early treatment for COVID-19 with SARS-CoV-2 neutralizing antibody sotrovimab. N Engl J Med. 2021;385(21):1941–50.
Dougan M, Azizad M, Chen P, Feldman B, Frieman M, Igbinadolor A, et al. Bebtelovimab, alone or together with bamlanivimab and etesevimab, as a broadly neutralizing monoclonal antibody treatment for mild to moderate, ambulatory COVID-19. medRxiv [Preprint]; 2022. Available from: https://doi.org/10.1101/2022.03.10.22272100.
Gottlieb RL, Vaca CE, Paredes R, Mera J, Webb BJ, Perez G, et al. Early remdesivir to prevent progression to severe COVID-19 in outpatients. N Engl J Med. 2022;386(4):305–15.
Hammond J, Leister-Tebbe H, Gardner A, Abreu P, Bao W, Wisemandle W, et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with COVID-19. N Engl J Med. 2022;386(15):1397–408.
Jayk Bernal A, Gomes da Silva MM, Musungaie DB, Kovalchuk E, Gonzalez A, Delos Reyes V, et al. Molnupiravir for oral treatment of COVID-19 in nonhospitalized patients. N Engl J Med. 2022;386(6):509–20.
Reis G, Dos Santos Moreira-Silva EA, Silva DCM, Thabane L, Milagres AC, Ferreira TS, et al. Effect of early treatment with fluvoxamine on risk of emergency care and hospitalisation among patients with COVID-19: the TOGETHER randomised, platform clinical trial. Lancet Glob Health. 2022;10(1):e42–51.
Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGEN-COV antibody combination and outcomes in outpatients with COVID-19. N Engl J Med. 2021;385(23):e81.
Gavriatopoulou M, Ntanasis-Stathopoulos I, Korompoki E, Fotiou D, Migkou M, Tzanninis IG, et al. Emerging treatment strategies for COVID-19 infection. Clin Exp Med. 2021;21(2):167–79.
Rabie AM. Teriflunomide: a possible effective drug for the comprehensive treatment of COVID-19. Curr Res Pharmacol Drug Discov. 2021;2:100055.
Rabie AM. Two antioxidant 2,5-disubstituted-1,3,4-oxadiazoles (CoViTris2020 and ChloViD2020): successful repurposing against COVID-19 as the first potent multitarget anti-SARS-CoV-2 drugs. New J Chem. 2021;45(2):761–71.
Gao YD, Ding M, Dong X, Zhang JJ, Kursat Azkur A, Azkur D, et al. Risk factors for severe and critically ill COVID-19 patients: a review. Allergy. 2021;76(2):428–55.
Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 cytokine storm; what we know so far. Front Immunol. 2020;11:1446.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (London, England). 2020;395(10223):497–506.
Mason RJ. Thoughts on the alveolar phase of COVID-19. Am J Physiol Lung Cell Mol Physiol. 2020;319(1):L115–l20.
Mossel EC, Wang J, Jeffers S, Edeen KE, Wang S, Cosgrove GP, et al. SARS-CoV replicates in primary human alveolar type II cell cultures but not in type I-like cells. Virology. 2008;372(1):127–35.
Said SI, Mutt V. Potent peripheral and splanchnic vasodilator peptide from normal gut. Nature. 1970;225(5235):863–4.
Bodanszky M, Klausner YS, Lin CY, Mutt V, Said SI. Synthesis of the vasoactive intestinal peptide (VIP). J Am Chem Soc. 1974;96(15):4973–8.
Bodansky M, Natarajan S, Gardner JD, Makhlouf GM, Said SI. Synthesis and some pharmacological properties of the 23-peptide 15-lysine-secretin-(5--27). Special role of the residue in position 15 in biological activity of the vasoactive intestinal polypeptide. J Med Chem. 1978;21(11):1171–3.
Mathioudakis A, Chatzimavridou-Grigoriadou V, Evangelopoulou E, Mathioudakis G. Vasoactive intestinal peptide inhaled agonists: potential role in respiratory therapeutics. Hippokratia. 2013;17(1):12–6.
Virgolini I, Kurtaran A, Raderer M, Leimer M, Angelberger P, Havlik E, et al. Vasoactive intestinal peptide receptor scintigraphy. J Nucl Med. 1995;36(10):1732–9.
Delgado M, Munoz-Elias EJ, Kan Y, Gozes I, Fridkin M, Brenneman DE, et al. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit tumor necrosis factor alpha transcriptional activation by regulating nuclear factor-kB and cAMP response element-binding protein/c-Jun. J Biol Chem. 1998;273(47):31427–36.
Sharma V, Delgado M, Ganea D. Granzyme B, a new player in activation-induced cell death, is down-regulated by vasoactive intestinal peptide in Th2 but not Th1 effectors. J Immunol (Baltimore, Md: 1950). 2006;176(1):97–110.
Li L, Luo ZQ, Zhou FW, Feng DD, Guang CX, Zhang CQ, et al. Effect of vasoactive intestinal peptide on pulmonary surfactants phospholipid synthesis in lung explants. Acta Pharmacol Sin. 2004;25(12):1652–8.
Temerozo JR, Sacramento CQ, Fintelman-Rodrigues N, Pão CRR, de Freitas CS, da Silva Gomes Dias S, et al. The neuropeptides VIP and PACAP inhibit SARS-CoV-2 replication in monocytes and lung epithelial cells, decrease the production of proinflammatory cytokines, and VIP levels are associated with survival in severe Covid-19 patients. bioRxiv [Preprint]; 2020. https://doi.org/10.1101/2020.07.25.220806.
Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6(10):a016295.
Said SI. VIP as a modulator of lung inflammation and airway constriction. Am Rev Respir Dis. 1991;143(3 Pt 2):S22–4.
Berisha H, Foda H, Sakakibara H, Trotz M, Pakbaz H, Said SI. Vasoactive intestinal peptide prevents lung injury due to xanthine/xanthine oxidase. Am J Physiol. 1990;259(2 Pt 1):L151–5.
Pakbaz H, Foda HD, Berisha HI, Trotz M, Said SI. Paraquat-induced lung injury: prevention by vasoactive intestinal peptide and related peptide helodermin. Am J Physiol. 1993;265(4 Pt 1):L369–73.
Said SI, Dickman KG. Pathways of inflammation and cell death in the lung: modulation by vasoactive intestinal peptide. Regul Pept. 2000;93(1-3):21–9.
Delgado M, Martinez C, Pozo D, Calvo JR, Leceta J, Ganea D, et al. Vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activation polypeptide (PACAP) protect mice from lethal endotoxemia through the inhibition of TNF-alpha and IL-6. J Immunol (Baltimore, Md: 1950). 1999;162(2):1200–5.
Berisha HI, Bratut M, Bangale Y, Colasurdo G, Paul S, Said SI. New evidence for transmitter role of VIP in the airways: impaired relaxation by a catalytic antibody. Pulm Pharmacol Ther. 2002;15(2):121–7.
Li L, She H, Yue SJ, Qin XQ, Guan CX, Liu HJ, et al. Role of c-fos gene in vasoactive intestinal peptide promoted synthesis of pulmonary surfactant phospholipids. Regul Pept. 2007;140(3):117–24.
Temerozo JR, Sacramento CQ, Fintelman-Rodrigues N, Pão CRR, de Freitas CS, Dias SSG, et al. VIP plasma levels associate with survival in severe COVID-19 patients, correlating with protective effects in SARS-CoV-2-infected cells. J Leukoc Biol. 2022;111(5):1107-21.
Youssef J, Lee R, Javitt J, Lavin P, Lenhardt R, Park D, et al. Intravenous aviptadil is associated with increased recovery and survival in patients with COVID-19 respiratory failure: results of a 60-day randomized controlled trial. SSRN [Preprint]; 2021. https://doi.org/10.2139/ssrn.3830051.
Prasse A, Zissel G, Lützen N, Schupp J, Schmiedlin R, Gonzalez-Rey E, et al. Inhaled vasoactive intestinal peptide exerts immunoregulatory effects in sarcoidosis. Am J Respir Crit Care Med. 2010;182(4):540–8.
Ran WZ, Dong L, Tang CY, Zhou Y, Sun GY, Liu T, et al. Vasoactive intestinal peptide suppresses macrophage-mediated inflammation by downregulating interleukin-17A expression via PKA- and PKC-dependent pathways. Int J Exp Pathol. 2015;96(4):269–75.
Anderson P, Gonzalez-Rey E. Vasoactive intestinal peptide induces cell cycle arrest and regulatory functions in human T cells at multiple levels. Mol Cell Biol. 2010;30(10):2537–51.
Gonzalez-Rey E, Fernandez-Martin A, Chorny A, Delgado M. Vasoactive intestinal peptide induces CD4+,CD25+ T regulatory cells with therapeutic effect in collagen-induced arthritis. Arthritis Rheum. 2006;54(3):864–76.
Frye BC, Meiss F, von Bubnoff D, Zissel G, Muller-Quernheim J. Vasoactive intestinal peptide in checkpoint inhibitor-induced pneumonitis. N Engl J Med. 2020;382(26):2573–4.
Petkov V, Mosgoeller W, Ziesche R, Raderer M, Stiebellehner L, Vonbank K, et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest. 2003;111(9):1339–46.
Leuchte HH, Baezner C, Baumgartner RA, Bevec D, Bacher G, Neurohr C, et al. Inhalation of vasoactive intestinal peptide in pulmonary hypertension. Eur Respir J. 2008;32(5):1289–94.
Javitt J, Youssef G, Javitt M. Treatment of sepsis-related acute respiratory distress syndrome with vasoactive intestinal peptide. Am J Respir Crit Care Med. 2021;203:A2490.
Mukherjee T, Behl T, Sharma S, Sehgal A, Singh S, Sharma N, et al. Anticipated pharmacological role of Aviptadil on COVID-19. Environ Sci Pollut Res Int. 2022;29(6):8109–25.
Levitt JE, Calfee CS, Goldstein BA, Vojnik R, Matthay MA. Early acute lung injury: criteria for identifying lung injury prior to the need for positive pressure ventilation. Crit Care Med. 2013;41(8):1929–37.
Gajic O, Dabbagh O, Park PK, Adesanya A, Chang SY, Hou P, et al. Early identification of patients at risk of acute lung injury: evaluation of lung injury prediction score in a multicenter cohort study. Am J Respir Crit Care Med. 2011;183(4):462–70.
Wu C, Chen X, Cai Y, Ja X, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934–43.
World Health Organization: WHO R&D Blueprint, novel coronavirus COVID-19 therapeutic trial synopsis. 2020. https://www.who.int/blueprint/priority-diseases/key-action/COVID-19_Treatment_Trial_Design_Master_Protocol_synopsis_Final_18022020.pdf. Accessed 29 Mar 2022.
Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A, et al. Compassionate use of remdesivir for patients with severe COVID-19. N Engl J Med. 2020;382(24):2327–36.
Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet (London, England). 2020;395(10236):1569–78.
Chow S, Shao J, Wang H. Sample size calculations in clinical research. 2nd ed. New York: Chapman & Hall; 2007.
R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2020. https://www.R-project.org/. Accessed 29 Mar 2022
Acknowledgements
The authors thank Dorian Bevec, Alexander Brett, Helmi van Hirtum, Wolfgang Hoppe, Andrea Kloetzer, Sabrina Maier, Katrin Schmelzle, Philip Tarr, Sandra Widmer, the team from the CTU Basel, and all study physicians for guidance, assistance, support, protocol-conform implementation, and valuable feedback.
Funding
This trial is mainly financed by AdVita Lifescience GmbH, Gundelfingen, Germany. In addition to financial and material support, AdVita Lifescience provides technical support for the used nebulizer devices. Qualified parties of the funding body share their prior experiences and knowledge about the investigational product with the study team in order to support with design-related and logistic decisions. However, the sponsor-investigator JDL takes full responsibility and ultimate authority for study design, data collection, data management, data analysis, interpretation of data, and decisions concerning publication. The Cantonal Hospital Baselland has full ownership of any study related patient data and results.
Furthermore, MBoe receives a personal research grant from the Swiss Academy of Medical Sciences within the program “Young Talents in Clinical Research.”
Author information
Authors and Affiliations
Contributions
Sponsor-investigator, responsibility for conception, design and supervision of the trial: JDL. Drafting of manuscript/corresponding author: MBoe. Study concept and design: MBoe, KA, PH, JDL. Global study coordination: MBoe, KA. Administrative supervision and submission to relevant authorities: KA. Local study supervision/principal investigators: JDL, MBra, RN, JCG, WP. Statistical analysis plan and sample size calculation: MBoe, SG. First version of protocol: JH, KA, ALT, JDL. Critical revision of manuscript for important intellectual content: MBoe, KA, MBra, MBru, EB, BCF, SG, JCG, PH, JH, FJ, ALT, GLC, JMQ, RN, WP, FR, JDL. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate {24}
Ethics approval to conduct this trial has been first granted for the sites Baselland and St. Gallen by the Ethics Commission for North-Western and Central Switzerland (EKNZ) and the Ethics Commission for Eastern Switzerland (EKOS) on December 3, 2020 (Project-ID: 2020-01902). Six amendments regarding additional study sites (Schwyz, Vienna, and Nieuwegein), eligibility criteria, structural changes of the CRF, study duration, and sample size have been approved by the EKNZ and EKOS between December 2020 and July 2022. Furthermore, we are currently awaiting ethics approval from the local ethics committees for the study sites in Vienna (Austria) and Nieuwegein (Netherlands).
The trial meets the criteria and principles of the Declaration of Helsinki and has been registered in the Clinicaltrials.gov database (Trial registration number: NCT04536350).
Informed consent to participate in the trial is obtained by the recruiting study physicians from all patients prior to study entry. Each patient will be informed that participation in the study is voluntary, that he/she may withdraw from the study at any time with no need for justification, and that withdrawal of consent will not affect his/her subsequent medical assistance and treatment. On the consent form, participants are asked for permission to use of their data should they choose to withdraw from the trial. Participants are also asked for permission for personal data being shared with regulatory authorities, when relevant. Furthermore, the patient will be informed on an obligatory basis that his/her medical records may be examined by authorized individuals other than their treating physician. All patients are covered by liability insurance for the total study duration.
Consent for publication {32}
No personal data of any patient is used in this manuscript, therefore a consent for publication is not needed.
Competing interests {28}
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Summary of statistical analysis plan.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Boesing, M., Abig, K., Brändle, M. et al. Inhaled aviptadil for the possible treatment of COVID-19 in patients at high risk for ARDS: study protocol for a randomized, placebo-controlled, and multicenter trial. Trials 23, 790 (2022). https://doi.org/10.1186/s13063-022-06723-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-022-06723-w