
Abstract
Background
Ventilator-associated pneumonia is a challenge in critical care and is associated with high mortality and morbidity. Although some consensuses on preventing ventilator-associated pneumonia are reached, it is still somewhat controversial. Meta-analysis has shown that postpyloric tube feeding may reduce the incidences of ventilator-associated pneumonia, which still desires high-quality evidence. This trial aims to evaluate the efficacy and safety profiles of postpyloric tube feeding versus gastric tube feeding.
Methods/design
In this multicenter, open-label, randomized controlled trial, we will recruit 924 subjects expected to receive mechanical ventilation for no less than 48 h. Subjects on mechanical ventilation will be randomized (1:1) to receive postpyloric or gastric tube feeding and routine preventive measures simultaneously. The primary outcome is the proportion of patients with at least one ventilator-associated pneumonia episode. Adverse events and serious adverse events will be observed closely.
Discussion
The VIP study is a large-sample-sized, multicenter, open-label, randomized, parallel-group, controlled trial of postpyloric tube feeding in China and is well-designed based on previous studies. The results of this trial may help to provide evidence-based recommendations for the prevention of ventilator-associated pneumonia.
Trial registration
Chictr.org.cn ChiCTR2100051593. Registered on 28 September 2021
Similar content being viewed by others

Background
Ventilator-associated pneumonia (VAP) is defined as an infection of the pulmonary parenchyma in patients exposed to invasive mechanical ventilation for at least 48 h. It is one of the significant nosocomial infections in intensive care units (ICUs), contributing to increased ICU stay, morbidity, and mortality [1, 2]. Zhang et al. [1] recently reported the standardized VAP incidence and mortality of 33.7% and 34.5% in China. Many host- and treatment-related colonization factors, such as the severity of the patient’s underlying disease, prior surgery, exposure to antibiotics, and exposure to invasive respiratory devices and equipment, are essential in the pathogenesis of VAP [3,4,5]. Intravenous antimicrobial therapy is the cornerstone of VAP treatment, emphasizing prompt empiric treatment and early initiation of pathogen-specific treatment with appropriate duration [6,7,8,9]. However, despite the advances in the understanding and management of VAP over the past decade, the disease is associated with a significant economic burden and poor outcomes in ICU patients. In a systematic review, VAP conferred a twofold attributable risk of dying in the ICU, with an assignable cost ranging from USD$10,000 to $13,000 per patient [10]. Therefore, preventing VAP before it occurs is also a patient safety priority.
In addition to avoiding intubation and speeding extubation as the prevention strategies of VAP, a bundle of measures, which include elevation of the head of the bed, daily sedation vacations, assessment of readiness to extubate, deep-vein thrombosis prophylaxis, and daily oral care, are also considered standard of care [11]. However, VAP prevention strategies are variably applied in clinical practice [12,13,14], underscoring the need for reliable, safe, effective, and available VAP reduction strategies. As we know, enteral nutrition has been considered a modifiable risk factor for VAP development, mainly because of an increased risk of aspiration of gastric contents [15, 16]. The stomach may be one of the potential reservoirs of nosocomial pathogens that contribute to bacterial colonization via aspiration into the lower respiratory tract [3, 17,18,19,20,21,22]. Emerging as a clinically plausible strategy, postpyloric tube feeding (PTF) may represent a novel approach to preventing VAP through influencing microbiota, enhancing gut barrier function, and reducing gastric reflux and aspiration of gastric content [23,24,25,26,27]. Systematic reviews suggest that PTF reduces VAP by 53–55% compared with gastric tube feeding (GTF) [26, 28]. Nevertheless, most previous randomized trials are small-sample-sized, leading to a less persuasive conclusion. Meta-analyses of small and weak-powered trials often yield implausibly large treatment effects [25, 27]. Hence, the clinical benefits of PTF may be underestimated, and a large, well-powered multicenter trial is needed.
Despite widely accepted recommendations for PTF in nutrition delivery in ICU patients, few studies have assessed the ability of this intervention for VAP prevention. We, therefore, designed this prospective, multicenter, open-label, randomized control trial to validate the efficacy and safety of enteral nutrition via PTF inserted by endoscopy versus GTF in lowering VAP incidence, shortening mechanical ventilation days, ICU or hospital stay, and improving patient outcomes including mortality.
Objectives
Primary objectives
The primary objectives of this trial are to:
-
1.
Determine the efficacy of PTF in lowering VAP incidence in critically ill patients receiving mechanical ventilation for more than 48 h
-
2.
Determine the safety of PTF in lowering VAP incidence in critically ill patients receiving mechanical ventilation for more than 48 h
Secondary objectives
The secondary objectives of this trial are to:
-
1.
Determine the efficacy of PTF in improving mortality in critically ill patients receiving mechanical ventilation for more than 48 h
-
2.
Determine the efficacy of PTF in shortening mechanical ventilation days, ICU or hospital stay, and if there is a benefit to decreasing the whole care cost in critically ill patients receiving mechanical ventilation for more than 48 h
-
3.
Determine the efficacy of PTF in improving nutrition deficit and immune ability to infection in critically ill patients receiving mechanical ventilation for more than 48 h
Methods/design
Study design
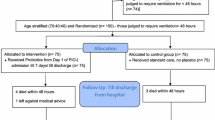
The VIP study is designed as a prospective, multicenter, open-label, randomized, parallel-group, GTF-controlled (PTF vs. GTF 1:1) superiority trial in 4 tertiary care hospitals. The participating sites in this study include medical/surgical, medical, surgical, or emergency ICUs (see Table 1 for details of participating centers). Patient enrollment is expected to last for up to 36 months. The end of the study is defined as the last follow-up of the last enrolled patient. The trial has been registered at chictr.org.cn (ChiCTR2100051593). We used the Standard Protocol Items: Recommendations for International Trials (SPIRIT) reporting guidelines [29], and the SPIRIT checklist is attached in the Supplementary appendixes.
Patient and public involvement
Unlike patients living with long-term medical conditions such as diabetes and hypertension, most ICU patients unanticipated being admitted into ICU. They are relatively elder and lack clinical research education and experience, making it challenging to determine who will be the potential beneficiaries in attending our trial. However, we have planned to get our participants involved in the communication and education during the research process to increase awareness and knowledge, build confidence and control with self-management, and finally prevent and cure VAP.
Recruitment
A well-trained study coordinator in each participating center that fulfills the requirement that a gastroscope can be performed at the bedside will be responsible for screening all potentially eligible patients based on the eligibility criteria. After confirming the patient’s eligibility for the trial, the study coordinator will obtain written informed consent from the patient or authorized representatives.
Inclusion criteria
Patients are eligible for the trial if they are expected to receive mechanical ventilation for no less than 48 h, are over the age of 18 years, and require GTF [30]. Patients will be enrolled within 6 h of their endotracheal intubation.
Exclusion criteria
Patients will be excluded if they are intubated for more than 6 h, have contradictions for feeding tube placement, or present conditions which will preclude the diagnosis, monitoring, and assessment of VAP development. Detailed exclusion criteria are summarized in Supplementary Appendix 1.
Drop-out criteria
Patients who meet eligibility criteria and write an informed consent form (see Supplementary Appendix) to participate but fail to accomplish the study process are regarded as drop-out cases if they meet any of the following criteria: (1) the participant or the legal representative requests withdrawal at any time, (2) the investigator considers it inappropriate to continue for safety purpose, and (3) additional reasons for the participant to discontinue from an investigator’s medical perspectives. The drop-out rate must be no more than 10% in this trial.
Diagnosis of ventilator-associated pneumonia and adjudication process
VAP should rather be suspected in patients with clinical signs of infection, such as at least two of the following criteria: new onset of fever, purulent endotracheal secretions, leukocytosis or leucopenia, increase in minute ventilation, a decline in oxygenation, or increased need for vasopressors to maintain blood pressure [31].
In cases of suspected VAP, the Clinical Pulmonary Infection Score (CPIS) and Sequential Organ Failure Assessment (SOFA) score are assessed. Bedside anteroposterior chest radiography, arterial blood gas analysis, blood cultures, and quantitative sampling of the lower respiratory tract (by either bronchoalveolar lavage or endotracheal aspiration, at the discretion of the attending physician) [32] are performed before any antibiotics are administered.
An adjudication committee composed of one senior radiologist and two senior intensivists unaware of the trial-group assignments reviewed all patients’ medical charts and adjudicated all respiratory tract infections. The intensivists have access to all monitored data, chest radiographs obtained during the ICU stay, and microbiologic documentation. Two of them analyzed data from every patient independently, and in case of disagreement, the third one arbitrated the diagnosis of VAP. Such infections are defined as early if they occurred within 7 days after randomization and as late if they occurred after 7 days, according to an adjudication chart (Supplementary Appendix 2) and the definition of VAP. All secondary infections that happened during the ICU stay are also recorded.
A diagnosis process to confirm reported clinical VAP is defined in a standardized approach with the use of criteria from the 2020 Food and Drug Administration guidance for diagnosis and confirmation of VAP, which relies on clinical, laboratory, and radiologic criteria (patients have to meet all three types of standards). The flow chart for diagnosis and confirmation of VAP is summarized in Supplementary Appendix 3.
Endotracheal tube, feeding tube, and enteral nutrients
According to Good Manufacturing Practice guidelines, all feeding tubes are prepared by Henan Anesthesia Medical Technology Co, Ltd. All endotracheal lines and enteral nutrients are provided by participating centers according to their routine practices. All studying materials aforementioned are shipped to each participating center by express service for a 14-day supply and stored in a cool, well-ventilated place to avoid direct sunlight.
Assignment of interventions
Sequence generation for allocation
Following informed consent and confirmation of inclusion and exclusion criteria, participants on mechanical ventilation will be randomly assigned in a 1:1 ratio to receiving either PTF or GTF. Allocation sequences are generated by computer-generated random numbers using the R package blockrand [33]. Stratified block randomization is employed to avoid inter-group differences (study or control group) due to differences in the source of patients in different hospitals. Each participating center is served as a stratification factor, and then the subjects in each ICU are randomized into other blocks. The subjects will be consecutively assigned when entering the trial. To reduce the predictability of a random sequence, blocking details are provided in a separate document that is unavailable to those who enroll participants or assign interventions.
Allocation concealment mechanism
A central randomization system is set up to conceal the allocation to investigators. To prevent early knowledge of treatment assignment and disruption of the assignment sequence, investigators must complete the clinical trial entry form attached to the case report form (CRF) and obtain the informed consent form before disclosing the unique sequence number and assignment group. Randomization methods and block sizes are blinded until all data analyses are completed.
Allocation implementation
Each center’s investigator and authorized clinicians will enroll subjects according to the protocol’s eligibility criteria and allocate the newly registered patient into the corresponding group according to the unique number for specific assignments acquired from a randomization squad. The squad fetches the assignment number through a prepared online central randomization system, which will be sent to the investigator via instant messaging software like WeChat once the informed consent form is signed.
Interventions
Patients who meet the enrollment criteria will be randomized 1:1 to either the PTF or the GTF group, receiving postpyloric or gastric tube feeding, respectively, with other treatments exactly the same.
Endoscopic feeding tube placement
A nasogastric feeding tube is inserted by nursing staff for the control group. Currently, in our clinical practice, the postpyloric feeding tube can be established by self-propelled dynamics from the gut with the assistance of prokinetic agents [34,35,36] and using a rescued bedside tube placement method by ICU physicians when necessary [37]. However, despite the short learning curve [38] and easy availability of the methods mentioned above, especially in the advantage of some decision support tools [39, 40], we otherwise decided to employ the endoscopic approach to establish the postpyloric feeding tube promptly and reliably for the study group. Hence, experienced endoscopists must be equipped when participating centers are recruited. In most ICUs currently in China, no special endoscopists are regularly prepared. In this case, a training course on endoscopic postpyloric tube placement is established to equip the physicians to become experienced endoscopists in the ICU.
Detailed training protocol is attached as Supplementary Appendix 4 in this manuscript. The essential procedures of endoscopic tube placement are as follows:
Patients are provided with oxygen inhalation, and ECG and blood oxygen saturation are monitored. After the patient is narcotized, they take the left lying position. During the operation, no secretion could be left in the mouth. A lubricated nasointestinal tube, approximately 25cm deep, is inserted through the nostrils and sent to the gastric lumen by gastroscopy. The esophagus, stomach, and duodenum are observed to confirm the absence of lesions or obstruction. The head end of the nasointestinal tube is clamped with foreign body forceps, and it is slowly pushed into the descending part of the duodenum to fix the nasointestinal tube. The gastroscope is returned to the gastric cavity, and the foreign body forceps are released into the gastric cavity. After about three times, they are transported to about 20–40 cm below the Treitz ligament. After the nasointestinal tube is fixed, the gastroscope and the nasointestinal tube guidewire are withdrawn.
Nutrition support protocol
In both groups, energy goals are set at 25 kcal per kg of ideal body weight per day, and the protein target is 1.2–2.0 g per kg of ideal body weight per day. Glucose control targets are set following international guidelines [41,42,43]. Incidences in which the patient develops an intolerance to EN (diagnosed when vomiting, diarrhea, abdominal pain, or abdominal distension occurred) are recorded. In these cases, the rate of feeding and EN are gradually reduced as tolerated. If the nutrition goal is not reached within 7 days, parenteral nutrition is provided.
Concomitant interventions
Routine use of a bundle of measures for the prevention of VAP (elevation of the head of the bed, daily sedation vacations and assessment of readiness to extubate, and deep-vein thrombosis prophylaxis) and daily oral care are highly recommended, and specific attention is given to standardize patient care [11].
Participants may receive management preventing from VAP following the 2016 Clinical Practice Guidelines for the Management of Adults with Hospital-acquired and Ventilator-associated Pneumonia by the Infectious Diseases Society of America and the American Thoracic Society, mainly including (1) manage patients without sedation whenever possible; (2) interrupt sedation daily; (3) assess readiness to extubate daily; (4) perform spontaneous breathing trials with sedatives turned off; (5) facilitate early mobility; (6) utilize endotracheal tubes with subglottic secretion drainage ports for patients expected to require more than 48 or 72 h of mechanical ventilation; (7) change the ventilator circuit only if visibly soiled or malfunctioning; and (8) elevate the head of the bed to 30–45. If any, the use of all the above maneuvers should be documented in the electronic case report form (eCRF).
Outcome measurements
Participants will be evaluated clinically and through laboratory testing according to Table 2. The following data will be recorded: demographics, VAP diagnosis, concurrent medical conditions and comorbidities, inclusion and exclusion criteria, the severity of illness and organ dysfunction scores, vital signs and laboratory results, potential confounding co-interventions (life-sustaining therapies and use of sedatives or vasopressors), and outcomes (vital status at ICU and hospital discharge, day 28 and day 90, ICU and hospital length of stay).
Primary efficacy endpoint
The primary efficacy endpoint of this study is the proportion of patients with at least 1 VAP episode.
Secondary efficacy endpoints
The secondary efficacy endpoints of this trial include the following:
-
(1)
Incidence analysis: including the cumulative VAP incidence and the total number of VAP episodes and numbers and percentages of microorganisms causing VAP
-
(2)
Mortality analysis: including ICU, hospital, and day-28 mortality rates
-
(3)
Time analysis: including time to VAP onset from mechanical ventilation, delayed time to first VAP occurrence, mechanical ventilation duration, the number of ventilator-free days until day 28, and ICU and hospital lengths of stay
-
(4)
Enteral feeding and nutritional status: including the proportions of patients with at least one vomiting episode, prokinetic treatment, and diarrhea; the proportion of patients given 100% of the calorie target; cumulative calorie deficit from day 0 to day 7; and variations in serum albumin during the first week of enteral nutrition
-
(5)
Organ functioning: including score variations in Sequential Organ Failure Assessment (SOFA)
-
(6)
Inflammatory level: including variations in serum C-reactive protein (CRP) levels during the first week of enteral nutrition
-
(7)
ICU-acquired infection: proportions of patients with ICU-acquired infections (bloodstream, urinary tract, catheter-related, and other infections); score variations in CPIS
We plan to collect data on organ dysfunction at baseline and at various time points during the study. All the six domains of the SOFA score, including respiratory, coagulation, hepatic, cardiovascular, renal, and central nervous system, will be documented. Vasopressors, sedatives, and renal replacement therapy will also be reported to assess organ dysfunction. VAP episodes are monitored during the whole study. New-onset VAP is diagnosed if new microbial culture results from distal respiratory specimens are different from those prior, and colonization is excluded.
Safety endpoints
The safety outcomes are major adverse tube-associated events (MATEs), including vital sign alert events (defined as HR, RR, or MAP fluctuating beyond the range of ± 15%, or pulse oxygen saturation declining to < 90%), the requirement for sedatives or analgesics during tube placement, vomiting, rhinorrhagia, misplacing into the thoracic cavity, gastrointestinal bleeding or perforation, and so forth.
Adverse events and serious adverse events
All treatment-related adverse events (AEs) and serious adverse events (SAEs) should be recorded on eCRF. SAEs will be reported to the Institutional Ethics Committee within 24 h of study staff becoming aware of the events. The participants are provided with commercial clinical research insurance by the manufacturer of the study product. Determination criteria for AE and SAE are detailed in Supplementary Appendix 5.
The treating physician will be responsible for determining the causal relationship of the SAE as either definitely, possibly, possibly not, or definitely not study treatment-related, as well as unclassified.
Data management
Trained staff will perform data management at each center using the Electronic Data Capture (EDC) system (https://www.mmphcrc.com/pdf/medicalHistory/vapcrf.html). The EDC system’s reliability, access control, and traceability will guarantee the quality of trial data management. Data collection will be restricted to those variables necessary to define baseline patient characteristics (demographics, VAP diagnosis, concurrent medical conditions and comorbidities, inclusion and exclusion criteria, severity of illness and organ dysfunction scores, vital signs, and laboratory results), the delivery of the nutrients, potential confounding co-interventions (life-sustaining therapies, and use of vasopressors or sedatives), and outcomes (vital signs at ICU and hospital discharge, day 28, length of stay in ICU and hospital). Randomized participants will be followed until either death or 28 days after randomization, whichever comes first. Study staff will attend follow-up by either direct contact with the patient or the next of kin. Participants who withdraw from the study will be followed up according to the follow-up schedule and analyzed on the ITT principle.
Data monitoring committee
A Data Monitoring Committee (DMC) will be responsible for data monitoring and blinded analysis. Members of the committee are experts in medicine, biometric statistics, and medical ethics who are independent and have no competing interests in the trial. No interim analysis is planned for this trial. The DMC will audit the trial regularly and advise the Research Ethics Committee in each center, who will also audit and determine to continue, modify, or discontinue the trial.
Ethics and dissemination
Protocol amendments
Protocol amendments will be documented with a brief description of the change and reference (date and number) when changes in the existing protocol significantly affect the safety of subjects, the scope of the investigation, or the scientific quality of the study. The coordinating investigator is obligated to notify this protocol amendments to all the investigators and the reviewing IRB and other relevant parties as appropriate.
Consent or assent
The clinical investigator ensures that informed consent is obtained from each research subject before that subject participates in the research study. When the research subject is disabled to consent for critical illness, informed consent is obtained from the authorized surrogates, for which the prior approval from the IRB is a prerequisite. We will only obtain consent to use data and samples for the research question described in this protocol. Thus, we do not intend to use participant data or biological samples in ancillary studies.
Access to data
Data from the VIP study will be made available in the future for collaborative research questions. Such requests must be authorized by the principal investigators and the appropriate Human Research Ethics Committees and Human Research Governance Safety Entities.
Ancillary and post-trial care
All patients would be treated, monitored, and routinely assessed regarding the VAP development and recovery process in each participating study center in China.
Dissemination policy
The findings from the data analysis will be disseminated in various ways, including abstracts, posters and presentations at conferences, and published manuscripts in peer-reviewed journals. These will also be reported to national, provincial, and local governments to inform policy and reports to funding bodies, institutes, and hospitals that participated in and supported the cohort study. Members of the study team will have publishing and authorship rights following the International Committee of Medical Journal Editors requirements for authorship and as described in research agreements.
Statistical analysis
Sample size estimation
Zhang et al. reported the incidence of 33.7% in a meta-analysis and systematic review of 334 publications concerning ICU-acquired pneumonia and VAP in China [1]. According to a meta-analysis by Ouyang et al. [28], a treatment effect with a relative risk reduction (RRR) of 51.4% is observed in ICU patients receiving postpyloric tube feeding (PTF) compared with receiving gastric tube feeding (GTF). A 10% inferiority margin is predetermined following previous guidelines and reviews. We calculate that enrollment of 924 participants will have a power of 80% to detect an absolute reduction of 17.3% (relative risk reduction of 51.4%) in the study group compared to 33.7% of the control group, allowing a loss of follow-up or withdrawal of 10%. Considering the prevalence of VAP in our patients and assuming the rate of patient recruitment of about ten per month per center (with three participating centers), this trial will be finished within 3 years.
Analyses set
The study protocol mention that analysis will be done considering the “intention to treat” principle. However, some randomized patients did not allow feeding tube insertion (gastric or postpyloric) during study conduction due to illness severity, or the attending physician decided not to insert the tubes. Those cases are not excluded from the analyses. Patients who complete the randomization, regardless of whether they completed the trial or received the treatment in the designated group, are retained in the original group for analysis. The randomization information is maintained to the maximum extent.
Intention-to-treat set (ITTS)
ITTS refers to the ideal population of subjects meeting the ITT principles. To best retain the randomized information, ensure that the differences in trial results are attributed to the differences in treatment, and make the effect of treatment (postpyloric feeding) best assessed, the principle of ITT analysis is adopted. ITT set (ITTS) included all randomized patients, regardless of whether they have received nasogastric tube placement, completed the trial, or received the treatment of this group, all of whom remain in the original group for analysis.
Per protocol set (PPS)
PPS refers to all participants who fulfill the eligibility criteria and achieve gastric or postpyloric tube placement per randomization. They have good compliance with the trial protocol, such as being ready to receive treatment and measurement for the primary efficacy endpoints, with the required contents in the CRF filled. PPS is used to analyze the primary efficacy endpoints.
Security set (SS)
The SS included all actual cases that have received at least one feeding tube placement after randomization, with the safety endpoints recorded. The incidence of adverse events is denominated by the number of patients in the safety set.
Data analysis
All analyses will be performed according to the ITT principle. A P value < 0.05 is considered statistically significant. All tests are two-sided with no adjustment for multiple comparisons. Continuous variables are reported as means and standard deviations or medians and interquartile ranges. Categorical variables are reported as proportions. Pearson’s chi-squared test and adjusted multivariable analysis will be applied for the primary outcome. We plan to perform subgroup analyses for the primary outcome for predefined variables: the proportion of patients with at least 1 VAP episode. All the other data, including age, gender, body mass index (weight, height), SOFA score, primary admission diagnosis, coexisting illness, and type of admission, will be presented as descriptive results. Pearson’s chi-squared test will compare incidence or mortality outcomes between groups. The t-test will compare total VAP episodes, SOFA scores, time to VAP onset from mechanical ventilation, mechanical ventilation duration, ICU and hospital lengths of stay, inflammatory biomarkers, and function indicators between groups. A paired t-test or repeated measures ANOVA will be used to compare baseline and changes during the intervention.
Interim analysis
No interim analysis is planned for this trial.
Discussion
VAP is one of the most frequent ICU-acquired infections associated with prolonged mechanical ventilation and ICU stay. The reported incidences vary widely from 5 to 40%, depending on the setting and diagnostic criteria. The estimated attributable mortality of VAP is around 10%, with higher mortality rates in surgical ICU patients and patients with mid-range severity scores at admission [31]. All the data above highlight reliable measures to prevent or limit VAP from occurring in the ICU.
The practices most consistently associated with lower mortality rates focus on limiting exposure to invasive mechanical ventilation by avoiding intubation and speeding extubation [44]. However, for those patients whose intubation is inevitable to save lives, many of our presumptions about how best to prevent VAP have recently been challenged. Oral care with chlorhexidine and stress ulcer prophylaxis may be harmful. New data affirm the long-held fear that selective oral and digestive decontamination may not be effective in ICUs with high baseline rates of antibiotic resistance. Subglottic secretion drainage may not shorten the duration of mechanical ventilation or ICU length-of-stay as is once thought [45,46,47,48,49]. Furthermore, two recent RCTs showed no significant difference in VAP development among ventilated ICU patients receiving probiotic or monoclonal antibody administration than placebo [50, 51].
Otherwise, PTF has been evaluated as a potentially promising management in preventing VAP due to not only reducing gastrointestinal and respiratory complications like vomiting and gastric distention in critically ill patients and ensuring that the nutritional goals are better achieved [52,53,54]. However, current evidence demonstrating the efficacy of PTF in the prevention of VAP, including RCTs, is mainly negative. Two studies [23, 27] showed no significant reduction in VAP incidence comparing PTF with GTF. There exist many critical limitations in those studies, including small-sample-sized and conducting in a single center. Statistically, the results from these studies are underpowered, and the conclusion is far less than robust. As a result, the effect of PTF on the incidence of VAP warrants validation by large, well-conducted RCTs in different settings [55].
To address this call, the VIP study is a large-sample-sized, rigorous multicenter randomized trial that aims to determine whether PTF is effective and safe in preventing VAP. In ethical consideration to maximize patients’ benefit, bundling practices, including elevation of the head of the bed, daily sedation vacations, assessment of readiness to extubate, deep-vein thrombosis prophylaxis, and daily oral care, are also implemented by trained investigators according to the study protocol [56,57,58,59]. This strategy might result in a relatively lower occurrence of VAP in the control group, which in turn required larger sample size to detect the potential difference in VAP incidence between the intervention and control groups. Based on an elaborate sample size calculation, we aimed to enroll nearly 1,000 patients to powerfully detect the possible underlying profit of PTF over GTF in improving a range of clinical outcomes for ICU patients.
VIP study has several strengths. First, the RCT design can limit the risk of biases related to the presence of confounding factors in evaluating the preventive effects on VAP and mortality. Second, the VIP study includes representation of persons aged from 18 years to 80 years in different hospitals located all over China to enhance the generalizability of the findings. Third, we are also documenting baseline organ malfunctions with coexisting diseases and SOFA scores to understand further the relationship between body frailty and critical care-associated infections. Furthermore, to shorten the time from endotracheal intubation to PTF placement and limit the effects of management heterogeneity among centers on outcome variables, all tubes are placed using endoscopic methods by well-trained intensivists in the present trial.
In conclusion, the VIP study is a large-sample-sized, multicenter, open-label, randomized, parallel-group, controlled trial of PTF in China and is well-designed based on previous studies. The results of this trial may help provide evidence-based recommendations for the prevention of VAP.
Trial status
This article is based on the study protocol version 2.8 of 30 August 2021. The VIP study started on 1 December 2021. Participants are currently being recruited and enrolled. Recruitment will probably continue until July 2025. Contact: Chunbo Chen, email: gghicu@163.com.
Availability of data and materials
All the compiled CRFs will be archived. After this study is complete, the final trial dataset and statistical codes will be available from the corresponding authors upon reasonable request, except for participants’ personal information.
Abbreviations
- AE:
-
Adverse event
- CPIS:
-
Clinical Pulmonary Infection Score
- CRF:
-
Case record form
- DMC:
-
Data Monitoring Committee
- eCRF:
-
Electronic case report form
- EDC:
-
Electronic Data Capture
- EN:
-
Enteral nutrition
- GTF:
-
Gastric tube feeding
- ICU:
-
Intensive care unit
- ITT:
-
Intention-to-treat
- PPS:
-
Per protocol set
- PTF:
-
Postpyloric tube feeding
- RCT:
-
Randomized controlled trial
- SAE:
-
Severe adverse event
- SOFA:
-
Sequential Organ Failure Assessment
- SS:
-
Security set
- VAP:
-
Ventilator-associated pneumonia
References
Zhang Y, Yao Z, Zhan S, Yang Z, Wei D, Zhang J, et al. Disease burden of intensive care unit-acquired pneumonia in China: a systematic review and meta-analysis. Int J Infect Dis. 2014;29:84–90.
Li G, Ke L, Tong Z, Li W, Ouyang X, Chen C. Is it necessary for all patients to use prokinetic agents to place a trans-pyloric tube? Intensive Care Med. 2019;45(5):751–2.
Craven DE, Steger KA. Nosocomial pneumonia in mechanically ventilated adult patients: epidemiology and prevention in 1996. Semin Respir Infect. 1996;11(1):32–53.
Johanson WG Jr, Pierce AK, Sanford JP, Thomas GD. Nosocomial respiratory infections with gram-negative bacilli. the significance of colonization of the respiratory tract. Ann Intern Med. 1972;77(5):701–6.
Kollef MH. The prevention of ventilator-associated pneumonia. N Engl J Med. 1999;340(8):627–34.
Chastre J, Wolff M, Fagon JY, Chevret S, Thomas F, Wermert D, et al. Comparison of 8 vs 15 days of antibiotic therapy for ventilator-associated pneumonia in adults: a randomized trial. JAMA. 2003;290(19):2588–98.
Kalil AC, Metersky ML, Klompas M, Muscedere J, Sweeney DA, Palmer LB, et al. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis. 2016;63(5):e61–e111.
Leone M, Bouadma L, Bouhemad B, Brissaud O, Dauger S, Gibot S, et al. Brief summary of French guidelines for the prevention, diagnosis and treatment of hospital-acquired pneumonia in ICU. Ann Intensive Care. 2018;8(1):104.
Torres A, Niederman MS, Chastre J, Ewig S, Fernandez-Vandellos P, Hanberger H, et al. International ERS/ESICM/ESCMID/ALAT guidelines for the management of hospital-acquired pneumonia and ventilator-associated pneumonia: guidelines for the management of hospital-acquired pneumonia (HAP)/ventilator-associated pneumonia (VAP) of the European Respiratory Society (ERS), European Society of Intensive Care Medicine (ESICM), European Society of Clinical Microbiology and Infectious Diseases (ESCMID) and Asociación Latinoamericana del Tórax (ALAT). Eur Respir J. 2017;50(3):1700582.
Safdar N, Dezfulian C, Collard HR, Saint S. Clinical and economic consequences of ventilator-associated pneumonia: a systematic review. Crit Care Med. 2005;33(10):2184–93.
Dodek P, Keenan S, Cook D, Heyland D, Jacka M, Hand L, et al. Evidence-based clinical practice guideline for the prevention of ventilator-associated pneumonia. Ann Intern Med. 2004;141(4):305–13.
Li Bassi G, Senussi T, Aguilera Xiol E. Prevention of ventilator-associated pneumonia. Curr Opin Infect Dis. 2017;30(2):214–20.
Zeng J, Wang CT, Zhang FS, Qi F, Wang SF, Ma S, et al. Effect of probiotics on the incidence of ventilator-associated pneumonia in critically ill patients: a randomized controlled multicenter trial. Intensive Care Med. 2016;42(6):1018–28.
Spapen H, van Laethem J, Hites M, Verdoodt A, Diltoer M, Honoré PM. Treatment of ventilator-associated pneumonia with high-dose colistin under continuous veno-venous hemofiltration. J Transl Int Med. 2019;7(3):100–5.
Tablan OC, Anderson LJ, Besser R, Bridges C, Hajjeh R, Cdc, et al. Guidelines for preventing health-care--associated pneumonia, 2003: recommendations of CDC and the Healthcare Infection Control Practices Advisory Committee. MMWR Recomm Rep. 2004;53(RR-3):1–36.
Pingleton SK, Hinthorn DR, Liu C. Enteral nutrition in patients receiving mechanical ventilation. Multiple sources of tracheal colonization include the stomach. Am J Med. 1986;80(5):827–32.
Cameron JL, Reynolds J, Zuidema GD. Aspiration in patients with tracheostomies. Surg Gynecol Obstet. 1973;136(1):68–70.
Valles J, Artigas A, Rello J, Bonsoms N, Fontanals D, Blanch L, et al. Continuous aspiration of subglottic secretions in preventing ventilator-associated pneumonia. Ann Intern Med. 1995;122(3):179–86.
Cook D, De Jonghe B, Brochard L, Brun-Buisson C. Influence of airway management on ventilator-associated pneumonia: evidence from randomized trials. JAMA. 1998;279(10):781–7.
Niederman MS, Craven DE. Devising strategies for preventing nosocomial pneumonia--should we ignore the stomach? Clin Infect Dis. 1997;24(3):320–3.
Bonten MJ, Bergmans DC, Ambergen AW, de Leeuw PW, van der Geest S, Stobberingh EE, et al. Risk factors for pneumonia, and colonization of respiratory tract and stomach in mechanically ventilated ICU patients. Am J Respir Crit Care Med. 1996;154(5):1339–46.
Bonten MJ, Gaillard CA, de Leeuw PW, Stobberingh EE. Role of colonization of the upper intestinal tract in the pathogenesis of ventilator-associated pneumonia. Clin Infect Dis. 1997;24(3):309–19.
Hsu CW, Sun SF, Lin SL, Kang SP, Chu KA, Lin CH, et al. Duodenal versus gastric feeding in medical intensive care unit patients: a prospective, randomized, clinical study. Crit Care Med. 2009;37(6):1866–72.
Jabbar A, McClave SA. Pre-pyloric versus post-pyloric feeding. Clin Nutr. 2005;24(5):719–26.
Kearns PJ, Chin D, Mueller L, Wallace K, Jensen WA, Kirsch CM. The incidence of ventilator-associated pneumonia and success in nutrient delivery with gastric versus small intestinal feeding: a randomized clinical trial. Crit Care Med. 2000;28(6):1742–6.
Liu Y, Wang Y, Zhang B, Wang J, Sun L, Xiao Q. Gastric-tube versus post-pyloric feeding in critical patients: a systematic review and meta-analysis of pulmonary aspiration- and nutrition-related outcomes. Eur J Clin Nutr. 2021;75(9):1337–48.
Zhu Y, Yin H, Zhang R, Ye X, Wei J. Gastric versus postpyloric enteral nutrition in elderly patients (age ≥ 75 years) on mechanical ventilation: a single-center randomized trial. Crit Care. 2018;22(1):170.
Ouyang X, Qu R, Hu B, Wang Y, Yao F, Lv B, et al. Is metoclopramide beneficial for the postpyloric placement of nasoenteric tubes? A systematic review and meta-analysis of randomized controlled trials. Nutr Clin Pract. 2022;37(2):316–27.
Chan AW, Tetzlaff JM, Gotzsche PC, Altman DG, Mann H, Berlin JA, et al. SPIRIT 2013 explanation and elaboration: guidance for protocols of clinical trials. BMJ. 2013;346:e7586.
Society AT, America IDSo. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171(4):388–416.
Papazian L, Klompas M, Luyt CE. Ventilator-associated pneumonia in adults: a narrative review. Intensive Care Med. 2020;46(5):888–906.
Pugin J, Auckenthaler R, Mili N, Janssens JP, Lew PD, Suter PM. Diagnosis of ventilator-associated pneumonia by bacteriologic analysis of bronchoscopic and nonbronchoscopic “blind” bronchoalveolar lavage fluid. Am Rev Respir Dis. 1991;143(5 Pt 1):1121–9.
Schulz KF, Grimes DA. Unequal group sizes in randomised trials: guarding against guessing. Lancet. 2002;359(9310):966–70.
Hu B, Ye H, Sun C, Zhang Y, Lao Z, Wu F, et al. Metoclopramide or domperidone improves post-pyloric placement of spiral nasojejunal tubes in critically ill patients: a prospective, multicenter, open-label, randomized, controlled clinical trial. Crit Care. 2015;19(1):61.
Xiao Y, He Z, Long Y, Chen W, Chen D, Chi R, et al. Simo decoction versus domperidone suspension for post-pyloric spiral nasoenteric tube placement: a multicenter, randomized, non-inferiority trial. Clin Nutr. 2020;39(8):2406–12.
Hu B, Ouyang X, Lei L, Sun C, Chi R, Guo J, et al. Erythromycin versus metoclopramide for post-pyloric spiral nasoenteric tube placement: a randomized non-inferiority trial. Intensive Care Med. 2018;44(12):2174–82.
Lv B, Hu L, Chen L, Hu B, Zhang Y, Ye H, et al. Blind bedside postpyloric placement of spiral tube as rescue therapy in critically ill patients: a prospective, tricentric, observational study. Crit Care. 2017;21(1):248.
Sun C, Lv B, Zheng W, Hu L, Ouyang X, Hu B, et al. The learning curve in blind bedside postpyloric placement of spiral tubes: data from a multicentre, prospective observational study. J Int Med Res. 2019;47(5):1884–96.
Chen W, Sun C, Wei R, Zhang Y, Ye H, Chi R, et al. Establishing decision trees for predicting successful postpyloric nasoenteric tube placement in critically ill patients. JPEN J Parenter Enteral Nutr. 2018;42(1):132–8.
Hu L, Nie Z, Zhang Y, Zhang Y, Ye H, Chi R, et al. Development and validation of a nomogram for predicting self-propelled postpyloric placement of spiral nasoenteric tube in the critically ill: mixed retrospective and prospective cohort study. Clin Nutr. 2019;38(6):2799–805.
Taylor BE, McClave SA, Martindale RG, Warren MM, Johnson DR, Braunschweig C, et al. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine (SCCM) and American Society for Parenteral and Enteral Nutrition (A.S.P.E.N.). Crit Care Med. 2016;44(2):390–438.
McClave SA, DiBaise JK, Mullin GE, Martindale RG. ACG clinical guideline: nutrition therapy in the adult hospitalized patient. Am J Gastroenterol. 2016;111(3):315–34 quiz 335.
Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017;43(3):304–77.
Legoff J, Zucman N, Lemiale V, Mokart D, Pène F, Lambert J, et al. Clinical significance of upper airway virus detection in critically ill hematology patients. Am J Respir Crit Care Med. 2019;199(4):518–28.
Wittekamp BH, Plantinga NL, Cooper BS, Lopez-Contreras J, Coll P, Mancebo J, et al. Decontamination strategies and bloodstream infections with antibiotic-resistant microorganisms in ventilated patients: a randomized clinical trial. JAMA. 2018;320(20):2087–98.
Huang HB, Jiang W, Wang CY, Qin HY, Du B. Stress ulcer prophylaxis in intensive care unit patients receiving enteral nutrition: a systematic review and meta-analysis. Crit Care. 2018;22(1):20.
Harris BD, Thomas GA, Greene MH, Spires SS, Talbot TR. Ventilator bundle compliance and risk of ventilator-associated events. Infect Control Hosp Epidemiol. 2018;39(6):637–43.
Klompas M. Oropharyngeal decontamination with antiseptics to prevent ventilator-associated pneumonia: rethinking the benefits of chlorhexidine. Semin Respir Crit Care Med. 2017;38(3):381–90.
Caroff DA, Li L, Muscedere J, Klompas M. Subglottic secretion drainage and objective outcomes: a systematic review and meta-analysis. Crit Care Med. 2016;44(4):830–40.
François B, Jafri HS, Chastre J, Sánchez-García M, Eggimann P, Dequin PF, et al. Efficacy and safety of suvratoxumab for prevention of Staphylococcus aureus ventilator-associated pneumonia (SAATELLITE): a multicentre, randomised, double-blind, placebo-controlled, parallel-group, phase 2 pilot trial. Lancet Infect Dis. 2021;21(9):1313–23.
Johnstone J, Meade M, Lauzier F, Marshall J, Duan E, Dionne J, et al. Effect of probiotics on incident ventilator-associated pneumonia in critically ill patients: a randomized clinical trial. JAMA. 2021;326(11):1024–33.
Jiyong J, Tiancha H, Huiqin W, Jingfen J. Effect of gastric versus post-pyloric feeding on the incidence of pneumonia in critically ill patients: observations from traditional and Bayesian random-effects meta-analysis. Clin Nutr. 2013;32(1):8–15.
Metheny NA, Stewart BJ, McClave SA. Relationship between feeding tube site and respiratory outcomes. JPEN J Parenter Enteral Nutr. 2011;35(3):346–55.
Heyland DK, Drover JW, MacDonald S, Novak F, Lam M. Effect of postpyloric feeding on gastroesophageal regurgitation and pulmonary microaspiration: results of a randomized controlled trial. Crit Care Med. 2001;29(8):1495–501.
Ouyang X, Qu R, Hu B, Wang Y, Yao F, Lv B, Sun C, Deng Y, Chen C. Is metoclopramide beneficial for the postpyloric placement of nasoenteric tubes? A systematic review and meta-analysis of randomized controlled trials. Nutr Clin Pract. 2022;37(2):316-27.
Hsieh SJ, Otusanya O, Gershengorn HB, Hope AA, Dayton C, Levi D, et al. Staged implementation of awakening and breathing, coordination, delirium monitoring and management, and early mobilization bundle improves patient outcomes and reduces hospital costs. Crit Care Med. 2019;47(7):885–93.
Pun BT, Balas MC, Barnes-Daly MA, Thompson JL, Aldrich JM, Barr J, et al. Caring for critically ill patients with the ABCDEF bundle: results of the ICU liberation collaborative in over 15,000 adults. Crit Care Med. 2019;47(1):3–14.
Pileggi C, Mascaro V, Bianco A, Nobile CGA, Pavia M. Ventilator bundle and its effects on mortality among ICU patients: a meta-analysis. Crit Care Med. 2018;46(7):1167–74.
Barnes-Daly MA, Phillips G, Ely EW. Improving hospital survival and reducing brain dysfunction at seven California community hospitals: implementing PAD guidelines via the ABCDEF bundle in 6,064 patients. Crit Care Med. 2017;45(2):171–8.
Acknowledgements
Not applicable.
Confidentiality
Each subject will be given a unique participant identification number after study enrollment. This participant identification number and quasi-identified initials will be used during randomization, drug distribution, eCRF filling, and sample transportation. The identification data, including full name, will be restricted in local medical charts and screening logs and must be preserved by researchers to maintain confidentiality.
Funding
Author Chunbo Chen is currently receiving a grant (#DFJH2020028) under the Major Program of Summit Project, Guangdong Province High-level Hospital Construction Project of Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, a grant (#number of MaoRenCaiBan[2020]24) from the Office of Talent Work Leading Group in Maoming, a grant (#201803010058) from the Guangzhou Science and Technology Program, a grant (#81671963) from the National Natural Science Foundation of China. Author Linhui Hu is currently receiving a grant (#2020YJ01) from the Emergent Science and Technology Project for Prevention and Treatment of Novel Coronavirus Pneumonia and a grant (#zx2020017) from the High-level Hospital Construction Research Project of Maoming People’s Hospital. The study is supported by the High-level Hospital Construction Research Project of Maoming People’s Hospital. The foundations have not been involved in any of the following parts of the study: study design, data collection, data management, analysis, and interpretation of the data. Also, the foundations have no ultimate authority over the aforementioned activities.
Author information
Authors and Affiliations
Contributions
CBC and LHH perceived and designed the protocol. KYP, XWH, ZW, QZW, YMX, YTH, YMH, XJZ, and CBC participated in the protocol design. LHH drafted the manuscript. CBC helped LHH draft and revised the manuscript. KYP, XWH, ZW, QZW, YMX, YTH, YMH, and XJZ were involved in the data collection. LHH, YMH, XJZ, and CBC analyzed the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The protocol and the informed consent document have been reviewed and approved by the Institutional Ethics Committee of all participating centers (Table 1). Study investigators will provide potential participants with verbal and written information before inclusion. Participants or their authorized representatives will provide informed consent.
Consent for publication
Not applicable. Results will be presented at relevant national and international conferences as well as being published in peer-reviewed journals.
Competing interests
All the other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Exclusion criteria for patient screening.
Additional file 2.
Adjudication chart for ventilator-associated pneumonia.
Additional file 3.
Criteria for diagnosis and confirmation of VAP.
Additional file 4.
Training protocol for bedside endoscopic tube placement.
Additional file 5.
Determination criteria for AE and SAE in VIP study.
Additional file 6.
SPIRIT checklist.
Additional file 7.
Informed Consent Form for Participants’ enrolment—English version.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hu, L., Peng, K., Huang, X. et al. Ventilator-associated pneumonia prevention in the Intensive care unit using Postpyloric tube feeding in China (VIP study): study protocol for a randomized controlled trial. Trials 23, 478 (2022). https://doi.org/10.1186/s13063-022-06407-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-022-06407-5