
Abstract
Background
Hydroxyethyl starch (HES) solutions are used for volume therapy to treat hypovolemia due to acute blood loss and to maintain hemodynamic stability. This study was requested by the European Medicines Agency (EMA) to provide more evidence on the long-term safety and efficacy of HES solutions in the perioperative setting.
Methods
PHOENICS is a randomized, controlled, double-blind, multi-center, multinational phase IV (IIIb) study with two parallel groups to investigate non-inferiority regarding the safety of a 6% HES 130 solution (Volulyte 6%, Fresenius Kabi, Germany) compared with a crystalloid solution (Ionolyte, Fresenius Kabi, Germany) for infusion in patients with acute blood loss during elective abdominal surgery. A total of 2280 eligible patients (male and female patients willing to participate, with expected blood loss ≥ 500 ml, aged > 40 and ≤ 85 years, and ASA Physical status II–III) are randomly assigned to receive either HES or crystalloid solution for the treatment of hypovolemia due to surgery-induced acute blood loss in hospitals in up to 11 European countries. The dosing of investigational products (IP) is individualized to patients’ volume needs and guided by a volume algorithm. Patients are treated with IP for maximally 24 h or until the maximum daily dose of 30 ml/kg body weight is reached.
The primary endpoint is the treatment group mean difference in the change from the pre-operative baseline value in cystatin-C-based estimated glomerular filtration rate (eGFR), to the eGFR value calculated from the highest cystatin-C level measured during post-operative days 1-3. Further safety and efficacy parameters include, e.g., combined mortality/major post-operative complications until day 90, renal function, coagulation, inflammation, hemodynamic variables, hospital length of stay, major post-operative complications, and 28-day, 90-day, and 1-year mortality.
Discussion
The study will provide important information on the long-term safety and efficacy of HES 130/0.4 when administered according to the approved European product information. The results will be relevant for volume therapy of surgical patients.
Trial registration
Similar content being viewed by others

Background
Hypovolemia is characterized by reduced circulating blood volume, which can be caused by a number of events, including blood loss due to surgery or trauma. Substantial loss of intravascular volume may lead to hemodynamic instability, tissue hypoperfusion, cellular hypoxia, organ damage, and ultimately death [1]. Accordingly, treatment aims at controlling further blood loss and restoring physiologic organ perfusion by providing infusion solutions targeting to match oxygen demands with delivery. While fluid therapy aims to substitute for protein-free fluid losses, the goal of volume therapy is substitution of blood volume, i.e., treatment of hypovolemia to maintain hemodynamics and vital functions [2, 3]. Crystalloid solutions (composed of water and electrolytes) and colloid solutions (containing macromolecules such as hydroxyethyl starch (HES), gelatin, or albumin) are routinely used for volume therapy [4]. While crystalloid solutions diffuse easily into the interstitial space, colloid solutions contain macromolecules, which are unable to pass intact semi-permeable biological membranes. In the surgical setting, an increased volume effect of colloids is observed compared to crystalloids [5,6,7,8,9,10]. This volume sparing effect is expected to reduce edema and associated complications [6,7,8,9].
The most relevant indication for the administration of colloids is volume replacement in patients undergoing general surgical procedures [6, 7]. Volume replacement during surgery aims for prompt and goal-directed colloid administration to optimize hemodynamic variables and to prevent fluid overload. Perioperative volume therapy should be guided by treatment algorithms that primarily use flow- or pressure-based target variables for hemodynamic optimization [9]. It has been demonstrated that post-operative outcomes may be improved with an algorithm-guided fluid administration; however, data are sparse [10].
HES represents one of the most frequently used colloids for volume replacement [11]. The HES-containing solution administered in this study, Volulyte 6% (HES 130/0.4), features a mean molecular weight of 130 kDa, a molar substitution of 0.38–0.45, and a substitution pattern of approximately 8:12.
Following the publication of investigator-initiated trials with methodological limitations indicating renal impairment and increased mortality upon HES administration in critically ill patients [12,13,14], the European Medicines Agency (EMA) started procedures to analyze the benefits and risks of HES-containing solutions. An Article 31 referral procedure (EMEA/H/A-31/1348) and an urgent union procedure under Article 107i of Directive 2001/83/EC (EMEA/H/A-107i/1376) for HES-containing medicinal products were initiated in 2012 and completed in 2013.
As part of the outcome of these referral procedures, the Marketing Authorization Holders of HES-containing solutions were requested to conduct, amongst others, a phase IV clinical trial to demonstrate the long-term safety of HES-containing solutions with regard to renal failure and mortality as well as efficacy in the perioperative setting. Scientific advice was given by the EMA’s Scientific Advice Working Party (SAWP) and adopted by the Committee for Medicinal Products for Human Use (CHMP) (EMA/CHMP/SAWP/544745/2014) regarding the design of this study. The current trial aims to study the elective abdominal surgery patients (> 40–≤ 85 years of age, ASA II–III) with an increased risk for post-operative complications. Administration of HES will be performed in accordance with the recently approved European HES product information, which is partly different to the volume regimen in the recently published FLASH study [15]. In the latter study, HES was used in patients with known risk factors (defined by an acute kidney risk index as an inclusion criterion) for the development of renal failure, and the dose limitations were not adhered to in a significant subset of patients. Consequently, 24% of patients in the HES group and 23% in the saline group had mild or moderate kidney dysfunction despite being a contraindication. In a recent multi-centric study in patients undergoing elective surgery, no signs for increased incidence of renal failure were found [16]. Moreover, a lower post-operative morbidity survey score was reported in a small-scale study when applying a goal-directed fluid management approach in surgical patients [17].
Study objective
The primary objective of this study is to assess non-inferiority regarding the safety of a 6% HES solution compared to an electrolyte solution in patients with acute blood loss during elective abdominal surgery. Secondary objectives are to further assess the efficacy and safety, e.g., combined mortality/major complications until day 90, renal function, coagulation parameters, inflammation, hemodynamic variables, length of hospital stay, major post-operative complications, and 28-day, 90-day, and 1-year mortality.
Methods/design
Trial design
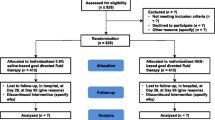
PHOENICS is a prospective, randomized, controlled, double-blind, multi-center, multinational phase IV study (in the Czech Republic and Serbia phase IIIb) performed in two parallel groups aiming to assess the safety of a 6% HES solution (Volulyte 6%, Fresenius Kabi Deutschland GmbH, Germany) versus an electrolyte solution (Ionolyte, Fresenius Kabi Deutschland GmbH, Germany) for the treatment of hypovolemia caused by acute blood loss in elective abdominal surgery. A total of 2280 eligible patients are randomly assigned to receive either Volulyte 6% (HES group) or Ionolyte (crystalloid group) in a 1:1 ratio, stratified by site. To achieve normovolemia, volume therapy will be given according to the clinical algorithms defined by each site before patient recruitment.
Participants
The study is conducted in a population of adult male and female patients > 40 and ≤ 85 years of age undergoing elective abdominal surgery with an expected blood loss of ≥ 500 ml. Eligible patients are patients with an ASA Physical status II–III and have to provide signed written informed consent to participate in this study. Women of childbearing potential must be tested negative for pregnancy (urine or serum) before inclusion. Reasons for exclusion are hypersensitivity to the active substances or to any of the other excipients of the investigational medicinal products, body weight ≥ 140 kg, sepsis, burns, renal impairment (AKIN stage ≥ 1 or chronic), acute and/or chronic renal replacement therapy (RRT), intracranial or cerebral hemorrhage, critically ill patients (typically admitted to the intensive care unit), hyperhydration, pulmonary edema, dehydration, hyperkalemia, severe hypernatremia, severe hyperchloremia, severely impaired hepatic function, congestive heart failure, severe coagulopathy, organ transplant patients, metabolic alkalosis, and simultaneous participation in another interventional clinical trial (drugs or medical devices studies). It is planned to activate study sites in up to 11 countries. A list of all participating sites is accessible at ClinicalTrials.gov.
Investigational products (IPs)
The investigational test product Volulyte 6% (Fresenius Kabi Deutschland GmbH, Germany) is a clear to slightly opalescent, colorless to slightly yellow (trial samples are colorless) 6% 130/0.4 hydroxyethyl starch solution in an isotonic, fully balanced electrolyte solution. The investigational reference product Ionolyte (Fresenius Kabi Deutschland GmbH, Germany) is a clear and colorless, aqueous, fully balanced electrolyte solution. Ionolyte is considered a suitable comparator since it has the identical electrolyte composition as Volulyte 6%. Both products are licensed solutions for infusion provided in a 500-ml polyolefin bag (freeflex) with overwrap.
Study phases
Enrollment (screening, randomization, and baseline)
Patients are screened within 1 week before surgery including verification of in- and exclusion criteria, demographic data, medical history, and anamnesis as well as the main indication for surgery, provision of informed consent, and pregnancy test in women of childbearing potential. The study starts with randomization (maximum of 1 day prior to surgery). After randomization and prior to induction of anesthesia, baseline variables will be determined (see Table 1).
Treatment phase
During the treatment phase, IP is administered intravenously to treat hypovolemia caused by surgery-induced acute blood loss. The administration of the IP and all defined safety and efficacy parameters are documented in an electronic case record form (see Tables 1 and 2). Perioperative IP administration has to be guided either by mean arterial pressure (MAP), stroke volume (SV), stroke volume variation (SVV), stroke volume index (SVI), or pulse pressure variation (PPV). The choice of the hemodynamic stabilization algorithm and the definition of volume responsiveness relies with the local investigator at each site at the beginning of the study but has to be followed for both groups during the whole study period within the study site.
The administration of IP starts during surgery and stops as soon as the patient is hemodynamically stabilized, but not exceeding the maximum daily IP dose of 30 ml kg−1 or the maximum treatment duration of 24 h.
As HES preparations for volume replacement rarely cause allergic reactions of varying severity, the first 10–20 ml of IP is infused slowly. In case of an allergic reaction, the infusion is stopped immediately, and appropriate treatment will be initiated.
If the patient is hemodynamically not stabilized after completion of the treatment phase, crystalloid solutions and/or albumin are to be administered for further volume therapy. The choice of the respective solution is at the discretion of the treating physician and is documented in the electronic case report form (eCRF) including applied volume. If needed, transfusion of blood products can be performed throughout the study period; blood products should be given in accordance with the current ESA guideline, recommending a target hemoglobin concentration of 7–9 g/dl during active bleeding.
Daily assessments
Patients are examined daily, including blood sampling, starting at POD 1 in the morning until POD 10 or hospital discharge, whatever occurs first. Safety and efficacy variables are recorded (see Tables 1 and 2).
Follow-up (FU)
Patients are invited to the hospital for additional visits on day 28 (± 5 days) and day 90 (± 14 days) after surgery. The patients will be invited for these follow-up visits to the hospital, by a follow-up letter, e-mail, or call. Alternatively, a qualified person (nurse or physician) may visit the patient at home. At each of these visits, renal function by means of serum cystatin-C, serum creatinine, cystatin-C-based and/or serum creatinine-based estimated glomerular filtration rate (eGFR), AKIN, and RIFLE scores are assessed. In addition, the occurrence of new RRT, (serious) adverse events, major post-operative complications, mortality, length of stay (hospital and ICU), and fit for discharge are recorded (see Tables 1 and 2). One year after surgery (± 30 days), mortality and renal outcome (i.e., new RRT after the 90-day visit) are re-assessed via a follow-up telephone call. Study phases and assessments are summarized in the PHOENICS study flow diagram (Table 1).
Concomitant medication
The following are the allowed concomitant medication/therapy:
-
Medication that is clinically required* (except other volume replacement (colloids or crystalloids) therapy during IP treatment period) by decision of the treating physician.
-
Vasoactive/inotropic treatment, starting earliest after third IP volume challenge.
-
Basal infusion of crystalloid solution (up to 4 ml/kg/h) as required. The choice of the crystalloid for the basal infusion and the required infusion rate is made by the treating physician.
-
Crystalloid solutions or albumin, if clinically required, after achieving the maximum daily dose of 30 ml/kg IP or maximum IP treatment period of 24 h, whatever occurs first.
-
If concomitant blood products are necessary, these should only be given according to the most current version of the ESA guideline on the management of severe perioperative bleeding, recommending a target hemoglobin concentration of 7–9 g/dl during active bleeding [20].
Administration of concomitant medication has to be provided via a separate infusion system independently from the IP infusion system. * e.g. basal vasoactive medication and initial crystalloid infusion to compensate for the effects of anesthetics for hemodynamic stabilization after induction of anesthesia (e.g., low doses of vasopressors).
The following are not allowed concomitant medication:
-
Any colloid (i.e., gelatin solutions, albumin, dextran, other HES solutions) during the treatment phase
-
Synthetic colloids (i.e., gelatin solutions, dextran, and other HES solutions) after the treatment phase until hospital discharge
-
Intravenous crystalloid solutions during treatment phase besides basal infusion (up to 4 ml−1kg−1 h−1)
Outcomes
Primary outcome
The primary outcome is the mean difference in cystatin-C-based eGFR calculated from the highest cystatin-C level measured during PODs 1–3. This difference is calculated for the change from the pre-operative baseline eGFR value. Measurements of cystatin-C in serum are performed in a central laboratory, blinded for patient allocation. Cystatin-C-based eGFR is calculated based on the equation developed by Inker et al. [21].
Secondary outcomes
Secondary variables (listed in Table 2) will provide further information on the safety and efficacy of the 6% HES solution. The date and time of assessment are documented for all variables. The assessments and corresponding time points are summarized in the PHOENICS study flow diagram (Table 1).
Laboratory analyses are performed in local laboratories of the sites, except for cystatin-C, which is determined in a central laboratory for all centers.
The investigator must record all (serious) adverse events ((S)AEs) occurring during the study on the appropriate eCRF page. All SAEs except those exempted from expedite reporting must be reported to the sponsor within 24 h (one working day) of the investigator becoming first knowledge. The sponsor will notify the competent authorities, IECs, and all concerned investigators about suspected unexpected serious adverse reactions in line with pertinent legal requirements.
Sample size
The primary objective of this study is to demonstrate non-inferiority of treatment with Volulyte 6% versus Ionolyte with respect to the prevention of relevant renal changes, quantified by the treatment group mean difference in change from the pre-operative baseline eGFR value to the eGFR value calculated from highest cystatin-C levels measured during post-operative days 1–3.
A t-test (α = 0.025 one-tailed) with a non-inferiority margin of δ = 8.1 mL/min/1.73 m2, corresponding to 9% of the lower limit of the normal range of GFR of 90mL/min/1.73 m2 (National Kidney Foundation 2002), was used for the sample size calculation. The EMA recommended in their final scientific advice (dated 22 January 2015) to use a non-inferiority margin lower than 10%. The standard deviation of the primary efficacy endpoint was assumed to be the same in both treatment arms and was estimated to be 52.5 mL/min/1.73 m2 based on data reported by Felicio and co-workers [22] and an estimated correlation coefficient of 0.5 for the correlation between pre-operative and post-operative eGFR values. It was further assumed that the real treatment group difference is zero.
Sample size estimation (power 1–β=0.9) resulted in 909 patients per group, i.e., 2280 patients in total, including a drop-out rate of 20% (SAS PROC POWER). This sample size also covers the secondary composite variable “mortality/major post-operative complications (including renal) until day 90.”
The drop-out rate is monitored during the study in order to adjust the sample size in case the drop-out rate exceeds 20%.
Randomization, blinding, and unblinding
Assignment to study treatment is randomized in a 1:1 ratio, stratified by site. An Interactive Response Technology System (IRTS) is used for randomization of patients (random permuted blocks of variable size) and IP supply.
The treatment group randomization list and the IP kits list are generated prior to the initiation of the study by the IRTS vendor and approved by an unblinded statistician (not involved in the study data analyses). Eligible patients are enrolled by the investigator and randomized by the IRTS.
Investigators and medical staff as well as study participants are blinded to the study treatment. Emergency unblinding will only be done via the IRTS by an investigator and/or dedicated authorized personnel (e.g., Pharmacovigilance Department of the Sponsor).
Statistical methods
All programming of tables, figures, listings, and statistical analyses will be performed using SAS® version 9.4 or higher. The planned statistics will be done in accordance with guideline ICH E9.
Primary endpoint
The mean difference in cystatin-C-based eGFR, calculated from the highest cystatin-C level measured during PODs 1–3, will be estimated with a two-sided 95% confidence interval by analyzing the change from the pre-operative baseline eGFR value in an analysis of covariance model, which includes treatment and study site as factors and baseline eGFR as a covariate. Non-inferiority of Volulyte 6% compared to Ionolyte will be tested with a one-sided contrast (α = 0.025) for a non-inferiority margin of δ = 8.1 mL/min/1.73 m2 (see the “Sample size” section for justification). The primary endpoint will be analyzed in the full analysis set (FAS) and in the per-protocol analysis set (PPS). Since the use of the FAS may not be conservative in a non-inferiority trial (ICH-E9 guideline) [23], the non-inferiority test performed in both datasets is considered as co-primary.
By definition, the FAS will comprise all patients reaching the post-operative period and monitored at least once with respect to the changes in cystatin-C-based eGFR. The results of both analyses (i.e., PPS and FAS) will be compared and possible differences assessed.
Secondary outcomes will be compared by means of descriptive statistics and appropriate statistical tests (time to event analyses (Kaplan-Meier plots, Cox regression), analysis of covariance, logistic regression, Mann-Whitney U test, χ2 test).
Strong efforts will be made to collect all data points in the study (e.g., by training of investigators and other authorized staff, regular monitoring of data entries). Missing pre-operative baseline values for serum creatinine-based eGFR will be imputed by assuming a numerical value at the lower end of the normal range (i.e., 75 ml/min per 1.73 m2). The corresponding missing serum creatinine value will be estimated based on the simplified “modification of diet in renal disease” formula based on age, race, and sex [24]. Details are outlined in the respective statistical analysis plan.
No interim analyses are planned.
Trial ethics and governance
This clinical study is being conducted in accordance with the Declaration of Helsinki and in compliance with the study protocol, Good Clinical Practice (GCP), designated SOPs, and local laws and regulations relevant to the country of conduct. The study protocol (version 4.5 for Germany, dated 5 November 2019) was approved by the respective competent authorities and ethic committees involved and used as the basis for this manuscript (country-specific protocol modifications not considered). The study is registered at the European clinical trial database EudraCT database, No.: 2016-002162-30, and in the ClinicalTrials.gov Protocol Registration and Results System, ClinicalTrials.gov ID: NCT03278548.
Written informed consent, in accordance with the origins of the Declaration of Helsinki and the applicable laws of the country, has to be obtained from all patients before entering the study. Informed consent forms for individual countries are available from the corresponding author upon request. The investigator explains the nature, purpose, and risks of the study and provides the patient with a copy of the patient information. The patient will be given sufficient time to consider the study’s implications before deciding whether to participate. There is no compensation for trial participation. All study participants are insured in accordance with the respective national legislations.
Information for the patients on the handling of their personal medical data and on the storage and handling of blood samples is part of the informed consent form. Blood samples that are collected specifically for this clinical study are kept until the end of the clinical study. Any remaining blood samples are destroyed after the completion of the clinical study. Storage of biological specimens for future research is not planned.
In case of any amendments to the protocol that would directly affect the patient’s participation in the study, the informed consent form will be amended, and the patient’s informed re-consent will be obtained, unless the patient has completed all procedures prior to the effectiveness of the corresponding amendments to the protocol.
Protocol amendments will be submitted to the concerned independent ethics committees (IECs) and competent authorities in line with pertinent regulatory requirements.
An independent audit at the study site may take place at any time during or after the study.
Any party (e.g., domestic and foreign regulatory authorities, the sponsor, and/ or authorized representatives of the sponsor such as monitors and auditors) with direct access takes all reasonable precautions within the constraints of the applicable regulatory requirements to maintain the confidentiality of patient identities and sponsor proprietary information. All patient data obtained in the context of the clinical trial are subject to data protection. The patient’s name in addition to other personal data (excluding age and sex) are not to be disclosed by the investigator. The storage of data for statistical assessment shall likewise be performed only under the patient’s study identification. Only the investigator will have the means to identify a patient’s name/other personal details via the study identification. If personal data and study-related documentation are stored and processed, the requirements of pertinent data protection legislation are to be observed.
Data generated in this study is recorded using a computerized system in accordance with applicable regulations. The system generates an individual eCRF for each patient participating in the study. Data entry is done by the respective site investigators, in part supported by study nurses. The responsibilities of the investigator, monitor, and sponsor of this clinical trial as regards to handling of data, storage of data, planning, assessment, and quality assurance are according to the recommendations on “International Conference on Harmonisation Topic E 6 Guideline for Good Clinical Practice.”
Authorized, qualified representatives of the sponsor will visit investigational sites in regular intervals as defined in the monitoring plan, to verify adherence to protocol and local legal requirements, to perform source data verification, and to assist the investigator in study-related activities. As a quality measure for monitoring, respective visits are reported to the study management (e.g., to define suitable corrective and preventive actions). In case the monitor identifies non-adherence to the protocol or legal requirements including data protection and data security requirements, a re-training will be performed, and its adherence will be subsequently strictly controlled.
Withdrawal of individual patients from treatment or from the study respectively could be caused by protocol deviation (e.g., dosing regimen, failure to comply with protocol).
A Data Safety Monitoring Board (DSMB), not involved in study conduct, consisting of two clinicians (one of them appointed as chair) and a biometrician, will monitor the progress of this study with a focus on safety as well as efficacy data.
The criteria for the study termination include unexpected safety concerns assessed by the DSMB.
Discussion
This study aims to provide data regarding the safety and efficacy of 6% HES 130/0.4 solutions in patients with hypovolemia due to surgery-induced acute blood loss. In contrast to previously published pragmatic studies comparing HES-containing solutions to crystalloid solutions in critically ill patients [25,26,27], the study design of this clinical trial aims to ensure that the HES 130 containing IP is administered in line with approved dosing recommendations and respecting contraindications. Hemodynamic stabilization is guided in accordance with established hemodynamic algorithms [13,14,15] and in accordance with the practice of the local investigator.
The primary endpoint of this study is defined as “treatment group mean difference in change from the pre-operative baseline cystatin-C-based eGFR to the eGFR that is calculated from the highest cystatin-C levels measured during post-operative days 1–3,” in line with the scientific advice obtained by the CHMP. eGFR has been shown to be a prognostic indicator of post-operative renal complications, associated with survival [28, 29]. In addition, a decline of eGFR was found to be indicative for acute kidney injury [28, 29]. eGFR can be calculated using defined formulas based on serum creatinine values or serum cystatin-C levels [21]. As cystatin-C was considered a more reliable marker for kidney function than creatinine, the calculation of eGFR in this study is based on serum cystatin-C levels. The primary endpoint of this study is thus a reliable indicator for post-operative renal complications and will allow to compare renal safety of HES 130 administration vs. crystalloid solutions in surgical patients after treatment of acute blood loss.
To demonstrate safety and efficacy for 6% HES 130/0.4 solutions, additional meaningful endpoints (e.g., coagulation, inflammation, hemodynamic variables, serum electrolytes, serum lactate levels, central venous oxygen saturation, length of hospital stay) will be compared between the groups. As major complications including renal impairment are frequently seen in high-risk surgery patients [10, 30], and long-term survival is strongly affected in patients with short-term surgical complications [31,32,33], a combined secondary endpoint of post-operative complications (including renal) and death will also be analyzed exploratively.
The patient population of this study comprises patients with an increased risk: the age of eligible patients is in the range of > 40– ≤ 85 years because it is known that several risk factors for post-operative morbidity and mortality increase with age. In particular, older age is associated with the development of acute kidney injury (AKI) [18, 34, 35]. Furthermore, we include only patients with ASA Physical Status II–III because it has been shown that concomitant diseases are independent predictors for post-operative morbidity and mortality [18, 19, 30, 36]. Post-operative outcome also depends on the type of surgery: the mortality rate after abdominal surgery is up to 8.4% compared to a general postsurgical mortality rate of up to 1.7% [30, 37, 38]. Moreover, the incidence of post-operative renal dysfunction after abdominal surgery was reported to be approximately 13% [39]. Other types of surgery that are also associated with increased post-operative acute kidney injury are cardiovascular and orthopedic surgeries. In cardiac surgery, however, kidney function is influenced extensively by the use of cardiopulmonary bypass[2, 40], and the mortality rate of older orthopedic surgical patients is usually lower compared to abdominal surgery [19].
In conclusion, this study will allow reliable assessment of the safety of HES 130 administration in surgical patients requiring volume therapy due to surgery-induced acute blood loss. Study results will substantially improve the availability of safety data on HES 130 solutions and allow to evaluate the safety of HES 130 administration in the surgical setting.
Trial status
This clinical study is currently in the recruitment phase. To update the patient information on regulatory procedures regarding HES products (referral procedure according to Art. 107i of Directive 2001/83/EC, in October 2018 to June 2019), a temporary halt was induced which lasted from January 2019 to July 2019. Recruitment began on September 28, 2017, and the end of recruitment is expected in April 2022.
Availability of data and materials
It is intended to publish the study results. They may be published as scientific literature in peer-reviewed journals and may also be used in submissions to regulatory authorities. Where necessary, the analyzed study datasets will be shared with regulatory authorities upon request.
Abbreviations
- AKD:
-
Acute kidney disease
- AKI:
-
Acute kidney injury
- aPTT:
-
Activated partial thromboplastin time
- ASA:
-
American Society of Anesthesiologists
- Ca2+ :
-
Calcium
- Cl- :
-
Chloride
- CVP:
-
Central venous pressure
- DAP:
-
Diastolic arterial pressure
- DSMB:
-
Data Safety Monitoring Board
- eCRF:
-
Electronic case report form
- eGFR:
-
Estimated glomerular filtration rate
- EMA:
-
European Medicines Agency
- FAS:
-
Full analysis set
- GCP:
-
Good Clinical Practice
- Hb:
-
Hemoglobin
- HCO3 − :
-
Hydrogen carbonate
- Hct:
-
Hematocrit
- HES:
-
Hydroxyethyl starch
- HR:
-
Heart rate
- ICU:
-
Intensive care unit
- IEC:
-
Independent ethics committee
- INR:
-
International norm ratio
- IP:
-
Investigational product
- IRTS:
-
Interactive Response Technology System
- K+ :
-
Potassium
- LOS:
-
Length of stay
- MAP:
-
Mean arterial pressure
- Na+ :
-
Sodium
- PADS:
-
Post-Anesthetic Discharge Scoring System
- pCO2 :
-
Carbon dioxide partial pressure
- pO2 :
-
Oxygen partial pressure
- POD:
-
Post-operative day
- PPS:
-
Per-protocol set
- PPV:
-
Pulse pressure variation
- RIFLE:
-
Risk, injury, failure, loss, end-stage renal disease
- RRT:
-
Renal replacement therapy
- SaO2 :
-
Oxygen saturation
- SAEs:
-
Serious adverse events
- SAP:
-
Systolic arterial pressure
- SCr:
-
Serum creatinine
- SOP:
-
Standard operating procedure
- SV:
-
Stroke volume
- SVI:
-
Stroke volume index
- SVV:
-
Stroke volume variation
References
Gutierrez G, Reines HD, Wulf-Gutierrez ME. Clinical review: hemorrhagic shock. Critical Care. 2004;8(5):373–81. https://doi.org/10.1186/cc2851.
Chappell D, Westphal M, Jacob M. The impact of the glycocalyx on microcirculatory oxygen distribution in critical illness. Current Opinion in Anaesthesiology. 2009;22(2):155–62. https://doi.org/10.1097/ACO.0b013e328328d1b6.
Chappell D, Jacob M. Flüssigkeits- und Volumentherapie 2013 Fluid and volume therapy in 2013. Notfall + Rettungsmedizin. 2013;16:617–24.
Heringlake M, Heinze H, Brauer K. Perioperatives Flüssigkeitsmanagement - Welches Volumen für welchen Patienten? Anasthesiologie Intensivmedizin Notfallmedizin Schmerztherapie. 2012;47(07/08):482–9. https://doi.org/10.1055/s-0032-1323570.
Yates DRA, Davies SJ, Milner HE, Wilson RJT. Crystalloid or colloid for goal-directed fluid therapy in colorectal surgery. British Journal of Anaesthesia. 2014;112(2):281–9. https://doi.org/10.1093/bja/aet307.
Martin C, Jacob M, Vicaut E, Guidet B, Van Aken H, Kurz A. Effect of waxy maize-derived hydroxyethyl starch 130/0.4 on renal function in surgical patients. Anesthesiology. 2013;118:387–94.
Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Critical Care Medicine. 2011;39(2):259–65. https://doi.org/10.1097/CCM.0b013e3181feeb15.
Sadaka F, Juarez M, Naydenov S, O’Brien J. Fluid resuscitation in septic shock: the effect of increasing fluid balance on mortality. J Intens Care Med. 2014;29(4):213–7. https://doi.org/10.1177/0885066613478899.
Payen D, de Pont AC, Sakr Y, Spies C, Reinhart K, Vincent JL. A positive fluid balance is associated with a worse outcome in patients with acute renal failure. Crit Care. 2008;12(3):R74. https://doi.org/10.1186/cc6916.
Bouchard J, Soroko SB, Chertow GM, Himmelfarb J, Ikizler TA, Paganini EP, et al. Fluid accumulation, survival and recovery of kidney function in critically ill patients with acute kidney injury. Kidney International. 2009;76(4):422–7. https://doi.org/10.1038/ki.2009.159.
Van Der Linden P, James M, Mythen M, Weiskopf RB. Safety of modern starches used during surgery. Anesthesia and Analgesia. 2013;116(1):35–48. https://doi.org/10.1213/ANE.0b013e31827175da.
Perner A, Haase N, Guttormsen AB, Tenhunen J, Klemenzson G, Åneman A, et al. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. New England Journal of Medicine. 2012;367(2):124–34. https://doi.org/10.1056/NEJMoa1204242.
Myburgh JA, Finfer S, Bellomo R, Billot L, Cass A, Gattas D, et al. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. New England Journal of Medicine. 2012;367(20):1901–11. https://doi.org/10.1056/NEJMoa1209759.
Brunkhorst FM, Engel C, Bloos F, Meier-Hellmann A, Ragaller M, Weiler N, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. New England Journal of Medicine. 2008;358(2):125–39. https://doi.org/10.1056/NEJMoa070716.
Futier E, Garot M, Godet T, Biais M, Verzilli D, Ouattara A, et al. Effect of hydroxyethyl starch vs saline for volume replacement therapy on death or postoperative complications among high-risk patients undergoing major abdominal surgery: the FLASH Randomized Clinical Trial. JAMA. 2020;323(3):1346–57. https://doi.org/10.1001/jama.2019.20833.
Kabon B, Sessler DI, Kurz A, Maheshwari K, Babazade R, Fiffick A, et al. Effect of intraoperative goal-directed balanced crystalloid versus colloid administration on major postoperative morbidity: a randomized trial. Anesthesiology. 2019;130(5):728–44. https://doi.org/10.1097/ALN.0000000000002601.
Joosten A, Delaporte A, Ickx B, Touihri K, Stany I, Barvais L, et al. Crystalloid versus colloid for intraoperative goal-directed fluid therapy using a closed-loop system: a randomized, double-blinded, controlled trial in major abdominal surgery. Anesthesiology. 2018;128(1):55–66. https://doi.org/10.1097/ALN.0000000000001936.
Turrentine FE, Wang H, Simpson VB, Jones RS. Surgical risk factors, morbidity, and mortality in elderly patients. Journal of the American College of Surgeons. 2006;203(6):865–77. https://doi.org/10.1016/j.jamcollsurg.2006.08.026.
Story DA, Leslie K, Myles PS, Fink M, Poustie SJ, Forbes A, et al. Complications and mortality in older surgical patients in Australia and New Zealand (the REASON study): a multicentre, prospective, observational study. Anaesthesia. 2010;65(10):1022–30. https://doi.org/10.1111/j.1365-2044.2010.06478.x.
Kozek-Langenecker SA, Ahmed AB, Afshari A, Albaladejo P, Aldecoa C, Barauskas G, et al. Management of severe perioperative bleeding: guidelines from the European Society of Anaesthesiology. Eur J Anaesthesiol. 2017;34(6):270–382. https://doi.org/10.1097/EJA.0000000000000630.
Inker LA, Schmid CH, Tighiouart H, Eckfeldt JH, Feldman HI, Greene T, et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. New England Journal of Medicine. 2012;367(1):20–9. https://doi.org/10.1056/NEJMoa1114248.
Felicio ML, de Andrade RR, Castiglia YMM, Silva MA de M, Vianna PTG, Martins AS. Cystatin C and glomerular filtration rate in the cardiac surgery with cardiopulmonary bypass. Brazilian J Cardiovasc Surg. 2009;24(3):305–11. https://doi.org/10.1590/S0102-76382009000400008.
European Medicines Agency. ICH Topic E 9 Statistical Principles for Clinical Trials. London; 1998.
Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P. Acute renal failure - definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8(4):R204–12. https://doi.org/10.1186/cc2872.
De Hert S, De Baerdemaeker L. Why hydroxyethyl starch solutions should not be banned from the operating room. Anaesthesiology Intensive Therapy. 2014;46(5):336–41. https://doi.org/10.5603/AIT.2014.0057.
Marx G, Schindler AW, Mosch C, Albers J, Bauer M, Gnass I, et al. Intravascular volume therapy in adults. European Journal of Anaesthesiology. 2016;33(7):488–521. https://doi.org/10.1097/EJA.0000000000000447.
Pearse RM, Harrison DA, MacDonald N, Gillies MA, Blunt M, Ackland G, et al. Effect of a perioperative, cardiac output-guided hemodynamic therapy algorithm on outcomes following major gastrointestinal surgery a randomized clinical trial and systematic review. JAMA. 2014;311(21):2181–90. https://doi.org/10.1001/jama.2014.5305.
Van Kuijk JP, Flu WJ, Chonchol M, Hoeks SE, Winkel TA, Verhagen HJM, et al. Temporary perioperative decline of renal function is an independent predictor for chronic kidney disease. Clin J Am Soc Nephrol. 2010;5(7):1198–204. https://doi.org/10.2215/CJN.00020110.
Welten GM, Schouten O, Chonchol M. Temporary worsening of renal function after aortic surgery is associated with higher long-term mortality. J Vasc Surg. 2008;47(2):219–28. https://doi.org/10.1016/j.jvs.2007.12.016.
Boyd O, Jackson N. Clinical review: how is risk defined in high-risk surgical patient management? Crit Care. 2005;9(4):390–6. https://doi.org/10.1186/cc3057.
Hamilton MA, Cecconi M, Rhodes A. A systematic review and meta-analysis on the use of preemptive hemodynamic intervention to improve postoperative outcomes in moderate and high-risk surgical patients. Anesthesia and Analgesia. 2011;112(6):1392–402. https://doi.org/10.1213/ANE.0b013e3181eeaae5.
Khuri SF, Henderson WG, DePalma RG, Mosca C, Healey NA, Kumbhani DJ. Determinants of long-term survival after major surgery and the adverse effect of postoperative complications. Ann Surg. 2005;242(3):326–41. https://doi.org/10.1097/01.sla.0000179621.33268.83.
Head J, Ferrie JE, Alexanderson K, Westerlund H, Vahtera J, Kivimäki M. Diagnosis-specific sickness absence as a predictor of mortality: the Whitehall II prospective cohort study. BMJ. 2008;337(oct02 2):a1469. https://doi.org/10.1136/bmj.a1469.
Calvert S, Shaw A. Perioperative acute kidney injury. Perioperative Medicine. 2012;1(1):6. https://doi.org/10.1186/2047-0525-1-6.
Kheterpal S, Tremper KK, Englesbe MJ, O’Reilly M, Shanks AM, Fetterman DM, et al. Predictors of postoperative acute renal failure after noncardiac surgery in patients with previously normal renal function. Anesthesiology. 2007;107(6):892–902. https://doi.org/10.1097/01.anes.0000290588.29668.38.
Silva JM, De Oliveira AMRR, Nogueira FAM, Vianna PMM, Pereira Filho MC, Dias LF, et al. The effect of excess fluid balance on the mortality rate of surgical patients: a multicenter prospective study. Crit Care. 2013;17(6):R288. https://doi.org/10.1186/cc13151.
Pearse RM, Harrison DA, James P, Watson D, Hinds C, Rhodes A, et al. Identification and characterisation of the high-risk surgical population in the United Kingdom. Critical Care. 2006;10(3):R81. https://doi.org/10.1186/cc4928.
Pearse RM, Moreno RP, Bauer P, Pelosi P, Metnitz P, Spies C, et al. Mortality after surgery in Europe: a 7 day cohort study. The Lancet. 2012;380(9847):1059–65. https://doi.org/10.1016/S0140-6736(12)61148-9.
O’Connor ME, Kirwan CJ, Pearse RM, Prowle JR. Incidence and associations of acute kidney injury after major abdominal surgery. Intensive Care Medicine. 2016;42(4):521–30. https://doi.org/10.1007/s00134-015-4157-7.
Sear JW. Kidney dysfunction in the postoperative period. British Journal of Anaesthesia. 2005;95(1):20–32. https://doi.org/10.1093/bja/aei018.
Acknowledgements
The authors would like to thank Reiner Tretter (Senior Manager Biostatistics, Fresenius Kabi, Germany) and Dr. Dirk Dormann (Vice President Medical Affairs I.V. Fluids, Fresenius Kabi, Germany) for supporting the preparation of this manuscript. The authors would like to thank all PHOENICS investigators for their contribution to the conduct of the study: Prof. Dr. Klimscha, Vienna, Austria; Dr. Kahn, Brussels, Belgium; Dr. Ongenae Gent, Belgium; Dr. Stessel Hasselt, Belgium; Dr. Baronica, Zagreb, Croatia; Dr. Persec, Zagreb, Croatia; Dr. Vymazal, Prague, Czech Republic; Dr. Tyll, Prague, Czech Republic; Dr. Novacek, Kolin, Czech Republic; Prof. Cholley, Paris, France; Dr. Mertes, Strasbourg, France; Dr. Boselli, Bourgoin Jallieu, France; Dr. Jaber, Montpellier, France; Prof. Constantin, Paris, France; Dr. Lasocki, Angers, France; Dr. Garnier, Paris, France; Dr. El Amine, Valenciennes, France; Dr. Kingler, Oldenburg, Germany; Prof. Weyland, Oldenburg, Germany; Prof. Dr. Gruenewald, Kiel, Germany; Prof. Wappler, Cologne, Germany; Prof. Zarbock, Münster, Germany; Prof Dr. Schäfer, Munich, Germany; Prof. Wulf, Marburg, Germany; Dr. Soukup, Cottbus, Germany; Prof. Dr. Scheeren, Groningen, the Netherlands; Prof. Dr Hollmann, Amsterdam, the Netherlands; Prof. Dr. Buhre, Maastricht, the Netherlands; Prof. Dr. Szczeklik, Krakow, Poland; Prof. Dr. Krzych, Katowice, Poland; Dr. Kudlinski, Zielona Gora, Poland; Prof. Gozdzik, Warsawa, Poland; Prof. Malas, Kielce, Poland; Prof. Owczuk, Gdansk, Poland; Dr. Kluzik, Poznan, Poland; Dr. Soro, Valencia, Spain; Prof. Diaz-Cambronero,Valencia, Spain; Dr. Garutti, Madrid, Spain; Dr. García del Valle, Alcorocn, Spain; Prof. Rodríguez Pérez, Gran Canaria, Spain; Dr. Martinez Castro, Madrid, Spain; Dr. Aldecoa, Valladolid, Spain; Dr. Felipe, Barcelona, Spain; Dr. De los Angeles Martin Pacetti, Malaga, Spain; and Dr. Ferrando, Barcelona, Spain.
Presentation
Not applicable
Funding
Fresenius Kabi Deutschland GmbH has prepared the scientific advice meetings at EMA’s Scientific Advice Working Party and provided the basis for the study design and protocol, supported by B. Braun Melsungen AG. B. Braun compiled the study protocol based on the EMA’s scientific advice document, contracted the academic contract research organization of the European Society of Anaesthesiology and Intensive Care (ESAIC CTN-CRO) for clinical trial application to ethic committees, monitoring, and collection/analysis of study data. B. Braun acted as the initial sponsor and Fresenius Kabi as collaborator until 2019. The process of sponsorship transfer from B. Braun to Fresenius Kabi took place until the end of March 2020. With the switch of sponsorship, the roles of collaborator, and sponsor changed accordingly. This clinical study is financed by Fresenius Kabi. Investigational test and reference products are produced and delivered by Fresenius Kabi to all participating sites.
Author information
Authors and Affiliations
Contributions
B. Braun und Fresenius Kabi were supported by AH and DS in designing the study and writing the study protocol. WB acts as coordinating investigator. WB, DdK, MGA, TS, MG, AZ, SD, MW, UB, TD, SSch, JFB, SDH, ZG, BC, TV, WS, HBC, MS, IG, RJ, and JB are responsible for conducting the study in all participating centers. WB, DdK, TS, MW, and UB drafted the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by Medizinische Ethikkommission der Carl von Ossietzky Universität Oldenburg, reference number 2017-008. Written, informed consent to participate will be obtained from all participants.
Competing interests
Wolfgang Buhre is a member of the Steering Committee of the Prodigy Study (sponsored by Medtronic), POSE Study (supported by ESAIC), Designation Study (supported with a grant from ZonMw), and PI of the AMAZONE study (supported with grants from the Dutch Cancer Society and ESAIC). Ute Brauer, Tamara Dehnhardt, and Sonja Schmier are employees of B. Braun Melsungen AG. Martin Westphal is and Jean-Francois Baron was an employee of Fresenius Kabi. Thomas Scheeren received research grants and honoraria from Edwards Lifesciences (Irvine, CA, USA) and Masimo Inc. (Irvine, CA, USA) for consulting and lecturing and from Pulsion Medical Systems SE (Feldkirchen, Germany) for lecturing (all payments made to the institution). Matthias Grunewald has received honoraria from CSL Behring, GE Healthcare, Gruenenthal, and Vifor Pharma (all unrelated to the PHOENICS study). Donald Spahn’s department is receiving grant support from Swiss National Science Foundation, the Swiss Society of Anesthesiology and Reanimation (SGAR), the Swiss Foundation for Anesthesia Research, Vifor SA, and Vifor (International) AG, Switzerland. Dr. Spahn is co-chair of the ABC-Trauma Faculty, sponsored by unrestricted educational grants from Novo Nordisk Health Care AG, Zurich, Switzerland; CSL Behring GmbH, Marburg, Germany; LFB Biomédicaments, Courtaboeuf Cedex, France; and Octapharma AG, Lachen, Switzerland. Dr. Spahn received honoraria/travel support for consulting or lecturing from Danube University of Krems, Austria; US Department of Defense, Washington, USA; European Society of Anesthesiology, Brussels, BE; Korean Society for Patient Blood Management, Seoul, Korea; Korean Society of Anesthesiologists, Seoul, Korea; Network for the Advancement of Patient Blood Management, Haemostasis and Thrombosis, Paris, France; Baxalta Switzerland AG, Volketswil, Switzerland; Bayer AG, Zürich, Switzerland; B. Braun Melsungen AG, Melsungen, Germany; Boehringer Ingelheim GmbH, Basel, Switzerland; Bristol-Myers-Squibb, Rueil-Malmaison Cedex, France and Baar, Switzerland; CSL Behring GmbH, Hattersheim am Main, Germany, and Berne, Switzerland; Celgene International II Sàrl, Couvet, Switzerland; Daiichi Sankyo AG, Thalwil, Switzerland; Haemonetics, Braintree, MA, USA; Instrumentation Laboratory (Werfen), Bedford, MA, USA; LFB Biomédicaments, Courtaboeuf Cedex, France; Merck Sharp & Dohme, Kenilworth, NJ, USA; PAION Deutschland GmbH, Aachen, Germany; Pharmacosmos A/S, Holbaek, Denmark; Pfizer AG, Zürich, Switzerland; Pierre Fabre Pharma, Alschwil, Switzerland, Portola Schweiz GmbH, Aarau, Switzerland; Roche Diagnostics International Ltd., Reinach, Switzerland; Sarstedt AG & Co., Sevelen, Switzerland and Nümbrecht, Germany; Shire Switzerland GmbH, Zug, Switzerland; Tem International GmbH, Munich, Germany; Vifor Pharma, Munich, Germany; Neuilly sur Seine, France and Villars-sur-Glâne, Switzerland; Vifor (International) AG, St. Gallen, Switzerland; and Zuellig Pharma Holdings, Singapore, Singapore. All other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Buhre, W., de Korte-de Boer, D., de Abreu, M.G. et al. Prospective, randomized, controlled, double-blind, multi-center, multinational study on the safety and efficacy of 6% Hydroxyethyl starch (HES) sOlution versus an Electrolyte solutioN In patients undergoing eleCtive abdominal Surgery: study protocol for the PHOENICS study. Trials 23, 168 (2022). https://doi.org/10.1186/s13063-022-06058-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-022-06058-6