
Abstract
Background/aims
Despite evidence that preferential use of balanced/buffered fluids may improve outcomes compared with chloride-rich 0.9% saline, saline remains the most commonly used fluid for children with septic shock. We aim to determine if resuscitation with balanced/buffered fluids as part of usual care will improve outcomes, in part through reduced kidney injury and without an increase in adverse effects, compared to 0.9% saline for children with septic shock.
Methods
The Pragmatic Pediatric Trial of Balanced versus Normal Saline Fluid in Sepsis (PRoMPT BOLUS) study is an international, open-label pragmatic interventional trial being conducted at > 40 sites in the USA, Canada, and Australia/New Zealand starting on August 25, 2020, and continuing for 5 years. Children > 6 months to < 18 years treated for suspected septic shock with abnormal perfusion in an emergency department will be randomized to receive either balanced/buffered crystalloids (intervention) or 0.9% saline (control) for initial resuscitation and maintenance fluids for up to 48 h. Eligible patients are enrolled and randomized using serially numbered, opaque envelopes concurrent with clinical care. Given the life-threatening nature of septic shock and narrow therapeutic window to start fluid resuscitation, patients may be enrolled under “exception from informed consent” in the USA or “deferred consent” in Canada and Australia/New Zealand. Other than fluid type, all decisions about timing, volume, and rate of fluid administration remain at the discretion of the treating clinicians. For pragmatic reasons, clinicians will not be blinded to study fluid type. Anticipated enrollment is 8800 patients. The primary outcome will be major adverse kidney events within 30 days (MAKE30), a composite of death, renal replacement therapy, and persistent kidney dysfunction. Additional effectiveness, safety, and biologic outcomes will also be analyzed.
Discussion
PRoMPT BOLUS will provide high-quality evidence for the comparative effectiveness of buffered/balanced crystalloids versus 0.9% saline for the initial fluid management of children with suspected septic shock in emergency settings.
Trial registration
PRoMPT BOLUS was first registered at ClinicalTrials.gov (NCT04102371) on September 25, 2019. Enrollment started on August 25, 2020.
Similar content being viewed by others

Background
Crystalloid fluid is part of the initial resuscitation for 25 million children with septic shock worldwide each year [1]. Options for crystalloid resuscitation include 0.9% saline or balanced/buffered fluids (lactated Ringer’s [LR], Hartmann’s solution, PlasmaLyte). Saline contains a supra-physiologic concentration of chloride and low strong ion difference (SID) that induce hyperchloremic metabolic acidosis, decrease renal blood flow, and promote inflammation [2, 3]. In contrast, balanced/buffered fluids have less chloride, some additional electrolytes, and a higher SID due to an anion buffer. Balanced/buffered fluids have been associated with better outcomes compared to 0.9% saline, including decreased acute kidney injury (AKI), need for renal replacement therapies (RRT), and death in adult [4,5,6,7,8,9,10] and pediatric [11] studies, while others reported no benefit [12] or harm [13]. Notably, 0.9% saline remains the most commonly used fluid for children with septic shock due to historical preference, universal availability, ease of use, drug compatibility, lower cost, and lack of definite benefit for alternatives [14,15,16].
The Isotonic Solutions and Major Adverse Renal Events Trial (SMART) found a small, but significant, reduction in major adverse kidney events within 30 days (MAKE30) with the use of balanced/buffered fluids compared to 0.9% saline in critically ill adults [17]. The largest benefits were in sepsis, with 4.2% lower mortality for balanced/buffered fluids. A parallel trial found similar benefits in non-critically ill adults [18]. In children, however, the two largest studies of fluid type in sepsis are observational and reached opposing conclusions [19, 20]. Consequently, recent guidelines issued a weak suggestion for balanced/buffered crystalloids while calling for studies to compare the effectiveness of different fluid types in pediatric septic shock [21, 22].
The primary aim of the Pragmatic Pediatric Trial of Balanced versus Normal Saline Fluid in Sepsis (PRoMPT BOLUS) is to determine if resuscitation with balanced/buffered fluids as part of usual care will improve outcomes compared to 0.9% saline for children with septic shock. Secondary aims are to compare the relative safety and determine the differential effect of each fluid type on kidney injury biomarkers in children with septic shock. We hypothesize that balanced/buffered fluid resuscitation will reduce MAKE30 (a composite of death, new RRT, or persistent kidney dysfunction) compared to 0.9% saline, in part through reduced kidney injury, without an increase in adverse effects. Notably, even a small clinical benefit would be important because sepsis is common, life-threatening, and the proposed practice change is inexpensive and readily available.
Methods
This manuscript was written in accordance with Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines (Additional file 1) and the 2010 CONSORT recommendations for pragmatic trials [23].
Trial design
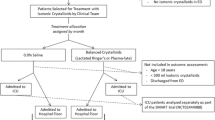
PRoMPT BOLUS is a multicenter, open-label, pragmatic interventional trial in which children with suspected septic shock are randomized to receive either 0.9% saline or balanced/buffered crystalloid fluids for initial resuscitation and maintenance fluids in addition to usual care. A pragmatic design [24] was selected to optimize cost-efficiency and generalizability (Table 1), and in a pilot study, was demonstrated to be feasible to implement within a busy pediatric emergency department [25]. Figure 1 shows an overview of the study design and Fig. 2 is the SPIRIT summary of study activities.

Schematic overview of the study design

SPIRIT summary of study activities
Study sites and timeline
This study is being conducted at > 40 sites from the Pediatric Emergency Research Networks, including the Pediatric Emergency Care Applied Research Network (PECARN) in the United States (US), Pediatric Emergency Research Canada (PERC), and Pediatric Research in Emergency Departments International Collaborative (PREDICT) in Australia/New Zealand. An international steering committee comprised of the overall study principal investigators (SLW, FB) and network-specific principal investigators from each geographic network will oversee all study activities. Enrollment started August 25, 2020, and is anticipated to continue for 5 years. Patients will only be enrolled in an emergency department (ED), where initial resuscitation of suspected septic shock will begin. Ongoing inpatient management of septic shock may occur on any hospital ward or intensive/critical care unit.
Participants
All patients > 6 months to < 18 years treated for suspected septic shock with parenteral antibiotics and at least one fluid bolus for abnormal perfusion in a study site ED will be eligible for study enrollment. The lower age was selected to adhere to the US Food and Drug Administration (FDA) warning that infants < 6 months may have reduced hepatic capacity to metabolize exogenous lactate in LR. “Suspected septic shock” is operationalized as a) the treating clinician’s diagnosis of septic shock and/or treatment that includes parenteral antibiotic and fluid resuscitation for abnormal perfusion or b) a sepsis alert on a site-specific screening tool with clinician confirmation to proceed with treatment for suspected septic shock. Abnormal perfusion is defined as the treating clinician’s judgment that hypotension or abnormal (either “flash” or “prolonged”) capillary refill is present. To reflect usual clinical practice, we do not define thresholds for hypotension or abnormal capillary refill but rather defer to the treating clinician’s discretion to differentiate abnormal from normal. Recognizing that fluid resuscitation may start prior to ED arrival, only those for whom total volume of crystalloid fluid administration can be confirmed as ≤40 mL/kg will be eligible.
Exclusion criteria are (1) clinician judgment that the patient’s condition deems it unsafe to administer either 0.9% saline or balanced/buffered fluids (see Additional file 2), (2) known pregnancy at time of assessment for eligibility, (3) known prisoner at time of assessment for eligibility (in USA, Australia/New Zealand), (4) patient is a ward of the state (in Australia/New Zealand), (5) known allergy to either fluid type, or (6) prior indication that patient would not agree to be enrolled in the study.
Research ethics approval
A single Institutional Review Board at The Children’s Hospital of Philadelphia (CHOP) will provide regulatory approval and oversight for all US study sites. In Canada, the University of Calgary will sponsor regulatory approval through Health Canada, while each site’s Research Ethics Board will oversee local human subjects’ protections (except sites in Ontario, for which centralized approval through Clinical Trials Ontario will be obtained). In Australia and New Zealand, a single ethics committee will provide regulatory oversight within each jurisdiction. Protocol modifications approved by the international steering committee will be first reviewed by the CHOP Institutional Review Board and, following approval, will be distributed by the network-specific principal investigators to all other regulatory bodies in the USA (local site Institutional Review Boards, FDA), Canada, Australia, and New Zealand, as well as to local investigators.
Consent
Prospective written informed consent will be sought from a legally authorized representative (LAR, e.g., at least one parent) prior to enrollment when sufficient time permits. However, because of the life-threatening nature of septic shock and narrow therapeutic window to commence fluid resuscitation, enrollment may proceed in the USA under the federal “Exception From Informed Consent” (EFIC, 21 CFR 50.24) for emergency research [26] and in Canada and Australia/New Zealand with “deferred consent” in compliance with the Canadian Tri-Council Policy Statement-2 [27] and National Statement on Ethical Conduct in Human Research [28], respectively. Such enrollment methods are ethically suitable for emergent, life-threatening conditions when patients may benefit from the research, available treatments are unproven/unsatisfactory, and obtaining prospective informed consent is not feasible [25, 29,30,31,32]. A waiver of assent will be requested due to the critical nature of illness in septic shock.
To comply with federal and ethical guidelines, community consultation and public disclosure were conducted in the USA (available at https://www.regulations.gov/docket/FDA-1995-S-0036) and community consultation was conducted in Australia/New Zealand to obtain input from and inform the local community prior to beginning enrollment [33, 34]. All EFIC studies are also subject to additional regulation through the US FDA, which approved the study protocol under an Investigational New Drug (IND #136978) application.
Participants enrolled under EFIC (USA) or deferred consent (Canada, Australia/New Zealand) will be notified of enrollment and study procedures as soon as feasible and offered the right to withdraw from or continue in the study. For study participants who die prior to the post-enrollment discussion in the USA or deferred consent in Canada or Australia/New Zealand, all reasonable attempts will be made to inform the LAR after the participant’s death, unless a waiver has been granted in local jurisdictions. However, successful LAR contact will not be required to include data in the analysis. At a subset of sites collecting biological samples, separate written informed consent will be obtained for the measurement of blood and urine biomarkers.
Randomization and allocation concealment
Patients will be allocated in equal numbers to groups by a permutated-block randomization sequence using blocks of 2, 4, or 6, stratified by site and generated by the lead trial statistician (JH). Block lengths of 2, 4, or 6 will be selected randomly with equal probability, and the treatment sequence within blocks will be randomly shuffled until a list of 300–600 is generated for each site. For large sites that expect enrollment from separate areas of the emergency department, separate randomization lists will be generated for each recruitment area. Study group allocation will be concealed using serially numbered, opaque envelopes, which provides an efficient process for quickly randomizing participants concurrent with ongoing clinical management. Treatment allocation to 0.9% saline or balanced/buffered fluids will be revealed after eligibility for enrollment has been confirmed.
Intervention
The intervention to be tested is predominant fluid resuscitation with balanced/buffered fluids (LR, Hartmann’s, or PlasmaLyte, as per local site preference) compared to the “usual practice” of predominant 0.9% saline resuscitation. Other than prescribing which crystalloid fluid type to use, the intervention will be implemented without efforts to standardize timing and volume of fluid resuscitation or other components of clinical care. Thus, all decisions to administer fluid, as well as timing, volume, and rate of fluid administration, remain at the discretion of the treating clinicians. Hospital fluid supplies will be used for this study without any change to packaging or labeling. Participants will be randomized to receive either balanced/buffered fluids or 0.9% saline for all bolus and maintenance fluid administration starting from randomization through 11:59 pm on the following calendar day. Maintenance fluids are included because these constitute a substantial proportion of total crystalloid fluid administration [35], including 40% of total crystalloids as maintenance fluids in our pilot study [25]. If the treating clinician deems it unsafe to continue study fluid or identifies a clinical indication that requires specific fluid or electrolyte therapy (e.g., hyponatremia, hyperkalemia, hypercalcemia, cerebral edema), non-study fluids are permitted. Use of non-isotonic fluids (e.g., 0.45% saline) is not recommended as standard maintenance fluids in children [36] and will be discouraged during the intervention phase. Colloid fluids and blood products may be administered at the discretion of the treating team.
Ending the intervention phase at 11:59 PM on the calendar day following randomization ensures all participants are eligible to receive study fluid for a minimum of 24 and a maximum of 48 h. This represents the time-window from the presentation during which most fluid resuscitation is completed for septic shock [17] and provides a pragmatic endpoint that signals the end of the intervention phase.
Adherence
All fluid administered through the end of the intervention window will be recorded. Adherence to the intervention is defined as receipt of ≥75% of total crystalloid fluid as the randomized fluid type during the interventional window. For example, participants randomized to balanced/buffered fluids are considered adherent to the intervention if they receive ≥75% of their total crystalloid as balanced/buffered fluids starting from the time of randomization through 11:59 PM on the following calendar day. Locally, all sites will establish a work-flow to communicate study enrollment and promote adherence across providers and hospital locations. Such strategies include verbal and/or written hand-offs between providers, use of a templated orderset indicating PRoMPT BOLUS study enrollment and treatment allocation, and automated electronic reminders to clinicians at the time an order is placed for non-study crystalloid fluids during the intervention window. Each participant’s adherence to the intervention will be monitored centrally, and the mean adherence to the intervention for all participants within each site will be regularly reported back to local investigators. Targeted education will be undertaken if < 80% of participants in either study arm meet the adherence criterion. If < 70% of participants meet the adherence criterion over two consecutive months, the site will be placed on monitored probation with additional efforts undertaken to understand and correct challenges to adherence. Sites that continue to fail to meet the adherence threshold in at least 80% of enrolled participants may be removed from study participation.
Blinding
Study participants, clinicians, and investigators will not be blinded to treatment allocation. Blinding study fluid is not pragmatic [17, 18] and unlikely to even be possible because available laboratory values overtly reflect the crystalloid being used [9]. However, randomization of individuals following study enrollment will minimize selection bias related to pre-enrollment awareness of study group assignment. The lead biostatistician (JH), however, will remain blinded to group assignment for purposes of data analysis.
Data collection and management
Data will be extracted from the medical record and recorded onto a standardized case report form. Each network will oversee data collection and quality from its respective sites. In the USA, data will be collected through FDA-compliant Advarra Electronic Data Capture (Columbia, MD) hosted at CHOP. In Canada and Australia/New Zealand, data will be collected in Research Electronic Data Capture (REDCap, Vanderbilt University; Nashville, TN) hosted at the University of Alberta and the Murdoch Children’s Research Institute, respectively [37]. However, all sites will collect the same data elements using a common data dictionary. All study data will be password-protected and coded with a study number to ensure confidentiality. Periodic verification of study data against source documents will be undertaken within each network to certify data accuracy. The US Data Coordinating Center (DCC) will centrally manage all data exported from Advarra and REDCap to ensure harmonization for analyses. Consistent with the tenets of a pragmatic trial, data collection will be brief, targeting key patient characteristics, fluid administration, and outcomes (Additional file 3).
Outcomes
The primary outcome will be MAKE30—a composite of death from any cause, initiation of RRT, or persistent kidney dysfunction—at either 30 days following study enrollment or hospital discharge, whichever comes first (Table 2) [38]. RRT will include treatment (or attempt to treat if the participant does not tolerate treatment) with any renal replacement modality during the hospitalization that was not a continuation of pre-hospital chronic therapy, censored at 30 days after randomization. Persistent kidney dysfunction will be measured as final creatinine ≥200% of baseline (i.e., at least a doubling of baseline creatinine) and a minimum absolute increase of ≥0.3 mg/dL. Baseline serum creatinine will be recorded as the lowest creatinine available between 12 months and 24 h prior to enrollment or, for participants without such data, an imputed value using established median values for age and sex [39]. Secondary efficacy, safety, and biologic outcomes are listed in Table 2.
The rationale for MAKE30 as the primary endpoint is that mediation of kidney injury provides the major mechanistic pathway through which balanced/buffered fluids are likely to improve patient outcomes. In addition, the only prior comparable randomized clinical trials demonstrated a reduction in MAKE30 with the use of balanced/buffered fluids in adult patients with sepsis [17, 18]. Finally, MAKE30 provides an objective and easily measured outcome endorsed as a patient-centered endpoint for clinical trials [40].
Adverse events
Adverse events (AEs) will be queried from the medical record at the end of the intervention phase and between study days 5–7. We will limit the time period for the determination of AEs because we are unaware of any biologically plausible reason to anticipate that either fluid type should lead to AEs remote from the immediate intervention period. AEs that are unexpected, serious, and at least possibly related to study fluid type will be reviewed by the international steering committee and reported promptly to regulatory bodies.
Sample size
A baseline incidence of MAKE30 in children with septic shock predominantly resuscitated with 0.9% saline was determined based on a prior pediatric study (9.6% MAKE30) [39], the proportion of patients aged 18–21 years enrolled in the SMART trial (9.7% MAKE30) [17], and institutional registry data from two US study sites (5.0% and 7.8% MAKE30). We anticipate a conservative MAKE30 incidence of 6% among children treated in an ED for suspected septic shock largely resuscitated with 0.9% saline. Enrollment of 8800 total participants will provide 95% power to detect an absolute risk reduction (ARR) in MAKE30 from 6.0% for children treated with 0.9% saline to 4.3% for balanced/buffered fluids with a standard type I error of 0.05. This ARR corresponds to the 28% relative risk reduction in MAKE30 observed in the youngest subset of patients (18–21 years) enrolled in SMART. Due to the small anticipated ARR with potential for type II error, we purposely selected a high power of 95% for the primary endpoint. This sample size will also provide 84% power to detect a decrease in mortality from 3% with saline to 2% with balanced/buffered fluids and > 99% power to detect a 1-day reduction in hospital-free days.
Recruitment of this large sample size requires collaboration across > 40 sites from three research networks. Based on the enrollment of 85% of eligible patients in our pilot study [25], sites are anticipated to enroll > 80% of eligible patients. Recruitment will be monitored, with targeted support provided to sites that consistently fall below this target. Targeted support will include local investigators meeting with the lead investigators from their respective networks to review the process for screening, determination of eligibility criteria, education of local emergency clinicians who assess for eligibility, location of enrollment materials relative to patient care in the emergency department, and tracking of reasons for missed enrollments. If enrollment fails to demonstrate improvement toward or above the > 80% target despite these efforts, the site will be considered for removal from study participation and a replacement site will be identified.
Data/safety monitoring and interim analysis
The study will be monitored by a Data Safety Monitoring Board (DSMB) comprised of 10 international members. Interim monitoring for the superiority of one treatment approach will be performed after enrollment of 15%, 40%, and 70% of total study participants. The significance levels at each interim analysis for the difference in MAKE30 between groups are 0.000000014, 0.00079, and 0.014, respectively, using the symmetric two-sided O’Brien-Fleming boundaries. The DSMB may also consider early termination of the trial if there is evidence of futility using an informal, conditional power approach [41].
Statistical analysis
The primary outcome of MAKE30 will be reported using the Mantel-Haenszel (MH) relative risk, stratified by the study site. The primary analysis will be performed using the intention-to-treat (ITT) principle, including all randomized participants for whom the three components of the MAKE30 outcome are not missing, regardless of whether study fluid was administered. To account for the three planned interim analyses, the final analysis will use a significance level of 0.044 for the primary outcome of MAKE30.
Secondary outcomes will also be analyzed using the ITT approach incorporating methods of Benjamini and Hochberg to control for multiple analyses [42, 43]. The analysis plan for secondary outcomes is described in Additional file 4.
A per-protocol analysis will include all participants who receive at least 75% of total crystalloid fluid volume (bolus and maintenance) during the intervention window as the fluid to which the participant was randomized. Because the study intervention (fluid type) is available outside of the study, participants who continue to receive the same fluid to which they were randomized after withdrawal from the study protocol will remain eligible for the per-protocol analysis if they meet the ≥75% threshold. Participants who are found to have been ineligible for enrollment (e.g., age incorrect at initial eligibility assessment) or who receive no crystalloid fluid after randomization will be excluded from the per-protocol analysis. To account for potential confounding introduced into the per-protocol analysis, we will use generalized mixed effect regression models to adjust for known covariate imbalances. We will also conduct an analysis using instrumental variables to adjust for potential bias created by treatment contamination [44].
Subgroup analyses
We will evaluate treatment differences among subgroups defined by the following stratification variables: age group, sex, cancer comorbidity, AKI at study enrollment, abnormal kidney function using day 0 biomarker measurements (Table 2), total fluid volume received during intervention window, and country of enrollment. If there is significant heterogeneity in treatment effect using the Breslow-Day test [45], we will report separate results within strata.
Missing Data
The primary ITT analysis will use complete case data if the percentage missing the primary outcome is ≤5% of total enrollment and all three country-specific percentages missing the primary outcome is ≤7% of country-specific enrollment. If these criteria are not met, we will use multiple imputation for the primary analysis with data assumed to be missing at random unless available data contradict this assumption. If the primary outcome is missing from between 1% and 5% of total participants, we will perform an adjunctive analysis using multiple imputation in addition to the complete case analysis. In addition, we will examine differences between participants with and without missing data and conduct a sensitivity analysis using the inverse-probability-weighting (IPW) method to conduct multiple imputation for the missing data [46, 47]. New analyses performed in addition to those described above will be delimitated as post hoc and considered hypothesis-generating.
Presentation of the results
Trial results will be communicated to the public through manuscript publication and ClinicalTrials.gov, including the full study protocol and statistical code. Authorship will adhere to the International Committee of Medical Journal Editors guidelines. To ensure widespread access to study findings, we will seek publication using “open access”. After the publication of main and secondary results, a public use dataset will be submitted to the Eunice Kennedy Shriver National Institute of Child Health and Development Data and Specimen Hub (DASH).
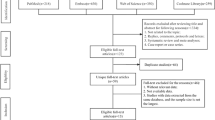
The flow of participants through the study will be presented as in Fig. 3. Baseline characteristics will be presented by the treatment group, as in Table 3. Total, bolus, and maintenance volumes of crystalloid and other fluids and blood products will also be presented by the treatment group, as in Table 4. Laboratory data will be presented in figures displaying initial and follow-up values. All outcomes will be reported by the treatment group with both unadjusted frequencies and proportions, as well as with any statistical adjustments. Heterogeneity of treatment effect for subgroup analyses will be displayed within forest plots.

Flow diagram of patients progressing through the trial
Discussion
PRoMPT BOLUS will provide high-quality evidence for the comparative effectiveness of buffered/balanced crystalloids versus 0.9% saline for the initial fluid management of children with suspected septic shock in emergency settings. Currently, there is insufficient evidence to support one crystalloid fluid type for pediatric shock resuscitation, which was acknowledged as a key knowledge gap by the 2020 Surviving Sepsis Campaign guidelines [21, 22]. Should balanced/buffered fluids improve outcomes compared to 0.9% saline, this practice change could be quickly implemented given that these fluids are readily accessible. Thus, the results of this trial could be rapidly incorporated into clinical practice with an immediate impact on the health of children worldwide. Conversely, because this trial is powered to detect small clinical differences, a finding of no difference between groups would signify that the initial choice of crystalloid fluid type is inconsequential for most children with septic shock during the initial 24-48 hour resuscitation period.
We selected a pragmatic design for several reasons [48, 49]. First, embedding enrollment into routine care will reduce cross-over by minimizing pre-enrollment exposure to non-study fluids. Second, a pragmatic protocol allows flexibility to execute the study across multiple sites, countries, and healthcare systems. Third, loosening reliance on a study team allows study procedures to continue across multiple hospital units and providers. Finally, generalizability is optimized by using a study protocol that mirrors the variance in clinical decision-making outside of this study.
The decision to use patient-level randomization rather than an alternative method based on systematic allocation (e.g., different fluid type in alternating months) or cluster randomization ensured that allocation concealment was maintained until after patient enrollment despite not blinding the study intervention. We were concerned that pre-enrollment awareness of treatment allocation could lead to selection bias. Importantly, there was no allocation strategy we believed could eliminate cross-over between groups. Selection of fluid type is dictated by multiple factors, including a potential lack of personal equipoise across providers, numerous clinical indications for specified fluid and electrolyte titration, ongoing debate about the utility of acid buffers in acutely ill patients [50, 51], and imperfect awareness of study enrollment. However, a tolerance of some use of non-study fluid after enrollment will mimic real-world conditions where cross-over between fluid types is common. As such, we defined adherence as receipt of ≥75% of total crystalloid administered by type randomized. Therefore, our study is most appropriately construed as the comparative effectiveness between the predominant—rather than exclusive—use of balanced/buffered fluids versus 0.9% saline. Even if exclusive use of a single fluid type could be executed under controlled experimental conditions, it is unlikely that such a practice could be generalized to real-world fluid management.
Several potential threats to the validity of our trial exist. First, enrollment of children with suspected septic shock exclusively in the ED will include a wide range of illness severity and fluid exposure. Prior studies have demonstrated that fluid type has a negligible clinical impact when exposure is limited to low volumes [17, 18]. Therefore, we only include children who require > 1 fluid bolus and the fluid is directed to treat abnormal perfusion rather than dehydration or hypovolemia. This approach balances early enrollment, when it may not yet be clear how much fluid a patient may require, with enrichment of the study population by children for whom fluid type is most likely to have a clinical impact. In our pilot study using the same enrollment criteria, 42% were admitted to the intensive care unit [25], which represents the population with the highest exposure to study fluid and the greatest risk of achieving the primary outcome. A second threat is the lack of blinding. This decision was a practical one to constrain costs of providing blinded fluid bags across all parts of the hospital and needing additional safeguards if the treating physician was blinded to fluid type. However, we chose objective endpoints unlikely to be influenced by knowledge of study group assignment and will maintain blinding of the lead study statistician. Third, frequent withdrawal after EFIC or deferred consent could limit exposure to the intervention and/or impede outcome assessment. Although studies indicate < 1% withdrawal after EFIC or deferred consent [32, 52] and only 2% withdrew after EFIC in our pilot study [25], we plan to modify the analysis if missing data exceeds expectations. Finally, MAKE30 represents a composite of endpoints that may not be of equivalent value to patients or clinicians.
Trial status
PRoMPT BOLUS is a pragmatic randomized interventional trial of the effectiveness and safety of fluid resuscitation with balanced/buffered fluids compared to 0.9% saline for children with septic shock as part of usual clinical care. Patient enrollment began on August 25, 2020, and is anticipated for 5 years (clinicalTrials.gov/NCT04102371) under this protocol version 1, dated May 25, 2021.
Availability of data and materials
The trial statisticians (HK, JH), lead data manager (KLH), and study principal investigators (SLW, FB, NK) will have full, unrestricted access to and be responsible for the final dataset. Members of the international steering committee will also have access to the final dataset upon request. All study investigators can request specific elements of the study data, which will require approval by the international steering committee. After the publication of main and secondary results, a public use dataset will be made available through the Eunice Kennedy Shriver National Institute of Child Health and Development Data and Specimen Hub (DASH). The study funders will not restrict access to study data.
References
Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–11. https://doi.org/10.1016/S0140-6736(19)32989-7.
Karakala N, Raghunathan K, Shaw AD. Intravenous fluids in sepsis: what to use and what to avoid. Curr Opin Crit Care. 2013;19(6):537–43. https://doi.org/10.1097/MCC.0000000000000028.
Myburgh JA, Mythen MG. Resuscitation fluids. N Engl J Med. 2013;369(13):1243–51. https://doi.org/10.1056/NEJMra1208627.
Annane D, Siami S, Jaber S, Martin C, Elatrous S, Declere AD, et al. Effects of fluid resuscitation with colloids vs crystalloids on mortality in critically ill patients presenting with hypovolemic shock: the CRISTAL randomized trial. JAMA. 2013;310(17):1809–17. https://doi.org/10.1001/jama.2013.280502.
Raghunathan K, Shaw A, Nathanson B, Sturmer T, Brookhart A, Stefan MS, et al. Association between the choice of IV crystalloid and in-hospital mortality among critically ill adults with sepsis*. Crit Care Med. 2014;42(7):1585–91. https://doi.org/10.1097/CCM.0000000000000305.
Rochwerg B, Alhazzani W, Gibson A, Ribic CM, Sindi A, Heels-Ansdell D, et al. Fluid type and the use of renal replacement therapy in sepsis: a systematic review and network meta-analysis. Intensive Care Med. 2015;41(9):1561–71. https://doi.org/10.1007/s00134-015-3794-1.
Rochwerg B, Alhazzani W, Sindi A, Heels-Ansdell D, Thabane L, Fox-Robichaud A, et al. Fluid resuscitation in sepsis: a systematic review and network meta-analysis. Ann Intern Med. 2014;161(5):347–55. https://doi.org/10.7326/M14-0178.
Shaw AD, Bagshaw SM, Goldstein SL, Scherer LA, Duan M, Schermer CR, et al. Major complications, mortality, and resource utilization after open abdominal surgery: 0.9% saline compared to Plasma-Lyte. Ann Surg. 2012;255(5):821–9. https://doi.org/10.1097/SLA.0b013e31825074f5.
Young JB, Utter GH, Schermer CR, Galante JM, Phan HH, Yang Y, et al. Saline versus Plasma-Lyte A in initial resuscitation of trauma patients: a randomized trial. Ann Surg. 2014;259(2):255–62. https://doi.org/10.1097/SLA.0b013e318295feba.
Yunos NM, Bellomo R, Hegarty C, Story D, Ho L, Bailey M. Association between a chloride-liberal vs chloride-restrictive intravenous fluid administration strategy and kidney injury in critically ill adults. JAMA. 2012;308(15):1566–72. https://doi.org/10.1001/jama.2012.13356.
Sharma A, Yadav M, Kumar BR, Lakshman PS, Iyenger R, Ramchandran G. A comparative study of Sterofundin and Ringer lactate based infusion protocol in scoliosis correction surgery. Anesth Essays Res. 2016;10(3):532–7. https://doi.org/10.4103/0259-1162.181425.
Young P, Bailey M, Beasley R, Henderson S, Mackle D, McArthur C, et al. Effect of a Buffered Crystalloid Solution vs Saline on Acute Kidney Injury Among Patients in the Intensive Care Unit: The SPLIT Randomized Clinical Trial. JAMA. 2015;314(16):1701–10. https://doi.org/10.1001/jama.2015.12334.
Ngo NT, Cao XT, Kneen R, Wills B, Nguyen VM, Nguyen TQ, et al. Acute management of dengue shock syndrome: a randomized double-blind comparison of 4 intravenous fluid regimens in the first hour. Clin Infect Dis. 2001;32(2):204–13.
Awad S, Allison SP, Lobo DN. The history of 0.9% saline. Clin Nutr. 2008;27(2):179–88. https://doi.org/10.1016/j.clnu.2008.01.008.
Long E, Babl F, Dalziel S, Dalton S, Etheridge C, Duke T, et al. Fluid resuscitation for paediatric sepsis: A survey of senior emergency physicians in Australia and New Zealand. Emerg Med Australas. 2015;27(3):245–50. https://doi.org/10.1111/1742-6723.12400.
Thompson GC, Macias CG. Recognition and Management of Sepsis in Children: Practice Patterns in the Emergency Department. J Emerg Med. 2015;49(4):391–9. https://doi.org/10.1016/j.jemermed.2015.03.012.
Semler MW, Self WH, Wanderer JP, Ehrenfeld JM, Wang L, Byrne DW, et al. Balanced Crystalloids versus Saline in Critically Ill Adults. N Engl J Med. 2018;378(9):829–39. https://doi.org/10.1056/NEJMoa1711584.
Self WH, Semler MW, Wanderer JP, Wang L, Byrne DW, Collins SP, et al. Balanced Crystalloids versus Saline in Noncritically Ill Adults. N Engl J Med. 2018;378(9):819–28. https://doi.org/10.1056/NEJMoa1711586.
Emrath ET, Fortenberry JD, Travers C, McCracken CE, Hebbar KB. Resuscitation With Balanced Fluids Is Associated With Improved Survival in Pediatric Severe Sepsis. Crit Care Med. 2017;45(7):1177–83. https://doi.org/10.1097/CCM.0000000000002365.
Weiss SL, Keele L, Balamuth F, Vendetti N, Ross R, Fitzgerald JC, et al. Crystalloid Fluid Choice and Clinical Outcomes in Pediatric Sepsis: A Matched Retrospective Cohort Study. J Pediatr. 2017;182:304–10 e10. https://doi.org/10.1016/j.jpeds.2016.11.075.
Weiss SL, Peters MJ, Alhazzani W, Agus MSD, Flori HR, Inwald DP, et al. Surviving Sepsis Campaign International Guidelines for the Management of Septic Shock and Sepsis-Associated Organ Dysfunction in Children. Pediatr Crit Care Med. 2020;21(2):e52–e106. https://doi.org/10.1097/PCC.0000000000002198.
Weiss SL, Peters MJ, Alhazzani W, Agus MSD, Flori HR, Inwald DP, et al. Surviving sepsis campaign international guidelines for the management of septic shock and sepsis-associated organ dysfunction in children. Intensive Care Med. 2020;46(Suppl 1):10–67. https://doi.org/10.1007/s00134-019-05878-6.
Zwarenstein M, Treweek S, Gagnier JJ, Altman DG, Tunis S, Haynes B, et al. Improving the reporting of pragmatic trials: an extension of the CONSORT statement. BMJ. 2008;337(nov11 2):a2390. https://doi.org/10.1136/bmj.a2390.
Thorpe KE, Zwarenstein M, Oxman AD, Treweek S, Furberg CD, Altman DG, et al. A pragmatic-explanatory continuum indicator summary (PRECIS): a tool to help trial designers. J Clin Epidemiol. 2009;62(5):464–75. https://doi.org/10.1016/j.jclinepi.2008.12.011.
Balamuth F, Kittick M, McBride P, Woodford A, Vestal N, Casper TC, et al. Pragmatic pediatric trial of balanced versus normal saline fluid in sepsis: the PRoMPT BOLUS pilot feasibility study. Acad Emerg Med. 2019;26(12):1346–56. https://doi.org/10.1111/acem.13815.
Watters D, Sayre MR, Silbergleit R. Research conditions that qualify for emergency exception from informed consent. Acad Emerg Med. 2005;12(11):1040–4. https://doi.org/10.1197/j.aem.2005.06.022.
Menon K, O'Hearn K, McNally JD, Acharya A, Wong HR, Lawson M, et al. Comparison of Consent Models in a Randomized Trial of Corticosteroids in Pediatric Septic Shock. Pediatr Crit Care Med. 2017;18(11):1009–18. https://doi.org/10.1097/PCC.0000000000001301.
The National Health and Medical Research Council tARCaUA. National Statement on Ethical Conduct in Human Research Commonwealth of Australia, Canberra2007 (updated 2018) [Available from: https://www.nhmrc.gov.au/about-us/publications/national-statement-ethical-conduct-human-research-2007-updated-2018.
Baren JM, Fish SS. Resuscitation research involving vulnerable populations: are additional protections needed for emergency exception from informed consent? Acad Emerg Med. 2005;12(11):1071–7. https://doi.org/10.1197/j.aem.2005.06.020.
Chamberlain JM, Okada P, Holsti M, Mahajan P, Brown KM, Vance C, et al. Lorazepam vs diazepam for pediatric status epilepticus: a randomized clinical trial. JAMA. 2014;311(16):1652–60. https://doi.org/10.1001/jama.2014.2625.
Inwald DP, Canter R, Woolfall K, Mouncey P, Zenasni Z, O'Hara C, et al. Restricted fluid bolus volume in early septic shock: results of the Fluids in Shock pilot trial. Arch Dis Child. 2018;104(5):426–31. https://doi.org/10.1136/archdischild-2018-314924.
Dalziel SR, Borland ML, Furyk J, Bonisch M, Neutze J, Donath S, et al. Levetiracetam versus phenytoin for second-line treatment of convulsive status epilepticus in children (ConSEPT): an open-label, multicentre, randomised controlled trial. Lancet. 2019;393(10186):2135–45. https://doi.org/10.1016/S0140-6736(19)30722-6.
Haggins AN, Harney D, Scott S, Silbergleit R. A systematic review of Federal Drug Administration Docket for community consultation and public disclosure in exception from informed consent trials. Clin Trials. 2018;15(1):29–35. https://doi.org/10.1177/1740774517737318.
Harvin JA, Podbielski JM, Vincent LE, Liang MK, Kao LS, Wade CE, et al. Impact of Social Media on Community Consultation in Exception From Informed Consent Clinical Trials. J Surg Res. 2019;234:65–71. https://doi.org/10.1016/j.jss.2018.09.007.
Van Regenmortel N, Verbrugghe W, Roelant E, Van den Wyngaert T, Jorens PG. Maintenance fluid therapy and fluid creep impose more significant fluid, sodium, and chloride burdens than resuscitation fluids in critically ill patients: a retrospective study in a tertiary mixed ICU population. Intensive Care Med. 2018;44(4):409–17. https://doi.org/10.1007/s00134-018-5147-3.
Feld LG, Neuspiel DR, Foster BA, Leu MG, Garber MD, Austin K, et al. Clinical Practice Guideline: Maintenance Intravenous Fluids in Children. Pediatrics. 2018;142(6)
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–81. https://doi.org/10.1016/j.jbi.2008.08.010.
Billings FT, Shaw AD. Clinical trial endpoints in acute kidney injury. Nephron Clin Pract. 2014;127(1-4):89–93. https://doi.org/10.1159/000363725.
Weiss SL, Balamuth F, Thurm CW, Downes KJ, Fitzgerald JC, Laskin BL. Major Adverse Kidney Events in Pediatric Sepsis. Clin J Am Soc Nephrol. 2019;14(5):664–72. https://doi.org/10.2215/CJN.12201018.
Palevsky PM, Molitoris BA, Okusa MD, Levin A, Waikar SS, Wald R, et al. Design of clinical trials in acute kidney injury: report from an NIDDK workshop on trial methodology. Clin J Am Soc Nephrol. 2012;7(5):844–50. https://doi.org/10.2215/CJN.12791211.
Lan KK, Simon R, Halperin M. Stochastically curtailed tests in long-term clinical trials. Commun Stat Sequential Anal. 1982;C1:207–19.
Benjamini Y, Cohen R. Weighted false discovery rate controlling procedures for clinical trials. Biostatistics. 2017;18(1):91–104. https://doi.org/10.1093/biostatistics/kxw030.
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125(1-2):279–84. https://doi.org/10.1016/S0166-4328(01)00297-2.
Dunn G, Maracy M, Tomenson B. Estimating treatment effects from randomized clinical trials with noncompliance and loss to follow-up: the role of instrumental variable methods. Stat Methods Med Res. 2005;14(4):369–95. https://doi.org/10.1191/0962280205sm403oa.
Breslow NE, Day NE. Statistical methods in cancer research. Volume I - The analysis of case-control studies. IARC Sci Publ. 1980;32:5–338.
Wooldridge J. Inverse probability weighted estimation for general missing data problems. J Econometrics. 2007;141(2):1281–301. https://doi.org/10.1016/j.jeconom.2007.02.002.
Horvitz DG, Thompson DJ. A generalization of sampling without replacement from a finite universe. J Am Stat Assoc. 1952;47(260):663–85. https://doi.org/10.1080/01621459.1952.10483446.
Patsopoulos NA. A pragmatic view on pragmatic trials. Dialogues Clin Neurosci. 2011;13(2):217–24. https://doi.org/10.31887/DCNS.2011.13.2/npatsopoulos.
Tosh G, Soares-Weiser K, Adams CE. Pragmatic vs explanatory trials: the pragmascope tool to help measure differences in protocols of mental health randomized controlled trials. Dialogues Clin Neurosci. 2011;13(2):209–15. https://doi.org/10.31887/DCNS.2011.13.2/gtosh.
Forsythe SM, Schmidt GA. Sodium bicarbonate for the treatment of lactic acidosis. Chest. 2000;117(1):260–7. https://doi.org/10.1378/chest.117.1.260.
Jaber S, Paugam C, Futier E, Lefrant JY, Lasocki S, Lescot T, et al. Sodium bicarbonate therapy for patients with severe metabolic acidaemia in the intensive care unit (BICAR-ICU): a multicentre, open-label, randomised controlled, phase 3 trial. Lancet. 2018;392(10141):31–40. https://doi.org/10.1016/S0140-6736(18)31080-8.
Klein L, Moore J, Biros M. A 20-year Review: The Use of Exception From Informed Consent and Waiver of Informed Consent in Emergency Research. Acad Emerg Med. 2018;25(10):1169–77. https://doi.org/10.1111/acem.13438.
Acknowledgements
We thank the global Pediatric Emergency Research Network (PERN) for endorsement of this study. We thank the National Center for Advancing Translational Sciences Trial Innovation Network for consultation in the study design/methods and for providing financial support of the PRoMPT BOLUS pilot and feasibility trial. We thank J. Michael Dean MD, Charlie Casper PhD, SallyJo Zuspan, and Melissa Metheney from the PECARN Data Coordinating Center and Rick Watts from the PERC Data Coordinating Center for their insightful comments and support of this trial. We acknowledge the contributions of Ed Oakley, MBBS FACEM and Sarah McNab, MBBS PhD as members of the Australian and New Zealand steering committee. We also acknowledge review and endorsement of the trial by the Pediatric Acute Lung Injury and Sepsis Investigators (PALISI) network, Society of Critical Care Medicine Discovery Network, Australian and New Zealand Intensive Care Society Paediatric Study Group, Australasian College of Emergency Medicine Clinical Trials Network, and the Children’s Inpatient Research Collaboration of Australia and New Zealand. Finally, we thank Andrew Costarino, MD for serving as the independent medical monitor for this study.
PRoMPT BOLUS INVESTIGATORS
Australia: E. Long, A. Williams, F. Babl (The Royal Children’s Hospital); M. Borland, S. O’Brien (Perth Children’s Hospital); S. Craig, E. Ramaga (Monash Children’s Hospital); A. Kochar, G. Nivea (Women’s and Children’s Hospital); S. Jani, D. Thosar (The Children’s Hospital at Westmead); A. Rao (Sydney Children’s Hospital, Randwick); N. Phillips (Queensland Children’s Hospital); S. George (Gold Coast University Hospital); A. Lithgow, C. Mitchell (Royal Darwin Hospital); Canada: G. Thompson, S. Freedman, S. Williamson-Urquhart, E. Gilad, S. Cooke (Alberta Children’s Hospital); P. Judge, S. Murthy, N. Kissoon (British Columbia Children’s Hospital); W. Alqurashi, F. Alnaji (Children’s Hospital of Eastern Ontario); G. Sangha (Children’s Hospital, London Health Sciences Centre); A. Mater, M, Brashaw (Jim Pattison Children’s Hospital); S. Curtis, A. Joffe (Stollery Children’s Hospital); Y. Shayan, M. Tucci (Centre Hospitalier Universitaire Sainte Justine); K. Gripp (The Children’s Hospital of Winnipeg); S. Berthelot, M. Weiss (Centre Hospitalier de l’Université Laval); A. Davis, E. Guifoyle, M. Moretti (Hospital for Sick Children); A. Kam, M. Parker, B. Rochwerg (McMaster Children’s Hospital); J. Emsley, N. Verma (IWK Health Centre); A. Sehgal (Kingston Health Sciences Centre); New Zealand: S. Dalziel, M. Bonisch (Starship Hospital); E. Tan, J. Neutze (Kidz First Children’s Hospital); United States: F. Balamuth, S. Weiss, E. Tsemberis, J. Huang, M. Cook, H. Katcoff, K. Hayes, C. Hickey (The Children’s Hospital of Philadelphia); M. Eisenberg, D. Lewander (Boston Children’s Hospital); C. Morris, D. Hurley; S. Baumer-Mouradian (Medical College of Wisconsin); L. Ambroggio, K. Grice (Children’s Hospital Colorado); A. Festekjian (Children’s Hospital Los Angeles); B. Hickey, R. Sada (Children’s Hospital Pittsburgh); J. Dodson, M. Badawy, C. Lebel, M. Elliott (Children’s Medical Center of Dallas); I. Koutralis, K. Hom (Children’s National Hospital); M. Eckerle, M. Singleton (Cincinnati Children’s Hospital Medical Center); A. Rogers, V. Cervantes (CS Mott Children’s Hospital); S. Duffy, I. Bahamon (Hasbro Children’s Hospital); L. Alpern, A. Sirizi (Ann & Robert H. Lurie Children's Hospital of Chicago); A. Haider Ahmad, A. Rubi Banegas (MD Anderson Cancer Center, Children’s Cancer Hospital); J. Lloyd, K. DiCostanzo (Nationwide Children’s Hospital); M. Kwok, J. Ochs (New York-Presbyterian Morgan Stanley Children’s Hospital); R. Lane, T. Harbour (Primary Children’s Hospital); N. Uspal, K. Cappetto (Seattle Children’s Hospital); L. Clukies, D. Robinson (St. Louis Children’s Hospital); J. McManemy, V. Gonzales (Texas Children’s Hospital); C. Vance, N. Kupperman, K. Pimenta (UC Davis Children’s Hospital), K. Mansour, L. Lavrisha (UCSF Benioff Children’s Hospital); M. Ramirez, J. Grad (NYU Langone Health)
Funding
Funding is provided by Eunice Kennedy Shriver National Institute of Child Health and Human Development R01HD101528, Commonwealth of Pennsylvania Department of Health SAP #4100085749, The Medical Research Futures Fund International Clinical Trial Collaboration (APP1190814), and the Canadian Institutes of Health Research 173498. Additional support is provided by The Children’s Hospital of Philadelphia Research Institute and Alberta Children’s Hospital Research Institute. The Pediatric Emergency Care Applied Research Network (PECARN) is funded through the Emergency Medical Services for Children Network Development Demonstration Program of the Maternal and Child Health Bureau, Health Resources and Services Administration, under cooperative agreement (awards U03MC00008, U03MC00001, U03MC00003, U03MC00006, U03MC00007, U03MC22684, and U03MC22685). The Pediatric Research in Emergency Departments International Collaborative (PREDICT) Network is funded, in part, through a National Health and Medical Research Council (NHMRC), Canberra, Australia, Centre of Research Excellence Grant (GNT1058560). SRD is partly funded by Cure Kids New Zealand. SF is supported by the Alberta Children’s Hospital Foundation Professorship in Child Health and Wellness. FEB’s time is part funded by an NHMRC Practitioner Fellowship and the Royal Children’s Hospital Foundation, Melbourne, Australia. The content and conclusions of this article are those of the authors and should not be construed as the official position or policy of, nor should any endorsements be inferred by, any funding or governmental agencies. The funding institutions had no role in (1) the conception, design, or conduct of the study; (2) the collection, management, analysis, interpretation, or presentation of the data; or (3) the preparation, review, or approval of the manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
SLW, FB, JH, and NK conceived of and designed the study. SLW, FB, EL, GCT, CPH, SRD, BG, SBF, FEB, and NK will execute the study. SLW, FB, EL, GCT, MC, ET, AW, SWU, MLB, SRD, BG, SBF, FEB, and NK will acquire data. KLH and HK will manage the data. SLW, FB, JH, and NK devised the statistical approach. SLW, FB, HK, JH, and NK drafted the initial manuscript. SLW, FB, EL, GCT, KLH, HK, MC, ET, CPH, AW, SWU, MLB, SRD, BG, SBF, FEB, JH, and NK critically revised and approved the manuscript. SLW, FB, EL, GCT, MLB, SRD, BG, SBF, FEB, and NK will supervise the study. SLW, FB, KLH, HK, JH, and NK will have complete access to the final trial dataset. All authors read and approved the final of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
A single Institutional Review Board at The Children's Hospital of Philadelphia is providing regulatory oversight for all US study sites (IRB #19-016484). In Canada, the University of Calgary will sponsor regulatory approval through Health Canada, while each site’s Research Ethics Board will oversee local human subjects’ protections, except sites in Ontario, for which centralized approval through Clinical Trials Ontario will be obtained (pending). A single ethics committee at The Royal Children’s Hospital Melbourne is providing regulatory oversight for Australia study sites (#68569). In New Zealand, a single ethics committee will provide regulatory oversight (pending).
Prospective written informed consent will be sought from a legally authorized representative (LAR, e.g., at least one parent) prior to enrollment when sufficient time permits. Alternatively, enrollment may proceed in the US under the federal “Exception From Informed Consent” (EFIC, 21 CFR 50.24) for emergency research and in Canada and Australia/New Zealand with “deferred consent” in compliance with the Canadian Tri-Council Policy Statement-2 and National Statement on Ethical Conduct in Human Research, respectively. A waiver of assent will be requested due to the critical nature of illness in septic shock.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
SPIRIT 2013 Checklist: Recommended Items to Address in a Clinical Trial Protocol and Related Documents
Additional file 2.
Suggested Unsafe Conditions for Study Enrollment
Additional file 3.
Data Collection Planned for the PRoMPT BOLUS Pragmatic Clinical Trial
Additional file 4.
Analysis Plan for Secondary Outcomes
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Weiss, S.L., Balamuth, F., Long, E. et al. PRagMatic Pediatric Trial of Balanced vs nOrmaL Saline FlUid in Sepsis: study protocol for the PRoMPT BOLUS randomized interventional trial. Trials 22, 776 (2021). https://doi.org/10.1186/s13063-021-05717-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-021-05717-4