
Abstract
Background
Major depressive disorder is the second leading cause of years lost to disability worldwide. Anyu Peibo Capsule has been shown to be effective and safe in phase II trials.
Methods
This clinical study is a multi-center, randomized, double-blinded, placebo-controlled, parallel-group, phase III trial of Anyu Peibo Capsule in China. The aim is to test whether the administration of Anyu Peibo Capsule compared to placebo improves clinical outcomes in adults (aged 18 to 65 years) with MDD. Patients will receive an 8-week treatment of Anyu Peibo Capsule 1.6 g per day or placebo. The primary outcome will be the change from baseline in the total score for the Montgomery-Asberg Depression Rating Scale at the end of the 8-week treatment.
Discussion
The trial aims to provide pivotal evidence for the efficacy and safety of Anyu Peibo Capsule in patients with major depressive disorder.
Trial registration
ClinicalTrials.govNCT04210973. Registered on December 26, 2019
Similar content being viewed by others

Background
Major depressive disorder (MDD) is a recurrent, disabling, and serious psychological disorder and the second leading cause of years lost to disability worldwide (World Health Organization [1]). Over the past two decades, China has experienced rapid changes which have had a huge impact on the lifestyles and mental health of individuals. Current epidemiological investigations have shown that the lifetime prevalence of MDD in China has reached 3.4% [2], meaning that there are about 50 million people with MDD requiring appropriate treatment.
Antidepressants play a prominent role in the treatment of MDD [3]. The Chinese guidelines for depressive disorders recommend antidepressants as a first-line treatment for MDD [4]. However, about 30% of patients fail to achieve remission despite treatment with multiple antidepressants [5]. Treatment continuity is one of the main challenges in treating patients with MDD [6]. A national survey in China has shown that the top reason (36.1%) for why patients with MDD discontinue their medication is concern about long-term side effects [7]. There is a need to develop novel treatments for the relief of the symptoms of depression and to increase patients’ tolerance for long-term treatment.
Anyu Peibo Capsule is a new antidepressant, extracted from Piper laetispicum C. DC. (Piperaceae), a climbing glabrous plant growing in the south of China which has been used for invigorating the circulation and reducing detumescence and stasis, as well as having analgesic properties [8,9,10,11]. The pharmacodynamics of Anyu Peibo Capsule have been assessed in healthy volunteers. A favorable safety profile, a trend that the curative effect was higher than that of placebo, and remission rate in MDD have been demonstrated for Anyu Peibo Capsule in randomized, blinded, and placebo-controlled phase IIa and IIb trials.
The ongoing clinical study is a Chinese multi-center, randomized, double-blinded, placebo-controlled, parallel-group, phase III trial of Anyu Peibo Capsule administration in adults (aged 18 to 65 years) with MDD (AYPB-MDD-III). Based on the results of a phase II study, this trial is designed to detect clinically relevant differences in clinical outcomes at the end of the 8-week treatment as the primary endpoint.
Methods
Trial design
AYPB-MDD-III is a multi-center, randomized, double-blinded, placebo-controlled, parallel-group phase III clinical trial of Auyu Peibo Capsule to treat adults with MDD. This clinical trial is sponsored by Su Zhou YiHua Biotechnology Co. Ltd. and conducted at Shanghai Mental Health Center and 13 other sites in China (Appendix 1).
Study objectives
The primary objective is to investigate the efficacy and safety of Anyu Peibo Capsule in comparison with placebo in adults with MDD. An exploratory objective is to explore potential predictive biomarkers for efficacy.
Participants and eligibility
Patients diagnosed with MDD at the time of screening will be enrolled across 14 sites in China. The trial period will be from July 2019 to December 2020. The inclusion, exclusion, and withdrawal criteria in AYPB-MDD-III (Table 1) have been chosen to exclude patients at risk of suicide.
Any of the following medications and treatments will be prohibited:
-
1.
Other psychoactive drugs (except drugs permitted in the protocol), including antipsychotics, antidepressants, mood stabilizers, anti-anxiety drugs, and nootropics
-
2.
Traditional Chinese medicines (including Chinese patent medicines and traditional Chinese medicine decoctions) which may have the effect of relieving depression
-
3.
Modified electroconvulsive therapy, transcranial magnetic stimulation, vagus nerve stimulation, light therapy, laser therapy, acupuncture, other traditional Chinese medicine treatments, biofeedback therapy, and other treatment methods
-
4.
Systematic psychotherapy, such as psychoanalysis and cognitive behavior therapy
-
5.
Other medications and treatments that may significantly affect the efficacy and safety of antidepressants
Some combination therapies will be permitted:
-
1.
For serious insomnia, zolpidem, zopiclone, S-Zopiclone, and zaleplon can be taken before sleep, as long as the dose does not exceed the upper limit specified in drug labels, and the cumulative dosing during the trial duration does not exceed 2 weeks.
-
2.
Medications used to treat physical diseases with efforts made to ensure the type and dosage do not change during the trial.
The following are the termination criteria of the trial:
-
1.
There is a major design error in the protocol which makes it difficult to evaluate the drug effect.
-
2.
During the implementation of the trial, a major deviation occurs or an unexpected, serious risk to the patients is found.
-
3.
The ethics committee or sponsor decides to terminate the clinical trial.
Justification for no concomitant therapy
This trial is designed according to Major Depressive Disorder: Developing Drugs for Treatment Guidance for Industry (FDA 2018) [12] to develop a new drug. Following this guidance, randomized, double-blind, placebo-controlled, parallel designs are recommended as the current standard for short-term efficacy trials in MDD. When this trial was to be designed as no concomitant therapy, actions for risk control and management were included in the first consideration. Therefore, we will exclude those who are at risk of suicide and ensure appropriate and sufficient informed consent process. What is more, when the patients’ condition declines due to no treatment, the condition will be assessed to decide whether to unblind or not, the patient could withdraw at any moment, and then standard treatment will be provided.
Randomization
Patients enrolled in the AYPB-MDD-III trial are allocated to treatment with Auyu Peibo Capsule or placebo in a 1:1 ratio via the clinical trial electronic central stochastic system (DAS) for interactive web response system (IWRS).
Trial interventions
Auyu Peibo is a capsule containing 20 mg of extracts (main ingredient: amide alkaloids); the taste- and visual-masked placebo is an identically shaped capsule containing no effective materials. Four tablets of the study drug or placebo will be administered orally, with water, 30 min after breakfast and supper, twice per day for 8 weeks. The patient’s adherence is monitored and assessed by the percentage of the number of practical drugs to the number of prescribed drugs.
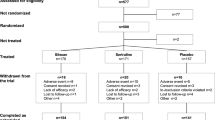
The patient flow is displayed in Fig. 1 according to the Consolidated Standards of Reporting Trials (CONSORT) [13] diagram, and the SPRIT [14] scheme of study procedures is shown in Fig. 2.

Participant flow through the AYPB-MDD-III

SPRIT scheme of study procedures
Blinding
AYPB-MDD-III is double-blinded. Patients, care providers, investigators, and outcome assessors will not know the treatment allocation. The investigated drug is prepared by the central pharmacy and will be provided as a ready-to-use drug or placebo, labeled with a random number and patient number. The placebo is taste- and visual-masked. Emergency unblinding can be conducted via DAS for IWRS. Unblinding under emergency will be permissible due to a serious adverse event, or for the purpose of notification to the Data and Safety Monitoring Board (DSMB).
Recruitment
Patients will be recruited from 14 sites (psychiatric hospitals) in China, where research psychiatrists and clinical staff from each site will invite potential patients to participate in the study.
Data management and quality assurance
Trained research psychiatrists from each site will record the collected data on a source document and a web-based eCRF (DAS for EDC V6.0, in Chinese). Verification and cross-checking of the eCRF will be conducted by the study monitor, who will be provided by the site clinical research organization as well as the sponsor. Omissions, errors, and values requiring further clarification will be reviewed. Corrections should be made only by authorized personnel and be documented in an audit trail.
Strategies to improve adherence to interventions
Reminder texts will be sent to patients before each study visit, and fees for public transportation will be reimbursed for attendance.
Provisions for post-trial care
There will be no special post-trial care, and patients will continue their usual health care when the trial has been finished.
Data collection methods
Study instruments
Chinese version of the Montgomery-Asberg Depression Rating Scale (MADRS)
The Chinese version of the MADRS [15, 16] has been translated from the original English version. It consists of 10 items rated on a 0–6 continuum (0 = no abnormality, 6 = severe) to assess the core symptoms of depression. Inter-rater reliability on the Chinese MADRS with different pairs of raters has been reported to be 0.954; reliability, validity, and sensitivity have been demonstrated to be good [17].
17-item Hamilton Depression Scale (HAMD-17) Chinese version
The Chinese version of the HAMD-17 has been translated from the original English version. It consists of 17 items that are rated to assess the symptoms of depression. The Chinese HAMD-17 Cronbach’s alpha coefficients are calculated to be over 0.70, and internal consistency and validity have been demonstrated to be good [18].
Chinese version of the Sheehan Disability Scale (SDS)
The Chinese SDS measures impairment in work/school, social life, and family life responsibilities. The Chinese version of the SDS has been reported to have good validity and reliability [19].
Primary outcome
The primary outcome is the change from baseline in the MADRS score following the 8-week treatment period.
Secondary outcomes
The following secondary efficacy endpoints will be evaluated:
-
1.
Clinical remission rate as assessed by the total MADRS score at the end of the study. Remission = at the end of the study, total MADRS score ≤ 10.
-
2.
Clinical remission rate as assessed by the total HAMD-17 score at the end of the study. Remission = at the end of the study, total HAMD-17 score ≤ 7.
-
3.
Clinical response rate according to the total HAMD-17 score at the end of the study. Response = at the end of the study, decreased rate (from baseline) for total MADRS score or total HAMD-17 score ≥ 50%.
-
4.
Change in total MADRS score over time.
-
5.
Change in total HAMD-17 score from baseline.
-
6.
Change in Hamilton Anxiety Scale (HAMA) [20] total score from baseline.
-
7.
Change in Clinical Global Impression-Severity of Illness (CGI-S) [21] score from baseline.
-
8.
Improvement in Clinical Global Impression (CGI-I) [22] score at different visits.
-
9.
Change in Discriminative Scale Space Tracker (DSST) [23] total score from baseline.
-
10.
Change in Trail Making Test (TMT) A and B [24] total scores from baseline.
- 11.
-
12.
Proportion of patients withdrawing due to poor efficacy. The investigators will assess patient efficacy according to his/her clinical status using rating scales, including MADRS, HAMD-17, HAMA, and CGI (already listed as outcomes).
-
13.
Proportion of patients using combined medication to treat insomnia.
Safety evaluation
Every patient enrolled in the trial will be assessed for the occurrence of adverse events (AEs). The incidence rate of AEs will be measured as the main safety outcome. The following measures will be used to evaluate the safety of the study drug:
-
1.
Breath rate per minute, pulse rate per minute, heartbeat rate per minute, diastolic and systolic blood pressure, and sitting position (mmHg)
-
2.
Electrocardiogram (ECG) and the number of patients with an abnormal 12-lead ECG report
-
3.
Assessment of the Columbia-Suicide Severity Rating Scale
-
4.
Assessment of the Arizona Sexual Experience Scale
-
5.
Number of patients with AEs resulting in early withdrawal
-
6.
Number of patients with serious adverse events (SAEs) resulting in early withdrawal
-
7.
Number of emerging AEs during the drug withdrawal period
Sample size determination
The sample size estimation was based on the results for an analysis of covariance (ANCOVA) comparing changes in the MADRS score from baseline to the last assessment in the 8-week treatment of the study drug or placebo. Reference was made to the results from the phase IIb trial in which the decrease in the MADRS total score in the study drug group was 16.04 and 12.75 in the placebo group. Using a significance level of 5%, a power of 80%, a SD of 8.5, and a dropout rate of 20%, the sample size was estimated to be 133 per group.
Statistical analysis
Full analysis set for clinical effectiveness analysis and compliance evaluation, and safety analysis set for safety evaluation will be performed in the study. The statistical analysis software will be SAS version 9.4 or above. All statistical tests will be double-sided, and P-values less than or equal to 0.05 will be considered statistically significant. The statistical analysis shall be carried out according to the statistical analysis plan. Descriptive statistics for cases will be the percentages (%); descriptive statistics for measurement data will describe the number of cases, mean, standard deviation, median, minimum, and maximum. Change score outcomes will be adjusted for baseline values to prevent regression to the mean.
Primary efficacy analysis
The treatment effect will be evaluated using ANCOVA with the study drug group, site, depressive episode, and baseline MADRS total score as the explanatory variables. The 95% confidence intervals and P-values will be presented. Missing data will be handled using the multiple imputation approach.
Secondary efficacy analysis
Clinical remission rate as assessed using MADRS and HAMD-17 at the end of the study will be analyzed using ordinal logistic regression as the (proportional) odds ratio of Anyu Peibo versus placebo with a two-sided Wald 95% confidence interval. Treatment, baseline MADRS (total score under beyond or under 30) or HAMD-17 (higher or lower than the median), site, and depressive episode (single episode or relapse) will be included in the model.
The t-tests with a two-sided alpha level of 0.05 will be used to analyze the following variables: total MADRS scores, MADRS scores over time, CGI-I scores at different visits, and the changes in total scores from baseline for the HAMD-17, HAMA, CGI-S, DSST, TMT A and B, and SDS. The mean differences in the scores and the 95% confidence intervals for Anyu Peibo compared to placebo will be used as the estimate of the treatment effect.
The estimate of the difference in the proportions of patients withdrawing due to poor efficacy and the proportions of patients using combined medication to treat insomnia (Anyu Peibo versus placebo), 95% confidence intervals, and chi-square P-values will be calculated. In the cases of low cell frequencies (< 5), the Fisher exact test will be used instead.
Trial management, monitoring, and auditing
An independent DSMB is monitoring the quality of the trial and has access to trial outcomes and accumulated safety data, including adherence, SAEs, suspected unexpected serious adverse reactions, and mortality. In addition, the DSMB will review the safety data from a clinical and safety point of view on an ongoing basis.
The sponsor will perform monitoring visits as frequently as necessary. Representatives from the sponsor’s quality assurance department can visit the trial site at any time during the study to conduct an audit of the study in compliance with the regulatory guidance.
Discussion
Antidepressants are the first-line recommended treatment for MDD, which is a serious psychological disorder with a high disease burden in adults [27, 28]. However, concerns over the long-term balance of benefits and harms arising from the currently marketed antidepressants point to the need for new antidepressants with improved efficacy and safety.
Piper laetispicum C. DC. has been used in Chinese traditional medicine as a sedative and analgesic and to treat toothache and snake bites [29]. The n-hexane extracts from Laetispicum C. DC. have been tested for their anti-inflammatory activity against cyclooxygenase-1 and 5-lipoxygenase, showing evidence that extracts of these species act as in vitro inhibitors of both enzymes [30,31,32]. Piper alkamides have been reported to possess various properties, including antidepressant effects. Its anxiolytic and antidepressant effects have been explained by its capability to inhibit monoamine oxidase activity and increase the levels of serotonin and noradrenaline in some regions of the mouse brain [33, 34].
Anyu Peibo Capsule includes the extracted Piper alkamides (amide alkaloids), and the administration of Anyu Peibo Capsule has been estimated to improve the MDD clinical outcome with a favorable safety profile in phase IIa and IIb trials. This phase III trial aims to demonstrate a beneficial effect of Anyu Peibo Capsule on patients with MDD, while minimizing the side effects, in particular those relating to sexual dysfunction and gastrointestinal discomfort.
Limitation
The long-term outcomes of Anyu Peibo were not included in this trial. A substantially larger effect will be demonstrated in a superiority trial.
Trial status
The trial is currently recruiting and enrolling patients according to version 1.2 of the protocol (August 2019). Recruitment began on January 23, 2020, and the approximate date for completion of recruitment will be in December 2020.
Availability of data and materials
Not applicable.
References
WHO. Depression: fact sheet. Geneva: World Health Organization, 2017. http://www.who.int/mediacentre/factsheets/fs369/en/ (accessed June 06, 2020).
Huang Y, Wang Y, Wang H, Liu Z, Yu X, Yan J, et al. Prevalence of mental disorders in China: a cross-sectional epidemiological study. Lancet Psychiatry. 2019;3:211–24.
Wang Z, Ma X, Xiao C. Standardized treatment strategy for depressive disorder. Adv Exp Med Biol. 2019;1180:193–9. https://doi.org/10.1007/978-981-32-9271-0_10.
Feng Y, Xiao L, Wang W, Ungvari GS, Ng CH, Wang G, et al. Guidelines for the diagnosis and treatment of depressive disorders in China: the second edition. J Affect Disorders. 2019;253:352–6. https://doi.org/10.1016/j.jad.2019.04.104.
Daly EJ, Singh JB, Fedgchin M, Cooper K, Lim P, Shelton RC, Thase ME, Winokur A, Van Nueten L, Manji H, Drevets WC. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):139–48.
Melartin TK, Rytsälä HJ, Leskelä US, Lestelä-Mielonen PS, Sokero TP, Isometsä ET. Continuity is the main challenge in treating major depressive disorder in psychiatric care. J Clin Psychiatry. 2005;66(2):220–7. https://doi.org/10.4088/JCP.v66n0210.
Zhu Y, Wu Z, Sie O, Cai Y, Huang J, Liu H, et al. Causes of drug discontinuation in patients with major depressive disorder in China. Prog Neuropsychopharmacol Biol Psychiatry. 2020;109755.
Zheng X, Xing F. Ethnobotanical study on medicinal plants around Mt.Yinggeling, Hainan Island, China. J Ethnopharmacol. 2009;124(2):197–210. https://doi.org/10.1016/j.jep.2009.04.042.
Xie H, Yan M, Jin D, Liu J, Yu M, Dong D, et al. Studies on antidepressant and antinociceptive effects of ethyl acetate extract from Piper laetispicum and structure–activity relationship of its amide alkaloids. Fitoterapia. 2011;82(7):1086–92. https://doi.org/10.1016/j.fitote.2011.07.006.
Li Q, Qu F, Gao Y, Jiang Y, Rahman K, Lee K, et al. Piper sarmentosum Roxb. produces antidepressant-like effects in rodents, associated with activation of the CREB-BDNF-ERK signaling pathway and reversal of HPA axis hyperactivity. J Ethnopharmacol. 2017;199:9–19. https://doi.org/10.1016/j.jep.2017.01.037.
Lopes JJ, Marx C, Ingrassia R, Picada JN, Pereira P, Ferraz ADBF. Neurobehavioral and toxicological activities of two potentially CNS-acting medicinal plants of Piper genus. Exp Toxicol Pathol. 2012;64(1-2):9–14. https://doi.org/10.1016/j.etp.2010.05.012.
FDA. Major depressive disorder: developing drugs for treatment guidance for industry, 2018. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/major-depressive-disorder-developing-drugs-treatment (accessed July 14, 2021)
http://www.consort-statement.org/consort-statement/flow-diagram(accessed June 06, 2020)
Chan A-W, Tetzlaff JM, Gøtzsche PC, Altman DG, Mann H, Berlin J, et al. SPIRIT 2013 Explanation and Elaboration: guidance for protocols of clinical trials. BMJ. 2013;346(jan08 15):e7586. https://doi.org/10.1136/bmj.e7586.
Perlman K, Benrimoh D, Israel S, Rollins C, Brown E, Tunteng J, et al. A systematic meta-review of predictors of antidepressant treatment outcome in major depressive disorder. J Affect Disord. 2019;243:503–15. https://doi.org/10.1016/j.jad.2018.09.067.
Liu J, Xiang YT, Lei H, Fang Y, Fang M, Li J, et al. Guidance on the conversion of the Chinese versions of the Quick Inventory of Depressive Symptomatology-Self-Report (C-QIDS-SR) and the Montgomery-Asberg Scale (C-MADRS) in Chinese patients with major depression. J Affect Disord. 2014;152-154:530-533.
Zhong BL, Wang Y, Chen HH, Wang XH. Reliability, validity and sensitivity of Montgomery-Asberg depression rating scale for patients with current major depression disorder. Chinese Journal of Behavioral Medicine and Brain Science. 2011;20(1):85–7. https://doi.org/10.3760/cma.j.issn.1674-6554.2011.01.032.
Sun XY, Li YX, Yu CQ, Li LM. Reliability and validity of depression scales of Chinese version: a systematic review. Chinese Journal of Epidemiology. 2017;38(1):110–6. https://doi.org/10.3760/cma.j.issn.0254-6450.2017.01.021.
Leu SH, Chou JY, Lee PC, Cheng HC, Shao WC, Hsien WL, et al. Validity and reliability of the Chinese version of the Sheehan Disability Scale (SDS-C). Asia-Pacific Psychiatry. 2015;7(2):215–22. https://doi.org/10.1111/appy.12182.
Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol. 1959;32(1):50–5. https://doi.org/10.1111/j.2044-8341.1959.tb00467.x.
Turkoz I, Fu DJ, Bossie CA, Sheehan JJ, Alphs L. Relationship between the clinical global impression of severity for schizoaffective disorder scale and established mood scales for mania and depression. J Affect Disord. 2013;150(1):17–22. https://doi.org/10.1016/j.jad.2013.01.047.
Mohebbi M, Dodd S, Dean OM, Berk M. Patient centric measures for a patient centric era: agreement and convergent between ratings on the Patient Global Impression of Improvement (PGI-I) scale and the Clinical Global Impressions - Improvement (CGI-S) scale in bipolar and major depressive disorder. Eur Psychiatry. 2018;53:17–22.
Danelljan M, Hager G, Khan FS, Felsberg M. Discriminative scale space tracking. IEEE Trans Pattern Anal Mach Intell. 2017;39(8):1561–75. https://doi.org/10.1109/TPAMI.2016.2609928.
Llinàs-Reglà J, Vilalta-Franch J, López-Pousa S, Calvó-Perxas L, Torrents Rodas D, Garre-Olmo J. The Trail Making Test. Assessment. 2017;24(2):183–96. https://doi.org/10.1177/1073191115602552.
Sheehan DV, Nakagome K, Asami Y, Pappadopulos EA, Boucher M. Restoring function in major depressive disorder: a systematic review. J Affect Disord. 2017;215:299–313. https://doi.org/10.1016/j.jad.2017.02.029.
Hobart M, Zhang P, Weiss C, Meehan SR, Eriksson H. Adjunctive brexpiprazole and functioning in major depressive disorder: a pooled analysis of six randomized studies using the Sheehan Disability Scale. Int J Neuropsychopharmacol. 2019;22(3):173–9. https://doi.org/10.1093/ijnp/pyy095.
Malhi GS, Mann JJ. Depression. Depression. Lancet. 2018;392(10161):2299–312. https://doi.org/10.1016/S0140-6736(18)31948-2.
GBD 2015 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1603–58. https://doi.org/10.1016/S0140-6736(16)31460-X.
Martins J. S B: Phytochemistry and pharmacology of anti-depressant medicinal plants: a review. Biomed Pharmacother. 2018;104:343–65. https://doi.org/10.1016/j.biopha.2018.05.044.
Ding D, Wang Y, Chen Y, Mei R, Yang J, Luo J, et al. Amides and neolignans from the aerial parts of Piper bonii. Phytochemistry. 2016;129:36–44. https://doi.org/10.1016/j.phytochem.2016.07.004.
Stöhr JR, Xiao P, Bauer R. Constituents of Chinese Piper species and their inhibitory activity on prostaglandin and leukotriene biosynthesis in vitro. J Ethnopharmacol. 2001;75:133–9. https://doi.org/10.1016/S0378-8741(00)00397-4.
Lopes JJ, Marx C, Ingrassia R, Picada JN, Pereira P, Ferraz ADBF. Neurobehavioral and toxicological activities of two potentially CNS-acting medicinal plants of Piper genus. Exp Toxicol Pathol. 2012;64:9–14. https://doi.org/10.1016/j.etp.2010.05.012.
Yao CY, Wang J, Dong D, Qian FG, Xie J, Pan SL. Laetispicine, an amide alkaloid from Piper laetispicum, presents antidepressant and antinociceptive effects in mice. Phytomedicine. 2009;16(9):823–9. https://doi.org/10.1016/j.phymed.2009.02.008.
Xie H, Yan M, Jin D, Liu J, Yu M, Dong D, Cai C, Pan S: Studies on antidepressant and antinociceptive effects of ethyl acetate extract from Piper laetispicum and structure–activity relationship of its amide alkaloids.
Acknowledgements
The authors would like to thank all the individuals involved in the protocol design and the members of the DSMB. Each author contributed substantially to this work.
Funding
Funding and drugs for this trial are provided by Su Zhou YiHua Biotechnology Co. Ltd. This trial is also supported by the China National Major Project for IND (2018ZX09734-005) and Shanghai Clinical Research Center for Mental Health (19MC1911100) in relation to the design of the study, collection of the data, and writing of the manuscript.
Author information
Authors and Affiliations
Contributions
JJH, YMY, and HFL developed the study concept and design and planned the analyses and manuscript preparation. HFL is the lead investigator. YJ and WC coordinated the funding and regulatory permissions. QSZ developed the statistical part of the protocol. JJH and HFL assisted in the further development of the protocol and manuscript preparation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The trial was approved by the Shanghai Mental Health Center Institutional Review Board (Central IRB, Approval No. 2019-39, No. 2019-39R1, IORG No. IORG0002202; FWA No. FWA00003065). All protocol amendments must be issued by the sponsor and signed by the investigator. Protocol amendments must not be implemented without IRB approval.
The procedures comply with the local ethical regulations, the institutional regulations, and the Declaration of Helsinki (2013) and its amendments. The procedures also comply with the currently approved international codes and norms regarding the Good Clinical Practice in Clinical Investigation.
Prior to enrollment, informed consent forms will be signed by each patient, as well as the investigator who provided the study information to the patient. Patients will be personally invited to participate when attending their regular visits to the outpatient clinic and will be informed about their freedom to withdraw consent at any time during the trial. The collection and processing of personal data from patients will be limited to those data that are necessary to fulfill the objectives of the study. These data will be collected with adequate precautions in place to ensure confidentiality and compliance with the applicable data privacy protection laws and regulations.
All information supplied by the sponsor to the investigator and the data generated as a result of this trial are considered confidential and remain the property of the sponsor. The investigators agree to maintain the data in confidence and will not use it for any other purpose than to accomplish this study without the sponsor’s consent.
Consent for publication
Patients will be asked to consent to the use of their anonymized data in scientific publications. An informed consent form template has been developed, and this has been approved by the Shanghai Mental Health Center Institutional Review Board.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, J., Yu, Y., Jiang, Y. et al. The efficacy and safety of Anyu Peibo Capsule in the treatment of patients with major depressive disorder in China: study protocol for a randomized placebo-controlled trial. Trials 22, 585 (2021). https://doi.org/10.1186/s13063-021-05550-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-021-05550-9