
Abstract
Background
The optimal analgesic strategy for surgical pain after lobectomy remains undefined. To compare the combination of flurbiprofen axetil and dezocine with flurbiprofen axetil alone and dezocine alone, in post-lobectomy patients.
Methods
A single-center, parallel-design double-blind superiority trial, with 5 groups (1:1:1:1:1 ratio) with different combinations of flurbiprofen and dezocine. Patients scheduled for lobectomy will be recruited. The primary outcome is total sufentanil use in patient-controlled intravenous analgesia within the first 24 postoperative hours. Secondary outcomes include pain numeric rating scales at 6th, 12th, 24th, 48th, and 72th postoperative hours, and on the 1st, 3rd, and 6th postoperative months at rest and during coughing, adverse effects from experimental drug treatment, sufentanil use at other time points, analgesia cost, time to chest tube removal, length of hospital stay, time to pass first flatus, and serum level of cytokines. Doctors, patients, and nurses are blinded, and only the manager is unblinded. Analysis is intention-to-treat. Statistical analysis is pre-specified. Statistical comparison of the treatment groups includes one-way analysis of variance followed by Tukey’s post hoc test.
Discussion
Trial did not begin to recruit. Participant recruitment start date is planned to be June 1, 2020. Approximate recruitment end date is May 31, 2021. If successful, the trial may shed light on the use of certain analgesic combinations in post-lobectomy pain control.
Trial registration
Chinese Clinical Trial Registry ChiCTR1800018563. Registered on September 25, 2018.
Similar content being viewed by others


Background
Postoperative pain is one of the most common complications after surgery [1]. As observed in the US population, 86% of patients experienced postoperative pain and 74% persisted after discharged [2]. Less than half of the patients reported adequate pain relief, which suggests that management is far from sufficient [3]. Specifically, postoperative pain adversely leads to insufficient coughing, immobility, and malnutrition among patients with lobectomy, which could result in prolonged functional recovery [4, 5].
Various perioperative interventions and management strategies have been developed to manage postoperative pain [6]. These include (1) patient-controlled intravenous analgesia (PCIA), which enables patient-oriented drug use [7]; (2) conventional intravenous injection [8]; (3) oral analgesic medications [9]; (4) epidural analgesia [10]; (5) paravertebral blockade [11]; (6) surgical wound infiltration [12]; (7) transdermal therapeutic system [13], etc. Opioids are among the mostly prescribed medications, whereas non-steroidal anti-inflammatory drugs (NSAIDs) are usually recommended for multimodal analgesic therapy [14, 15]. Over the past few years, the combined use of different analgesics for postoperative pain management has increasingly attracted the attention of surgeons and researchers [16]. In China, dezocine is one of the most widely used opioids for postoperative pain relief, far superseding use of morphine for pain control [17]. Wu et al. conducted a randomized controlled trial (RCT) recruiting 60 thoracotomy patients and found that the combination of dezocine and morphine had better analgesic effects and less adverse effects over using dezocine alone [18]. Flurbiprofen axetil is one of the NSAIDs applied in this clinical setting as well [19]. As Wu et al. demonstrated in patients with cancer, opioids incorporating with flurbiprofen axetil yielded better pain relief and reducing drug consumption in comparison to opioids alone [20]. Flurbiprofen axetil and dezocine have different analgesia mechanisms. The former inhibits prostaglandins release to reduce the sensitivity of local pain receptors, while the latter acts on the κ opioid receptor in the central nerve system [17, 19]. However, the benefit of this combined regimen was not tested in pain control after lobectomy. On the basis of our preliminary clinical observations, we proposed that a combination of flurbiprofen axetil and dezocine may elicit a synergistic antinociceptive effect, which allows for reduced drug intake of each constituent drug and ultimately less adverse effects. Thus, we conduct this RCT to identify the efficacy and safety of flurbiprofen axetil combined with dezocine on pain management after lobectomy.
Methods and analysis
Trial design
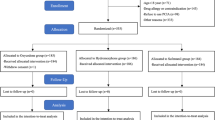
This is a prospective, randomized, double-blind, parallel-controlled, and superiority trial (Fig. 1), which follows the SPIRIT reporting guideline (see Additional file 1). The time schedule of enrolment, intervention, and follow-up were summarized in the SPIRIT figure (Fig. 2).

The flow chart of this study

Schedule of enrolment, interventions, and assessments
Study objective
The primary objective of this study is to compare the synergistic antinociceptive efficacy and the resultant decrease of adverse effects among different combinations of flurbiprofen axetil and dezocine in patients after lobectomy. The analgesia efficacy was assessed with the consumption of sufentanil in PCIA (evaluated at 6th, 12th, 24th, 48th, and 72th postoperative hours) and the score of numeric rating scales (evaluated at the 6th, 12th, 24th, 48th, and 72th postoperative hours and 1st, 3rd, and 6th postoperative months). The adverse effects were assessed by the incidence of treatment-related adverse effects.
Study setting
This study will be conducted on patients undergoing lobectomy in the Department of Thoracic Surgery, West China Hospital, Sichuan University. This institution is a public academic hospital located in the western China, which ranked at the 1st (in China) and the 25th (all over the world) in the Nature Index 2020 Tables in Healthcare Institutions [21].
Recruitment
Participants
Patients eligible for the trial must comply with all the inclusion criteria and do not meet any exclusion criterion before enrolment. To achieve adequate participant enrolment, all physicians in the thoracic department of the hospital are informed of the trial, and to contact the trial coordinator if they encounter potential eligible patients. All the included patients will sign an informed consent form, which should contain (1) detailed information of the study design, (2) benefits of participating, (3) risk of respiratory and circulatory depression, and (4) remedial action for patients who experienced any harms of the intervention. A trained individual researcher will obtain informed consent from the patient, in a separate setting from the trial coordinator.
Inclusion criteria
Patients who, at the time of enrolment, meet all the following criteria, are eligible for the study:
-
1.
Patients aged between 18 and 70
-
2.
Patients undergoing planned lobectomy
-
3.
ASA risk class of I–II
-
4.
Ability to sign an informed consent form
-
5.
Ability to understand all the study procedures and communicate clearly with the researchers and staff
Exclusion criteria
Patients who, at the time of enrolment, meet any of the following criteria are not eligible for the study:
-
1.
Body mass index (BMI) > 30 kg/m2
-
2.
Last smoked < 2 weeks prior to surgery for current smokers
-
3.
Pregnant or breastfeeding females (females aged 18 to 55 will receive pregnancy test)
-
4.
Hepatic (presence of ≥ 3 times the reference value of alanine aminotransferase/aspartate aminotransferase), renal (undergoing renal replacement therapy before surgery), or cardiac (the left ventricular ejection fraction < 30%) dysfunction [22, 23]
-
5.
History of gastrointestinal disease (e.g., peptic disease, intestinal perforation)
-
6.
Severe or unstable mental illness (presence of mental illness in International Classification of Diseases-10 diagnosis groups F2, F3, F4, F5, or F6) [24]
-
7.
Severe or unstable physical conditions in which the selective surgery would not be considered (e.g., cerebral stroke, cardiac ischemia, congestive heart failure, hepatic failure, grade 3 hypertension)
-
8.
Patient with known allergies to opioids or NSAIDs
-
9.
Current or recent (within 1 month prior to surgery) administration of opioids or NSAIDs.
-
10.
Intake of any analgesics within 48 h prior to surgery
-
11.
History of alcohol, opioids or other drugs abuse, or chronic use of opioids
-
12.
Patients with contradictions to the use of PCIA
-
13.
Preoperative pain diagnosis (e.g., chronic pain, cancer-causing pain in site of surgery)
-
14.
Emergency surgery
-
15.
Reoperations for severe postoperative complications (e.g., severe bleeding, bronchopleural fistula)
Drop-out criteria
Patients will be able to drop out at any time of this study under the following circumstance:
-
1.
After recruitment, the condition of the patient changes, and the surgeon in charge confirms that emergency surgery is needed.
-
2.
Patients can request a withdrawal to terminate the experimental treatment due to any personal reasons at any stage after recruitment.
-
3.
Any protocol violations occur.
Randomization and blinding
We will perform the randomization by assigning candidates to 5 groups (Fig. 1) based on computer-generated random numbers shortly prior to the surgery. In order to maintain allocation concealment, the randomly-generated group name will be printed and placed in consecutively numbered, separate sealed opaque envelopes which will be only opened once a patient is deemed eligible. When receiving a patient who meets the inclusion criteria, an unblinded trial manager will assign the participant to a group according to the number in the envelope.
All the drugs will be packed with the same outlook by a research pharmacist who is blind to the allocation of patients. The investigators, treating doctors and nurses, as well as the study participants are blinded to the allocation of the participants, while the project manager remains unblinded. In case of unexpected emergency circumstances, the randomization and allocation will be disclosed to the investigator. The blinding will be discontinued after all data analyses are completed. If unblinding occurs, the participants will discontinue the study medication and a complete report will be made.
Sample size
We estimated the sample size based on one-way analysis of variance (ANOVA) of total consumption of sufentanil in PCIA within the first 24 postoperative hours in group PD and group P0. The estimated consumptions of sufentanil in PCIA within the first 24 postoperative hours were 63 and 71 μg in group PD and group P0, respectively [25]. Standard deviations of each group were estimated as 1.15, and effect size of 0.67 was calculated [25]. The numbers of participants will allow a 5% chance of type I error and have 95% power. The sample size of 49 in each group was calculated by using G*Power (Software Version 3.1.3, University of Düsseldorf, Germany). Based on prior experience at our center, we set the dropout rate as 10% to account for the inability to complete the treatment, data errors, loss to follow-up, and other unanticipated study problems. The adjusted sample size for each group is 55. Thus, this study needs to recruit a total of 275 participants.
Intervention
A total of 275 subjects who are scheduled for lobectomy will be recruited in our study according to inclusion and exclusion criteria. All patients are equally allocated into 5 groups: Group P0: only PCIA, Group PF: PCIA + flurbiprofen axetil, Group PD: PCIA + dezocine, Group PFD1: PCIA + flurbiprofen axetil+ dezocine, and Group PFD2: PCIA + flurbiprofen axetil (half-dose) + dezocine (half-dose).
All the participants will receive electronic PCIA as the basic component of postoperative pain management once awake after anesthesia. The PCIA drug formulation is 1 μg/mL of sufentanil citrate (Yichang Humanwell Pharmaceutical Co., Ltd., Yichang, China), 15 mg of tropisetron hydrochloride (Jiangsu Hengrui Medicine Co., Ltd., Jiangsu, China), and 200 μg of dexmedetomidine (Taiji group·Southwest Pharmaceutical Co., Ltd., Chongqing, China) combined with normal saline into 200 mL solution. The PCIA drug will be administered as a single 2-mL dose at a 10-min locking time, with a maximum dose of 10 mL every hour and a background input quantity of 0.5 mL/h. Other than PCIA, all drugs are administered intravenously by nurses coordinated by the unblinded manager in the trial. None of the experimental drugs are taken orally, eliminating the need to monitor adherence. All the following study analgesics, including PCIA, are discontinued at 72 h after operation. If pain cannot be controlled after 72 postoperative hours, PCIA is continued as a “rescue” analgesic strategy to achieve a pain score of numeric rating scales (NRS) ≤ 5. An extra 5 mg of dezocine [Yangtze River Pharmaceutical (Group) Co., Ltd., Yichang, China] via intramuscular injection will be administered when intolerant pain happens. The dosage of added analgesics and administration timepoints will be recorded. If the above analgesic regimens cannot relieve pain, the patient will be withdrawn from the study and allowed additional analgesic regimens which are deemed appropriate, and a complete report will be made.
Group P0: only PCIA
Group PF: PCIA + flurbiprofen axetil
In addition to PCIA, patients will receive 50 mg flurbiprofen axetil via intravenous injection (Beijing Tide Pharmaceutical Co., Ltd., Beijing, China) when surgery begins as well as on the evening of the day of operation. From the postoperative day (POD) 1, the dosage will be given every 12 h.
Group PD: PCIA+ dezocine
In addition to PCIA, 5 mg dezocine (Yangtze River Pharmaceutical (Group) Co., Ltd., Yichang, China) will be intravenous injected when surgery begins as well as on the evening of the day of operation. From POD1, dosage will be given every 12 h.
Group PFD1: PCIA + flurbiprofen axetil + dezocine
In addition to PCIA, patients will receive 50 mg flurbiprofen axetil combined with 5 mg dezocine via intravenous injection when surgery begins as well as on the evening of the day of operation. From POD1, the dosage will be given every 12 h.
Group PFD2: PCIA + flurbiprofen axetil (half-dose) + dezocine (half-dose)
In addition to PCIA, patients will receive of 25 mg flurbiprofen axetil combined with 2.5 mg dezocine via intravenous injection when surgery begins as well as on the evening of the day of operation. From POD1, the dosage will be given every 12 h.
Anesthesia and operations
All operations will be performed under general anesthesia with double-lumen tracheal intubation. Five-lead electrocardiogram (ECG), pulse oxygen saturation (SpO2), bispectral index (BIS), end-tidal carbon dioxide tension (EtCO2), and blood pressure (BP) will be recorded during surgery. All surgeons participating in this study have a surgical experience with over 200 lung cancer cases annually. Patients will receive mechanical ventilation with a tidal volume (VT) of 6–8 mL/kg of predicted body weight and a positive end-expiratory pressure (PEEP) of 6–8 cm of water. The fluid infusion speed and volume will be controlled according to urine output, and blood transfusion will be given when necessary. Intraoperative explorations include the examination for pleural effusion, lung fissure, adhesion, tumor invasiveness, etc. The surgery type will be restricted to lobectomy and the choice of thoracotomy or thoracoscopic surgery will be made by the surgeon-in-charge. One 28F chest tube (Yangzhou Hanjiang Huafei Medical Device Factory, Co., Ltd., Hanzhou, China) will be placed in the thoracic cavity from the 7th intercostal space to drain the excessive air and fluid. All patients will be applied the same conventional water-sealed drainage bottle without negative pressure. During the surgery, if the surgeon decides that a patient is no longer a candidate for lobectomy, alternative therapy will be provided, and the patient will be withdrawn from the trial, and a complete report will be made.
Study dropouts
All enrolled participants have the right to withdraw from the study for any reason at any time without any negative consequence for current or future treatment. All investigators have the right to terminate the participation of any subject at any time if deemed to be in the participant’s best interest. All reasons and circumstances of subject termination or withdrawal will be recorded timely in the case report form (CRF). Any participants who withdraw from this study will continue to be monitored as a part of this protocol, and efficacy and safety data will continue to be recorded. All data will be analyzed according to the intention-to-treat (ITT) principle.
Patient and public involvement
Patients and public are not involved in the trial design. The final results of this trial will be available to all the patients via letters, phone calls as well as email communication.
Statistical analysis
The statistical analysis will follow the ITT principle and a data analysis plan will be confirmed prior to the data being locked for final analysis, including the statistical methods, description of the missing data, and further quality control to guarantee the robustness of the findings reported. The main contents of the analysis are for the effectiveness and safety of the treatment groups. The analysis of all continuous variables will be presented as mean, standard deviation, median, quartile spacing, and maximum and minimum values. The analysis of all dichotomous variables will be presented as rate, constituent ratio, and hazard ratio. Statistical comparisons among five groups will be mainly conducted with one-way analysis of variance followed by post hoc Tukey’s tests, described data [26]. Dichotomous data will be compared with chi-squared tests. Univariate and multivariate logistic regression analysis will also be applied. The factors (p < 0.05) in univariate analysis will be used for multivariate analysis. Only when overall difference is significant, two-to-two comparison will be considered. All the data will be checked twice by two independent researchers to guarantee facticity. If dropout rate is higher than the expected 10%, multiple imputation will be used to avoid pitfalls involved with listwise deletion of cases that have missing values [27]. We will perform explanatory subgroup analysis based on age, gender, comorbidities, surgery type, and so on. The difference between groups will be considered statistically significant if p < 0.05. No interim analyses are planned. Analysis will only be conducted after enrolment close. All data will be analyzed using SPSS (software version 25.0, Chicago, IL).
Study organization
Data collection and management
JZ and WC designed the patient-reported outcomes (PRO) content of the trial protocol, and are responsible for PRO contents. All patients’ demographic and clinical data will be recorded by investigators independently. Preoperative data will be collected within 3 days after recruitment. Surgery data will be collected within 2 days after the operation. Postoperative data will be collected within 3 days after discharge. For patients discharged home, we will retrieve follow-up information by phone calls, and these data will be recorded within 3 days. The organizational structure of the trial is as follows. The steering committee, which comprises JZ, LL, and administrators at the West China Hospital, has full oversight over the design of the trial. We have a data management safety committee (DMSC) comprised of 5 independent investigators. They will supervise the study protocol adherence and confirm that the CRF is correctly completed and consistent with the original data. If there are any errors or omissions, the investigator will correct them immediately. The raw data will be marked clearly when revising and signed by the investigator with date when the modifications are made. All the data can only be obtained by the study researchers who have signed the confidential disclosure agreement. We do not plan to collect personal information about potential and enrolled participants beyond what is collected during normal hospitalization. To maintain confidentiality, electronic health information will be encrypted as per hospital protocol. After the trial, personally identifiable information will be omitted and placed in a separate database before any data analysis will be performed. The adherence to the study protocol, data collection, statistical analysis, and related safety issues will be strictly monitored by the Institutional Ethics Committee of West China Hospital, Sichuan University.
Adverse effects
Drug adverse effects including nausea, vomiting, dizziness, fatigue, drowsiness, pruritus, abdominal distention, bloating, constipation, euphoria, respiratory depression, and indigestion will be recorded timely. Medical treatment will be applied as necessary. Unexpected severe drug adverse effects will be reported to the ethics committee.
Primary and secondary outcomes
The primary outcome is total consumption of sufentanil in PCIA within the first 24 postoperative hours. PCIA sufentanil consumption will be measured automatically by the PCIA pump, and recorded by nurses trained for this trial. The secondary outcomes include (1) the NRS at the 6th, 12th, 24th, 48th, and 72th postoperative hours, and on the 1st, 3rd, and 6th postoperative months at rest and during coughing; data collection timepoints are pre-specified. The NRS, ranging from 0 (no pain) to 10 (worst pain imaginable), will be utilized to assess the severity of postoperative pain for all participants. The NRS is a patient-reported subjective scale, with evidence of its use and interpretation shown elsewhere [28]. Only the Chinese language version of the NRS question will be used. To minimize avoidable missing data, nurses are required to key in the results into an electronic chart directly after the patient encounter. NRS will be collected by an oral survey administrated by trained nurses to ask in a standardized manner, in the setting of the hospital ward, until discharge. At the 1st, 3rd, and 6th post-operative month, NRS will be asked with phone-call by the follow-up team; (2) the total consumption of sufentanil in PCIA within the first 6, 12, 48, 72 postoperative hours, and the total consumption of sufentanil in “rescue” PCIA; (3) incidence of treatment-related adverse effects: nausea, vomiting, dizziness, fatigue, drowsiness, pruritus, abdominal distention, bloating, constipation, euphoria, respiratory depression, and indigestion, and any adverse events observed by a nurse will also be assessed by an on-site investigator; (4) the cost of analgesia, time to chest tube removal, length of hospital stay, time to pass first flatus; and (5) the serum level of interleukin-1β (IL-1β), IL-6, IL-8, IL-2, tumor necrosis factor-α (TNF-α), C-reactive protein (CRP), prostaglandin E2 (PGE2), and 5-hydroxytryptamine (5-HT). Collection of blood samples for cytokine studies would be performed by trained nurses. The laboratory evaluation would be conducted by technicians in the Department of Laboratory Medicine, blinded to treatment groups. Laboratory results will be placed in the electronic chart. Specimens will be destroyed and not stored for ancillary studies.
Adverse event
In this study, the adverse event is defined as any untoward treatment-related medical occurrence, including any treatment-related unfavorable and unintended sign, symptom, or disease. Within 30 days after the last treatment of this protocol, any newly occurring events or previous conditions (level of postoperative pain) increased in intensity or frequency would be considered as the adverse event. Lack of efficacy before the study end would not be considered an adverse event. The adverse event would be evaluated and reported to the principal investigator according to Common Terminology Criteria for Adverse Events (v4.03) [29].
Auditing
The ethics committee will conduct local monitoring of trial quality after the first patients have been enrolled. The trial database provides on-going data quality checking, including the recruitment rate, data quality, and adverse event reporting.
Discussion
Sufficient postoperative pain control is essential to surgical outcomes and postoperative recovery [30]. Dezocine and flurbiprofen axetil, representatives of opioids and NSAIDs, respectively, are important components of perioperative pain management [20, 31, 32], and the combined application of the two differentially acting drugs is becoming widely accepted. Our center generally adopted a combination of PCIA sufentanil with intravenous flurbiprofen or parecoxib sodium. Oral opioids are only used when needed. Adverse effects of dezocine, including nausea, vomiting, respiratory depression, and opioid withdrawal, are frequently reported and can be life-threatening in severe cases. Likewise, flurbiprofen axetil use can be complicated by nausea, vomiting, diarrhea, and perforation of gastrointestinal tracts. Nevertheless, our study indicated a minimization of adverse effects with maximization of analgesic effects by incorporating flurbiprofen axetil to dezocine. A paucity of data investigating the synergistic antinociceptive effect and the resultant decrease of adverse effects left the optimal regimen undefined. To fill this gap of knowledge, we will conduct this clinical trial to find the optimal analgesic regimen for the pain management after lobectomy. This study will investigate the pain score-NRS, the incidence of drug-related adverse effects, and the precise total consumption of sufentanil in PCIA.
Several limitations of this trial should be noted. First, the characteristics of single-center trial, along with the limited availability of dezocine, will limit the generalizability of the study. A multiple-center large-sample clinical trial may be warranted in the future. Second, a diverse group of participants may be enrolled which could potentially affect the final results. Third, individual threshold of pain perception and the duration of surgery demanded by individual conditions may have an impact on the severity of postoperative pain. Thus, we should cautiously interpret our results.
To our knowledge, this study will be the first RCT on the analgesic effects and drug-related adverse events of combined dezocine and flurbiprofen axetil after lobectomy. This study may contribute to decision-making when an individualized pain control is requested and achieve the optimal regimen with the least complications and the most satisfactory.
Trial status
Enrolment is not begun. Participant recruitment start date is planned to be June 1, 2020. The approximate recruitment end date is May 31, 2021. The protocol version is Version 2.0, dated August 24, 2018.
Availability of data and materials
Results of this study are going to be published in a peer-reviewed journal. There are plans to publish the full statistical code in the final manuscript of the RCT after the trial has ended. There are no concrete plans to publish a participant-level dataset at this time.
Abbreviations
- PCIA:
-
Patient-controlled intravenous analgesia
- NRS:
-
Numeric rating scales
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- RCT:
-
Randomized controlled trial
- ECG:
-
Electrocardiogram
- SpO2:
-
Pulse oxygen saturation
- BIS:
-
Bispectral index
- EtCO2:
-
End-tidal carbon dioxide tension
- BP:
-
Blood pressure
- VT:
-
Tidal volume
- PEEP:
-
Positive end-expiratory pressure
- CRF:
-
Case report form
- ITT:
-
Intention-to-treat
- PRO:
-
Patient-reported outcomes
- DMSC:
-
Data management safety committee
- IL-:
-
Interleukin-
- TNF-α:
-
tumor necrosis factor-α
- CRP:
-
C-reactive protein
- PGE2:
-
Prostaglandin E2
- 5-HT:
-
5-hydroxytryptamine
References
Alzahrani T. Pain relief following thoracic surgical procedures: a literature review of the uncommon techniques. Saudi J Anaesth. 2017;11(3):327–31.
Gan TJ, Habib AS, Miller TE, White W, Apfelbaum JL. Incidence, patient satisfaction, and perceptions of post-surgical pain: results from a US national survey. Curr Med Res Opin. 2014;30(1):149–60.
Apfelbaum JL, Chen C, Mehta SS, Gan TJ. Postoperative pain experience: results from a national survey suggest postoperative pain continues to be undermanaged. Anesthesia Analgesia. 2003;97(2):534–40 table of contents.
Ried M, Schilling C, Potzger T, Ittner KP, Rupp A, Szoke T, et al. Prospective, comparative study of the On-Q(R) PainBuster(R) postoperative pain relief system and thoracic epidural analgesia after thoracic surgery. J Cardiothorac Vasc Anesth. 2014;28(4):973–8.
Offner KA, Loop T. Pathophysiology and epidemiology of pain in thoracic surgery. Anasthesiol Intensivmed Notfallmed Schmerzther. 2012;47(9):568–75.
Rawal N. Current issues in postoperative pain management. Eur J Anaesthesiol. 2016;33(3):160–71.
Zhou Y, Huang JX, Lu XH, Zhang YF, Zhang W. Patient-controlled intravenous analgesia for non-small cell lung cancer patient after thoracotomy. J Cancer Res Ther. 2015;11(Suppl 1):C128–30.
Esme H, Apiliogullari B, Duran FM, Yoldas B, Bekci TT. Comparison between intermittent intravenous analgesia and intermittent paravertebral subpleural analgesia for pain relief after thoracotomy. Eur J Cardio Thoracic Surg. 2012;41(1):10–3.
Bottiger BA, Esper SA, Stafford-Smith M. Pain management strategies for thoracotomy and thoracic pain syndromes. Semin Cardiothorac Vasc Anesth. 2014;18(1):45–56.
Baidya DK, Khanna P, Maitra S. Analgesic efficacy and safety of thoracic paravertebral and epidural analgesia for thoracic surgery: a systematic review and meta-analysis. Interact Cardiovasc Thorac Surg. 2014;18(5):626–35.
Liu F, Zhang J, Zhang HK, Zhao YQ, Liang P, Zuo YX. Thoracic paravertebral block in the PACU for immediate postoperative pain relief after video-assisted thoracoscopic surgery. Zhonghua Yi Xue Za Zhi. 2017;97(2):119–22.
Zhang X, Shu L, Lin C, Yang P, Zhou Y, Wang Q, et al. Comparison between intraoperative two-space injection thoracic paravertebral block and wound infiltration as a component of multimodal analgesia for postoperative pain management after video-assisted thoracoscopic lobectomy: a randomized controlled trial. J Cardiothorac Vasc Anesth. 2015;29(6):1550–6.
Osipova NA, Petrova VV, Lastukhin AV, Kudriavtsev SB, Vashakmadze LA, Khomiakov VM. Prevention and treatment of postoperative pain syndrome in extensive thoracoabdominal oncological surgery. Anesteziol Reanimatol. 2010;3:29–33.
Lovich-Sapola J, Smith CE, Brandt CP. Postoperative pain control. Surg Clin North Am. 2015;95(2):301–18.
Xiao X, Zhang Q, Ouyang Z, Guo X. Comparison of perioperative flurbiprofen axetil or celecoxib administration for pain management after total-knee arthroplasty: a retrospective study. Medicine. 2018;97(37):e12391.
Elmore B, Nguyen V, Blank R, Yount K, Lau C. Pain management following thoracic surgery. Thorac Surg Clin. 2015;25(4):393–409.
Wang YX, Mao XF, Li TF, Gong N, Zhang MZ. Dezocine exhibits antihypersensitivity activities in neuropathy through spinal mu-opioid receptor activation and norepinephrine reuptake inhibition. Sci Rep. 2017;7:43137.
Wu L, Dong YP, Sun L, Sun L. Low concentration of dezocine in combination with morphine enhance the postoperative analgesia for thoracotomy. J Cardiothorac Vasc Anesth. 2015;29(4):950–4.
Yin Y, Yi Y, Yu J, Sun X, Liu C, Xu F. Effects of flurbiprofen on serum level of interleukin-6, prostacyclin and corticosteroid A2 in patients with bone metastases of cancer. Oncol Lett. 2018;15(2):1545–8.
Wu TT, Wang ZG, Ou WL, Wang J, Yao GQ, Yang B, et al. Intravenous flurbiprofen axetil enhances analgesic effect of opioids in patients with refractory cancer pain by increasing plasma beta-endorphin. Asian Pacific J Cancer Prev. 2014;15(24):10855–60.
Nature S. Nature Index 2020 Annual Tables-Top healthcare institutions 2020 [Available from: https://www.natureindex.com/annual-tables/2020/institution/healthcare/all. Accessed 20 Nov 2020.
de Leeuw TG, Mangiarini L, Lundin R, Kaguelidou F, van der Zanden T, Pasqua OD, et al. Gabapentin as add-on to morphine for severe neuropathic or mixed pain in children from age 3 months to 18 years - evaluation of the safety, pharmacokinetics, and efficacy of a new gabapentin liquid formulation: study protocol for a randomized controlled trial. Trials. 2019;20(1):49.
Ni C, Masters J, Zhu L, Yu W, Jiao Y, Yang Y, et al. Study design of the DAS-OLT trial: a randomized controlled trial to evaluate the impact of dexmedetomidine on early allograft dysfunction following liver transplantation. Trials. 2020;21(1):582.
Mueller-Stierlin AS, Meixner F, Kohlmann A, Schumacher M, Hansel A, Pouwels M, et al. Effectiveness and cost-effectiveness of a community-based mental health care programme (GBV) for people with severe mental illness in Germany: study protocol for a randomised controlled trial. Trials. 2020;21(1):598.
Zhou L, Zhang Y, Sun H, Hu R, Wang J, Xu G. Effect of preemptive dezocine before general anesthesia on postoperative analgesia in patients undergoing laparoscopic cholecystectomy: a prospective observational study. Medicine. 2018;97(39):e12533.
Liu Z, Lu H, He G, Ma H, Wang J. Non-steroidal anti-inflammatory drugs reduce the stress response during sevoflurane anesthesia. Acta Anaesthesiol Scand. 2012;56(7):890–5.
Sterne JA, White IR, Carlin JB, Spratt M, Royston P, Kenward MG, et al. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ. 2009;338:b2393.
Farrar JT, Portenoy RK, Berlin JA, Kinman JL, Strom BL. Defining the clinically important difference in pain outcome measures. Pain. 2000;88(3):287–94.
Institute NC. Common Terminology Criteria for Adverse Events (CTCAE) v4.03 2010 [Available from: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm. Accessed 20 Nov 2020.
Chou R, Gordon DB, de Leon-Casasola OA, Rosenberg JM, Bickler S, Brennan T, et al. Management of postoperative pain: a clinical practice guideline from the American Pain Society, the American Society of Regional Anesthesia and Pain Medicine, and the American Society of Anesthesiologists’ Committee on Regional Anesthesia, Executive Committee, and Administrative Council. J Pain. 2016;17(2):131–57.
Zhou X, Zhang C, Wang M, Yu L, Yan M. Dezocine for preventing postoperative pain: a meta-analysis of randomized controlled trials. PLoS One. 2015;10(8):e0136091.
Zhang L, Zhu J, Xu L, Zhang X, Wang H, Luo Z, et al. Efficacy and safety of flurbiprofen axetil in the prevention of pain on propofol injection: a systematic review and meta-analysis. Med Sci Monit. 2014;20:995–1002.
Acknowledgements
Not applicable.
Funding
This work was supported by Key Science and Technology Program of Sichuan Province, China (2016FZ0118) (to Dr. Lunxu Liu), 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYGD18021, ZYJC18009) (to Dr. Lunxu Liu), and 1.3.5 Project for Disciplines of Excellence-Clinical Research Incubation Project, West China Hospital, Sichuan University (2019HXFH060) (to Dr. Qiang Pu). The funder played no role in the study design or drafting of the protocol and will have no authority over the collection, management, analysis, or interpretation of data.
Author information
Authors and Affiliations
Contributions
To be eligible for authorship, individuals must have critically contributed to the design and/or execution of the study, have oversight of the study, and are responsible for successive revisions of the manuscript. Professional writers will not be employed for the preparation of the manuscript. LL and JZ conceived of the study, finished its design and coordination. JZ, QP, LL, WC, and LL conducted reference collection and developed the protocol. JZ and BC drafted the manuscript. WC and YH provided critical changes to successive versions of the manuscript. All authors read and approved the final manuscript. JZ and WC designed the patient-reported outcomes (PRO) content of the trial protocol, and are responsible for PRO contents. The allocation sequence will be generated by JZ. The trial coordinator, QP will enroll participants. The unblinded trial manager, WC will assign participants to interventions. JZ, QP, and LL will participate in statistics analysis.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval has been obtained from the Institutional Ethics Committee for Clinical Research of West China Hospital, Sichuan University (NO. 2018-289). Only participants who have provided informed consent in writing will be recruited.
The protocol is made available in the Chinese CTR database. Important protocol modifications, such as changes to eligibility criteria or measured outcomes, will be reported to investigators and ethics committees before the change is implemented. If approved, the protocol changes will then be communicated to trial registries, journals, and external regulators.
Consent for publication
Not applicable.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
SPIRIT 2013 checklist: recommended items to address in a clinical trial protocol and related documents.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, J., Pu, Q., Lin, L. et al. Effect of patient-controlled intravenous analgesia combined with flurbiprofen axetil and dezocine on postoperative analgesia for lobectomy (EPIC-FAD): a trial protocol. Trials 22, 175 (2021). https://doi.org/10.1186/s13063-021-05108-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-021-05108-9