
Abstract
Background
Arterial hypertension is the most prevalent risk factor for cardiovascular disease in sub-Saharan Africa. Only a few and mostly small randomized trials have studied antihypertensive treatments in people of African descent living in sub-Saharan Africa.
Methods
In this open-label, three-arm, parallel randomized controlled trial conducted at two rural hospitals in Lesotho and Tanzania, we compare the efficacy and cost-effectiveness of three antihypertensive treatment strategies among participants aged ≥ 18 years. The study includes patients with untreated uncomplicated arterial hypertension diagnosed by a standardized office blood pressure ≥ 140/90 mmHg. The trial encompasses a superiority comparison between a triple low-dose antihypertensive drug combination versus the current standard of care (monotherapy followed by dual treatment), as well as a non-inferiority comparison for a dual drug combination versus standard of care with optional dose titration after 4 and 8 weeks for participants not reaching the target blood pressure. The sample size is 1268 participants with parallel allocation and a randomization ratio of 2:1:2 for the dual, triple and control arms, respectively. The primary endpoint is the proportion of participants reaching a target blood pressure at 12 weeks of ≤ 130/80 mmHg and ≤ 140/90 mmHg among those aged < 65 years and ≥ 65 years, respectively. Clinical manifestations of end-organ damage and cost-effectiveness at 6 months are secondary endpoints.
Discussion
This trial will help to identify the most effective and cost-effective treatment strategies for uncomplicated arterial hypertension among people of African descent living in rural sub-Saharan Africa and inform future clinical guidelines on antihypertensive management in the region.
Trial registration
Clinicaltrials.govNCT04129840. Registered on 17 October 2019 (https://www.clinicaltrials.gov/).
Similar content being viewed by others

Background
Cardiovascular morbidity and mortality in low- and middle-income countries—particularly sub-Saharan Africa—are rising [1, 2]. The most important risk factor for cardiovascular disease in sub-Saharan Africa is arterial hypertension with a prevalence of 30–46% [3,4,5,6,7,8,9] and an age-standardized mean systolic blood pressure (BP) being 5–20 mmHg higher compared to North America or Europe [1]. Black ethnicity has been associated with elevated BP [10] due to genetic factors, epigenetic adaptation to climate [11, 12], and increased susceptibility to salt intake [13, 14]. Moreover, complications of arterial hypertension such as stroke, chronic kidney disease, and myocardial infarction have shown to be more prevalent in black compared to white populations [15]. Despite the high burden of arterial hypertension in sub-Saharan Africa, less than 40% of hypertensive patients are aware of their diagnosis. Among those who are aware of their diagnosis, less than 30% are receiving antihypertensive medications and less than 20% of those being treated have a controlled BP [6, 16].
Most patients need a combination of at least two antihypertensive drugs to achieve BP control [17,18,19]. The latest American and European guidelines recommend starting a combination pharmacologic treatment with at least two classes of antihypertensive medications for patients with a BP ≥ 140/90 mmHg [20]. However, the World Health Organization (WHO) guidelines still recommend a sequential treatment approach starting with a calcium channel blocker (CCB) or a thiazide diuretic (TZD), and combining both drugs only in case of inadequate response [21,22,23]. From sub-Saharan Africa, there is very little evidence supporting the WHO approach: Only five, mostly small randomized trials comparing the effectiveness of different antihypertensive regimens were conducted in sub-Saharan Africa [24,25,26,27,28]. A recent trial performed in ten centers in six African countries found amlodipine-containing regimens with either hydrochlorothiazide or perindopril to be superior to perindopril plus hydrochlorothiazide in controlling BP at 6 months [29].
The control Arterial Hypertension in sub-Saharan Africa (coArtHA) trial aims at comparing three treatment strategies to achieve rapid BP control with widely available drugs within 12 weeks in participants of African descent in rural sub-Saharan Africa. In addition, it assesses hypertension-mediated organ damage and compares the cost-effectiveness of the three treatment strategies considered.
Methods
Study setting
The coArtHA trial is conducted at the St. Francis Referral Hospital in Ifakara, Southwestern Tanzania, and Mokhotlong District Hospital, Mokhotlong town, Northern Lesotho. In Tanzania, the STEP survey 2013 showed a prevalence of arterial hypertension of 25.9% in individuals aged 24–65 years of age [30]. At the Chronic Diseases Clinic of Ifakara (CDCI) of the St. Francis Referral Hospital, participants of the Kilombero and Ulanga Antiretroviral Cohort (KIULARCO) [31, 32] were hypertensive at enrolment in 12% [33]. Among HIV-positive patients on stable ART prevalence of arterial hypertension was even higher with 27% overall and 44% among patients aged ≥ 50 years [34]. The CDCI cares for about 4500 patients with an HIV infection, while the general outpatient department sees 36,000 patients a year [35].
In Lesotho, prevalence of arterial hypertension in the general population is around 31% among persons aged 25 to 64 years [36] and 28% and 22% among HIV-positive females and males, respectively [37]. Mokhotlong Hospital serves the district of Mokhotlong, which is situated in northeast of Lesotho and has about 120,000 habitants, the majority living in remote villages scattered over a mountainous area of 4075 km2. The hospital has 110 beds and its outpatient clinic serves 4500–7500 adult patients per month.
Study design
The coArtHA trial is an investigator-initiated, open-label, three-arm randomized controlled two-country trial to compare the effectiveness and cost-effectiveness of three antihypertensive treatment strategies in HIV-positive and HIV-negative participants with uncomplicated arterial hypertension in rural Tanzania and Lesotho.
The trial is designed for a superiority comparison between the triple drug combination regimen versus control, and a non-inferiority comparison between the dual drug combination regimen versus control. Allocation is 2:1:2 for the dual combination, triple combination, and control arms, respectively, with parallel assignment.
Control and intervention arms
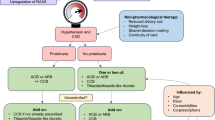
Treatment strategies are shown in Fig. 1. The control arm follows standard of care, i.e., national guidelines of Lesotho and Tanzania, which recommend a CCB or a TZD as first line, and if insufficient, both drugs are combined [10, 20, 38, 39]. For this trial, participants in the control arm start treatment with amlodipine 10 mg. Participants randomized to the dual arm receive a combination of half-dose amlodipine (5 mg) and losartan (50 mg). Participants randomized to the triple arm receive a combination of quarter-dose amlodipine (2.5 mg), hydrochlorothiazide (6.25 mg), and losartan (12.5 mg). The choice of amlodipine, losartan, and hydrochlorothiazide is based on their broad availability and low cost. All of the three drugs are part of the essential drug list by the WHO [40]. Participants in all three arms follow a prespecified dose titration after 4 and 8 weeks if target BP values are not met (Fig. 1).

Study interventions and drug dosing according to study arm. OD, once daily. $ Increases in dosages only if target BP is not reached (see above). * In case of orthostatic hypotension or adverse events, medication will be reduced to the prior step—or to half of the initial dosage. ** if regimen shows insufficient effect, individualized adaptation possible according the treating physician
Study procedures
Screening and eligibility criteria
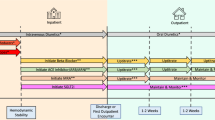
During routine care at the HIV clinic or the outpatient department, a BP measurement is done, which serves as pre-screening for the study. Individuals with a pre-screening BP ≥ 140/90 mmHg are referred to the study nurse. The study nurse informs the individual about the study, obtains written informed consent, and checks eligibility criteria (Table 1). Screening of participants is a stepwise procedure starting with a questionnaire to ensure absence of acute disease, followed by a standardized office BP measurement (see below). A urine pregnancy test is performed in all women of childbearing age (18–45 years) to exclude pregnancy. From a fingerpick blood sample, an HIV test is done if the participant is not known positive or has not been tested during the last 3 months with a documented result. A point of care creatinine is done to exclude severe renal impairment (creatinine clearance < 30 ml/min) (Fig. 2).

Study visit schedule. SV, screening visit; ER, enrolment; FUP, follow-up; EOS, end of study; ALT, alanine aminotransferase; Alb/Crea, albumin/creatinine; HIV, human immunodeficiency virus. *Day 0 (screening) and day 1 (enrolment) are the same day for participants not enrolled in the 24-h ambulatory BP study, £for all women of reproductive age (18–45 years), ** in 100 participants from Ifakara (nested study)
Enrolment and randomization
Immediately after screening, enrolment is done with a detailed history and a clinical exam. All information is entered into an electronic questionnaire (MACRO®, Elsevier). By venipuncture 5 ml of blood is withdrawn and sent to the laboratory for full-blood count, serum creatinine, and alanine aminotransferase. Urine is analyzed for albumin-creatinine ratio. A 12-lead-Electrocardiogram (ECG), a focused echocardiography using Lumify® device (Philips), and a retinal picture (iExaminer®, Welch-Allyn) are performed (Fig. 2). The results of these analyses are stored electronically for later interpretation by a cardiologist and ophthalmologist.
Randomization is stratified by site (Lesotho, Tanzania), HIV status (negative, positive), and age (< 65/≥ 65 years), using permuted blocks with varying block sizes. The randomization list was prepared in advance by an independent statistician and is stored securely on a server with restricted access. The allocation is concealed by using opaque, sealed, and labeled envelopes prepared by independent persons based on the randomization list. The envelopes are labeled on the outside with the stratification information and a sequential identification number and contain the randomized allocation and subject identification number. The first five randomizations in each stratum are checked in real-time, and subsequent regular checks are performed to ensure that the randomization sequence is respected. The nurse opens the envelope according the stratification, and the study physician fills in an electronic drug prescription according to the arm. The nurse dispenses the drugs accordingly and provides pre-packed and labeled medication for 1 month to the patient. Handing out of study drugs goes along with clear instructions on intake, adherence, and appointment for the next follow-up visit.
Follow-up clinic visit procedures
Follow-up visits are scheduled at 4, 8, 12, and 24 weeks after enrolment. During these visits, the study nurse evaluates adherence to the study drugs, asks for symptoms relating to side effects and other adverse events, and performs standardized BP measurements. In women of childbearing age, a pregnancy test is repeated at every visit. The study doctor examines the participant and prescribes study drugs according to the treatment arm (Fig. 1). Participants who reach the target BP and do not report side effects are prescribed the same medication at weeks 4, 8, and 12. In participants, who do not reach the target BP, the drug prescription is adapted by dosage increase or addition of other drugs as per protocol (Fig. 1). Additional visits can be scheduled if clinically indicated. Participants missing their appointment are tracked within a week of the missed scheduled appointment—first by a phone call, and if the participant is not reachable by tracking with the help of community health workers or a person blinded to the allocation going to the participant’s house.
On the last follow-up visit at 24 weeks, participants undergo again examinations to quantify surrogate markers of end-organ damage (Fig. 2). After successful completion of the study, participants are referred to the local medical team for continued management including further prescription of drugs. Participants do not receive any payment to be part of the study besides compensation for transport expenses caused by additional clinic visits.
Standardized blood pressure measurement
For the determination of BP, we use a standard operating procedure based on the European Society of Cardiology/European Society of Hypertension (ESC/ESH) guidelines 2018 [41], which has been used in several recent clinical trials and epidemiological studies [42,43,44]. In brief, arm circumference is measured to determine the cuff size according to the recommendations of the BP device manufacturer (Omron M6 Comfort [HEM-7321-E] [45]. BP measures are taken in sitting position after 5 min of rest with feet on floor; back supported; no caffeine, exercise, or smoking in the 30 min before measurement; emptied bladder; no talking during measurement; comfortable clothes; and arms supported (e.g., on table). At the screening visit, the reference arm is determined by measuring BP on both arms. The reference arm (with higher BP) is noted and used for all further BP measurements. The BP is calculated as the mean value of the last two out of three consecutive measurements, spaced 1–2 min apart.
Endpoints
The primary endpoint is the proportion of participants reaching target BP (≤ 130/80 mmHg in participants aged < 65 years and ≤ 140/90 mmHg in participants aged ≥ 65 years) at 12 weeks. We chose this target BP in line with updated European guidelines and the documented beneficial effect on cardiovascular outcomes [41]. The secondary endpoints are defined in Table 2.
Sample size calculation
We hypothesize that the proportion of participants reaching the primary endpoint will be higher in the triple combination arm compared to the control arm. Additionally, we hypothesize that the dual combination arm will be non-inferior to the control arm (Table 3). We assumed a response rate in the control arm of 40%, an improvement in the triple combination arm of 15 percentage points (two-sided alpha of 0.05) for the superiority comparison between the triple combination and control arms, and a non-inferiority margin of 10% (one-sided alpha of 0.025) for the non-inferiority comparison between the dual combination and control arms. Based on these assumptions, we calculated a sample size of 431 participants in each of the control and dual combination arms, and 216 participants in the triple combination arm (power of 85% for the non-inferiority comparison and 95% for the superiority comparison). The overall sample size is therefore 1078 participants, with the randomization ratio of 2:1:2 for the dual combination, triple combination, and control arms, respectively. Assuming 15% of participants will become lost-to-follow-up [29] brings the total required sample size to 1268 individuals.
Data collection and management
Baseline information containing demographics and clinical evaluation are filled into a standardized electronic data management system (MACRO®, Elsevier) using password-protected laptops. Participants are assigned a unique identifier at screening and randomization which is used on all study documentation.
Data are checked by the principal investigator and the data manager to ensure complete and accurate data, with queries raised within the electronic data capture system to clarify inconsistencies and missing data. At each site, a master list linking the participant’s unique identifier to the participant’s details such as name is kept in a locked cupboard. Data will be stored in Swiss Tropical and Public Health Institute (Swiss TPH) servers which are located in Basel, Switzerland, with a defined policy in place for server set-up, maintenance, and security. Data are kept in compliance with local legal requirements, for a minimum of 10 years after completion of the study.
Analyses
Analyses and reporting will follow CONSORT guidelines [54,55,56] and intention-to-treat (ITT) principles, that is including participants as randomized. A flowchart will describe the inclusion and follow-up of participants by study arm. Baseline characteristics will be described by study arm with summary statistics such as median and interquartile range or number and percentage; no formal testing between arms will be performed [57]. Outcomes will be described by arm using summary statistics. The primary outcome, the proportion of participants reaching the target BP within 12 weeks, will be assessed using a logistic regression model, reporting odds ratios and risk differences with standard errors estimated using the delta method [58]. Binary secondary outcomes will be evaluated in the same way. Continuous secondary outcomes will be assessed using linear regression models, reporting mean differences. Time to event outcomes will be assessed using Kaplan-Meier estimation and Cox proportional hazards models. Estimates will be reported with 95% confidence intervals (CI). All models will be adjusted for baseline BP and the stratification factors of site, HIV status, and age [59]. Effect modification of the primary outcome by site and HIV status will be assessed by incorporating an interaction between arm and site or HIV status, respectively, acknowledging that power will be low. Appropriate methods such as multiple imputation will be considered to account for participants with missing outcome data. We will compare each of the intervention arms versus control. For the non-inferiority comparison between the dual combination and control arms, a CI approach will be used. A figure illustrating the CIs and the non-inferiority margin will be presented. Primary analyses for the non-inferiority comparison will be performed on both the ITT and per protocol sets [60]. If the dual combination is found to be non-inferior to the control, then we will assess for superiority using the ITT set. The trial statistician will perform the statistical analyses using Stata (version 15, Stata Corporation, Austin, TX, USA). A full statistical analysis plan will be developed.
Nested studies and additional analyses
In a subset of 100 consenting participants (with a separate informed consent) living close to the CDCI in Ifakara, 24-h ambulatory BP and standardized unattended BP measurement will be offered, to assess the proportion of participants with white coat hypertension [29]. In consenting participants, the 24-h BP measurement is started immediately after enrolment, before randomization and study drug dispensing. The device is programmed to take measurements every 20 min between 6:00 and 22:00 and every 30 min between 22:00 and 6:00 [61]. At the end of the 24-h ambulatory BP measurement, an unattended automated office BP measurement is done using a Dräger Infinity Delta® monitor, which is programmed to take five consecutive measurements after 5 min of rest, spaced 1 min apart with a calculation of the mean out of all measurements [61,62,63,64]. After completing both 24-h and unattended BP measurements, the participant is randomized and receives study drugs as described above. Both, the 24-h ambulatory BP measurement and the unattended BP measurement are repeated at 12 weeks. Results have no influence on randomization but will help to evaluate unattended office blood pressure as a tool to investigate white coat hypertension in low resource environments, where ambulatory blood pressure measurement is not widely available. Participants are informed of the results at the end of the study.
For the cost-effectiveness analysis, we follow the JAMA guidelines and calculate incremental cost-effectiveness of the three regimens from both a health-systems and a societal perspective [65]. Health systems cost will include total medication cost as well as staff time and a fixed cost for each facility visit, which will be compared to the total health benefits achieved by the three arms [66]. Medical cost will be directly collected at the facility level in the two sites; we will obtain WHO reference prices for the respective drugs and treatments for comparison. For the societal perspective, we will include additional private cost of participants, with a particular focus on out-of-pocket expenditure for visits to facilities (transport, overnight stays) as well as costs for additional medication needed and days of work lost due to sickness [65]. To compute incremental cost-effectiveness ratios, we will use the control arm as our reference case and then compute the additional costs and benefits of the two intervention arms relative to this baseline scenario. Health outcomes will directly be observed over a 24-week period; reduced morbidity will be converted to disability-adjusted life years using the 2013 Global Burden of Disease disability weight estimates [67]. A separate analysis plan will be developed.
Monitoring and independent data monitoring committee
Monitoring is done by the Quality Management team of the Ifakara Health Institute (IHI) in Tanzania and by the Clinical Operations Unit, Swiss TPH in Lesotho. The study sites are visited by the trial monitoring team for site initiation, during the trial and at study closure. An independent data monitoring committee (IDMC) has been established to monitor the trial for efficacy and safety in accordance with an IDMC charter consisting of five members, including clinical experts from both countries and a statistician. An interim analysis to monitor the trial for efficacy and safety is planned after 50% of the target sample size has completed their primary outcome assessment at 12 weeks, which is expected to be approximately 1 year after the start of the trial. Only IDMC will have access to unblinded efficacy and safety data. Whether further analyses are needed and the timing of such analyses will be determined by the IDMC. Furthermore, the IDMC will recommend that the trial continues, be modified, or be terminated based on their review.
Safety
All trial drugs have a well-established safety profile. Safety outcomes are assessed by adverse events (AE) and serious adverse events (SAE), which are captured at every visit and are documented at the earliest possible time point. (S) AEs are documented, graded according to the common terminology criteria for adverse events (CTCAE), and reported according to ethics regulations of Tanzania, Lesotho, and Switzerland. The study physician is responsible for management and documentation of all (S)AEs. If a participant develops an AE of grade 2 or higher at the last study visit, he/she remains under observation by the study physicians beyond study termination, until the AE is resolved or stabilized.
Discussion
Worldwide, around 41 million people die annually from non-communicable diseases (NCDs). Arterial hypertension is the most prevalent risk factor for cardiovascular diseases and claims approximately 7.5 million lives annually [68]. Africa has the highest burden of arterial hypertension with an estimated prevalence of 40% [3]. This adds to the burden of the health care systems which are already overwhelmed with the management of a high number of infectious diseases in the region, including long-term care for patients with HIV [68,69,70,71]. Despite the high burden in Africa, there are still few clinical trials evaluating the best treatment for arterial hypertension in sub-Saharan Africa [41].
Since more than two thirds of patients need a combination therapy of antihypertensive medications to reach optimal BP targets [17, 18, 20]—which will be even more so in the light of new tighter targets—the question is less about the optimal first-line drug class but rather the optimal combination and strategy to reach the target in the shortest time frame. A recent randomized controlled trial found that CCB-containing regimens were superior compared to a combination of diuretics and ACE-inhibitors among Africans [29]. Additionally, BP control has not been studied with respect to possible interactions with ART in people living with HIV, likely affecting treatment response [72,73,74,75]. This is of particular public health importance since more than 60% of HIV-infected patients worldwide live in sub-Saharan Africa [76].
With the coArtHA trial, we aim to address these gaps by investigating three different regimens of widely available antihypertensive drugs listed in the WHO essential drug list in HIV-positive and HIV-negative participants with uncomplicated arterial hypertension in rural Tanzania and Lesotho. Furthermore, the trial evaluates surrogate markers of end-organ damage such as renal impairment, cardiac function, and ocular manifestations.
We foresee some limitations; firstly, that 24-h BP measurement is not feasible for all participants posing a risk that we miss white coat hypertension in participants living remotely from the facility [77, 78]. We chose a pragmatic approach using a highly standardized, stringent technique of office BP measurement and plan a nested study to compare in a subset of participants the office BP with a 24-h ambulatory BP measurement. Secondly, since this is an open-label study, both participants and study staff are aware of the treatment allocation. However clinical endpoints such as ECG, echocardiography, retinal picture, and 24-h ambulatory BP measurements will be interpreted by clinicians blinded to the study arm.
In summary, the coArtHA trial will inform on the best treatment strategies for uncomplicated arterial hypertension in people living in sub-Saharan Africa. The trial aims to inform future guidelines, to assess hypertension-mediated end-organ damage, and to determine the cost-effectiveness of different arterial hypertension treatment strategies.
Timeline
The study duration of the study is planned to be 18 months, with a 12-month recruitment period followed by 6 months of follow-up. Recruitment started at Mokhotlong District Hospital on March 06, 2020, and at St. Francis Referral Hospital, Ifakara, on March 24, 2020. Due to national lockdown measures during the SARS-Cov-2 pandemic, recruitment was interrupted in Lesotho from March 29, 2020, and in Tanzania from March 30, 2020. Recruitment was resumed in Lesotho on May 20, 2020, and in Tanzania on June 08, 2020. In case of slow recruitment, it might be extended to nearby hospitals. As of 28.07.2020, the number of participants recruited was 142.
Availability of data and materials
A minimal verified and anonymized dataset will be made available to a public data repository.
Abbreviations
- ACE:
-
Angiotensin converting enzyme
- AE:
-
Adverse events
- AIDS:
-
Acquired immunodeficiency syndrome
- ARB:
-
Angiotensin receptor blocker
- ART:
-
Antiretroviral treatment
- BP:
-
Blood pressure
- CCB:
-
Calcium channel blocker
- CDCI:
-
Chronic Diseases Clinic of Ifakara
- CI:
-
Confidence intervals
- CKD-EPI:
-
Chronic Kidney Disease Epidemiology Collaboration
- CoArtHA :
-
Control Arterial Hypertension in sub-Saharan Africa
- CTCAE :
-
Common terminology criteria for adverse events
- ECG:
-
Electrocardiogram
- eCRF:
-
Electronic case report forms
- ESC:
-
European Society of Cardiology
- ESH:
-
European Society of Hypertension
- HIV:
-
Human immunodeficiency virus
- IDMC:
-
Independent data monitoring committee
- IHI:
-
Ifakara Health Institute
- IHI-IRB:
-
Ifakara Health Institute – Institutional Review Board
- ITT:
-
Intention-to-treat
- JAMA:
-
Journal of the American Medical Association
- KIULARCO:
-
Kilombero and Ulanga Antiretroviral Cohort
- NCDs:
-
Non-communicable diseases
- NIMR:
-
National Institute for Medical Research
- SAE:
-
Serious adverse event
- Swiss TPH:
-
Swiss Tropical and Public Health Institute
- TMDA:
-
Tanzania Medicines and Medical Devices Authority
- TZD:
-
Thiazide diuretic
- WHO:
-
World Health Organization
References
Forouzanfar MH, Liu P, Roth GA, Ng M, Biryukov S, Marczak L, et al. Global burden of hypertension and systolic blood pressure of at least 110 to 115 mm hg, 1990-2015. JAMA. 2017;317:165–82 https://doi.org/10.1001/jama.2016.19043.
Mokdad AH, Forouzanfar MH, Daoud F, Mokdad AA, El Bcheraoui C, Moradi-Lakeh M, et al. Global burden of diseases, injuries, and risk factors for young people’s health during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2016;387:2383–401 https://doi.org/10.1016/S0140-6736(16)00648-6.
WHO | Raised blood pressure. WHO n.d. https://www.who.int/gho/ncd/risk_factors/blood_pressure_prevalence_text/en/. (Accessed 26 Mar 2020).
Vlado P, Rachel H, Wu Y, Dorairaj P, Stephen MM. The burden of blood pressure-related disease. Hypertension. 2007;50:991–7 https://doi.org/10.1161/HYPERTENSIONAHA.107.095497.
Srinath RK, Salim Y. Emerging epidemic of cardiovascular disease in developing countries. Circulation. 1998;97:596–601 https://doi.org/10.1161/01.CIR.97.6.596.
Addo J, Smeeth L, Leon DA. Hypertension in sub-Saharan Africa: a systematic review. Hypertension. 2007;50:1012–8 https://doi.org/10.1161/HYPERTENSIONAHA.107.093336.
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365:217–23 https://doi.org/10.1016/S0140-6736(05)17741-1.
Opie LH, Seedat YK. Hypertension in sub-Saharan African populations. Circulation. 2005;112:3562–8 https://doi.org/10.1161/CIRCULATIONAHA.105.539569.
Ataklte F, Erqou S, Kaptoge S, Taye B, Echouffo-Tcheugui JB, Kengne AP. Burden of undiagnosed hypertension in sub-saharan Africa: a systematic review and meta-analysis. Hypertension. 2015;65:291–8 https://doi.org/10.1161/HYPERTENSIONAHA.114.04394.
Flack JM, Sica DA, George B, Brown Angela L, Ferdinand Keith C, Grimm Richard H, et al. Management of high blood pressure in Blacks. Hypertension. 2010;56:780–800 https://doi.org/10.1161/HYPERTENSIONAHA.110.152892.
Adeyemo A, Gerry N, Chen G, Herbert A, Doumatey A, Huang H, et al. A genome-wide association study of hypertension and blood pressure in African Americans. PLoS Genet. 2009;5:e1000564 https://doi.org/10.1371/journal.pgen.1000564.
Young JH, Chang Y-PC, Kim JD-O, Chretien J-P, Klag MJ, Levine MA, et al. Differential susceptibility to hypertension is due to selection during the out-of-Africa expansion. PLoS Genet. 2005;1:e82 https://doi.org/10.1371/journal.pgen.0010082.
Tiffin N, Meintjes A, Ramesar R, Bajic VB, Rayner B. Computational analysis of candidate disease genes and variants for salt-sensitive hypertension in indigenous Southern Africans. PLoS One. 2010;5:e12989 https://doi.org/10.1371/journal.pone.0012989.
Falkner B, Kushner H. Effect of chronic sodium loading on cardiovascular response in young blacks and whites. Hypertension. 1990;15:36–43.
Howard G, Lackland DT, Kleindorfer DO, Kissela BM, Moy CS, Judd SE, et al. Racial differences in the impact of elevated systolic blood pressure on stroke risk. JAMA Intern Med. 2013;173:46–51 https://doi.org/10.1001/2013.jamainternmed.857.
Geldsetzer P, Manne-Goehler J, Marcus M-E, Ebert C, Zhumadilov Z, Wesseh CS, et al. The state of hypertension care in 44 low-income and middle-income countries: a cross-sectional study of nationally representative individual-level data from 1·1 million adults. Lancet. 2019;394:652–62 https://doi.org/10.1016/S0140-6736(19)30955-9.
Chow CK, Thakkar J, Bennett A, Hillis G, Burke M, Usherwood T, et al. Quarter-dose quadruple combination therapy for initial treatment of hypertension: placebo-controlled, crossover, randomised trial and systematic review. Lancet. 2017;389:1035–42 https://doi.org/10.1016/S0140-6736(17)30260-X.
Wald DS, Law M, Morris JK, Bestwick JP, Wald NJ. Combination therapy versus monotherapy in reducing blood pressure: meta-analysis on 11,000 participants from 42 trials. Am J Med. 2009;122:290–300 https://doi.org/10.1016/j.amjmed.2008.09.038.
Cushman WC, Ford CE, Cutler JA, Margolis KL, Davis BR, Grimm RH, et al. Original papers. Success and predictors of blood pressure control in diverse north American settings: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). J Clin Hypertens. 2002;4:393–404 https://doi.org/10.1111/j.1524-6175.2002.02045.x.
Whelton PK, Carey RM, Aronow WS, Casey DE, Collins KJ, Cheryl DH, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71:1269–324 https://doi.org/10.1161/HeYP.0000000000000066.
Organization WH. Prevention of cardiovascular disease : guidelines for assessment and management of total cardiovascular risk: World Health Organization; 2007. https://apps.who.int/iris/handle/10665/43685. (Accessed 23 Dec 2020)
Organization WH, Group IS of HW. 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J Hypertens. 2003;21:1983–92.
Unger T, Borghi C, Charchar F, Khan NA, Poulter NR, Prabhakaran D, et al. 2020 International Society of Hypertension Global Hypertension Practice Guidelines. J Hypertens. 2020;38(6):982–1004.
Seedat YK. Trial of atenolol and chlorthalidone for hypertension in black South Africans. Br Med J. 1980;281:1241–3. https://doi.org/10.1136/bmj.281.6250.1241.
Sareli P, Radevski IV, Valtchanova ZP, Libhaber E, Candy GP, Hond ED, et al. Efficacy of different drug classes used to initiate antihypertensive treatment in black subjects: results of a randomized trial in Johannesburg, South Africa. Arch Intern Med. 2001;161:965–71 https://doi.org/10.1001/archinte.161.7.965.
M’Buyamba-Kabangu JR, Anisiuba BC, Ndiaye MB, Lemogoum D, Jacobs L, Ijoma CK, et al. Efficacy of newer versus older antihypertensive drugs in black patients living in sub-Saharan Africa. J Hum Hypertens. 2013;27:729–35 https://doi.org/10.1038/jhh.2013.56.
Odili AN, Ezeala-Adikaibe B, Ndiaye MB, Anisiuba BC, Kamdem MM, Ijoma CK, et al. Progress report on the first sub-Saharan Africa trial of newer versus older antihypertensive drugs in native black patients. Trials. 2012;13:59 https://doi.org/10.1186/1745-6215-13-59.
Djoumessi RN, Noubiap JJN, Kaze FF, Essouma M, Menanga AP, Kengne AP, et al. Effect of low-dose spironolactone on resistant hypertension in type 2 diabetes mellitus: a randomized controlled trial in a sub-Saharan African population. BMC Res Notes. 2016;9:187 https://doi.org/10.1186/s13104-016-1987-5.
Ojji DB, Mayosi B, Francis V, Badri M, Cornelius V, Smythe W, et al. Comparison of dual therapies for lowering blood pressure in black Africans. N Engl J Med. 2019;380:2429–39 https://doi.org/10.1056/NEJMoa1901113.
Mary - TANZANIA STEPS SURVEY REPORT 2012.pdf [Internet]. https://www.who.int/ncds/surveillance/steps/UR_Tanzania_2012_STEPS_Report.pdf?ua=1. (Accessed 23 Dec 2020).
Vanobberghen F, Letang E, Gamell A, Mnzava DK, Faini D, Luwanda LB, et al. A decade of HIV care in rural Tanzania: trends in clinical outcomes and impact of clinic optimisation in an open, prospective cohort. PLoS One. 2017;12:e0180983 https://doi.org/10.1371/journal.pone.0180983.
Letang E, Kalinjuma AV, Glass TR, Gamell A, Mapesi H, Sikalengo GR, et al. Cohort profile: the Kilombero and Ulanga Antiretroviral Cohort (KIULARCO) - a prospective HIV cohort in rural Tanzania. Swiss Med Wkly. 2017;147:w14485 https://doi.org/10.4414/smw.2017.14485.
Rodríguez-Arbolí E, Mwamelo K, Kalinjuma AV, Furrer H, Hatz C, Tanner M, et al. Incidence and risk factors for hypertension among HIV patients in rural Tanzania - a prospective cohort study. PLoS One. 2017;12:e0172089 https://doi.org/10.1371/journal.pone.0172089.
Albrecht S, Franzeck FC, Mapesi H, Hatz C, Kalinjuma AV, Glass TR, et al. Age-related comorbidities and mortality in people living with HIV in rural Tanzania: data from a prospective cohort study. AIDS. 2019; https://doi.org/10.1097/QAD.0000000000002171.
Mchomvu E, Mbunda G, Simon N, Kitila F, Temba Y, Msumba I, et al. Diagnoses made in an emergency department in rural sub-Saharan Africa. Swiss Med Wkly. 2019;149:w20018 https://doi.org/10.4414/smw.2019.20018.
Lesotho_2012_STEPS_fact_sheet.pdf. https://www.who.int/ncds/surveillance/steps/Lesotho_2012_STEPS_fact_sheet.pdf. (Accessed on 23 Dec 2020).
Labhardt ND, Müller UF, Ringera I, Ehmer J, Motlatsi MM, Pfeiffer K, et al. Metabolic syndrome in patients on first-line antiretroviral therapy containing zidovudine or tenofovir in rural Lesotho, Southern Africa. Tropical Med Int Health. 2017;22:725–33 https://doi.org/10.1111/tmi.12872.
Whitworth JA, World Health Organization, International Society of Hypertension Writing Group. 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J Hypertens. 2003;21:1983–92 https://doi.org/10.1097/00004872-200311000-00002.
Khetsi MT. Standard treatment guidelines and essential medicines list for Lesotho. 2005. https://www.who.int/selection_medicines/country_lists/lso_2005_STGs_EML.pdf?ua=1. (Accessed on 23 Dec 2020).
WHO | WHO Model Lists of Essential Medicines. WHO n.d. http://www.who.int/medicines/publications/essentialmedicines/en/. (Accessed 14 July 2020).
Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, et al. 2018 ESC/ESH guidelines for the management of arterial hypertension. Eur Heart J. 2018;39:3021–104 https://doi.org/10.1093/eurheartj/ehy339.
Appel LJ, Wright JT, Greene T, Agodoa LY, Astor BC, Bakris GL, et al. Intensive blood-pressure control in hypertensive chronic kidney disease. N Engl J Med. 2010;363:918–29 https://doi.org/10.1056/NEJMoa0910975.
Beaney T, Burrell LM, Castillo RR, Charchar FJ, Cro S, Damasceno A, et al. May Measurement Month 2018: a pragmatic global screening campaign to raise awareness of blood pressure by the International Society of Hypertension. Eur Heart J. 2019;40:2006–17 https://doi.org/10.1093/eurheartj/ehz300.
Williams B, MacDonald TM, Morant S, Webb DJ, Sever P, McInnes G, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386:2059–68 https://doi.org/10.1016/S0140-6736(15)00257-3.
Belghazi J, El Feghali RN, Moussalem T, Rejdych M, Asmar RG. Validation of four automatic devices for self-measurement of blood pressure according to the International Protocol of the European Society of Hypertension. Vasc Health Risk Manag. 2007;3:389–400.
Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. 1949. Ann Noninvasive Electrocardiol. 2001;6:343–68 https://doi.org/10.1111/j.1542-474x.2001.tb00129.x.
Jain A, Tandri H, Dalal D, Chahal H, Soliman EZ, Prineas RJ, et al. Diagnostic and prognostic utility of electrocardiography for left ventricular hypertrophy defined by magnetic resonance imaging in relationship to ethnicity: the Multi-Ethnic Study of Atherosclerosis (MESA). Am Heart J. 2010;159:652–8 https://doi.org/10.1016/j.ahj.2009.12.035.
Nascimento BR, Beaton AZ, Nunes MCP, Tompsett AR, Oliveira KKB, Diamantino AC, et al. Integration of echocardiographic screening by non-physicians with remote reading in primary care. Heart. 2019;105:283–90 https://doi.org/10.1136/heartjnl-2018-313593.
Cuspidi C, Rescaldani M, Sala C. Prevalence of echocardiographic left-atrial enlargement in hypertension: a systematic review of recent clinical studies. Am J Hypertens. 2013;26:456–64 https://doi.org/10.1093/ajh/hpt001.
Su G, Cao H, Xu S, Lu Y, Shuai X, Sun Y, et al. Left atrial enlargement in the early stage of hypertensive heart disease: a common but ignored condition. J Clin Hypertens (Greenwich). 2014;16:192–7 https://doi.org/10.1111/jch.12282.
Maamari RN, Keenan JD, Fletcher DA, Margolis TP. A mobile phone-based retinal camera for portable wide field imaging. Br J Ophthalmol. 2014;98:438–41 https://doi.org/10.1136/bjophthalmol-2013-303797.
Jamerson K, Weber MA, Bakris GL, Dahlöf B, Pitt B, Shi V, et al. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med. 2008;359:2417–28 https://doi.org/10.1056/NEJMoa0806182.
Iyalomhe GBS, Omogbai EKI, Isah AO, Iyalomhe OOB, Dada FL, Iyalomhe SI. Efficacy of initiating therapy with amlodipine and hydrochlorothiazide or their combination in hypertensive Nigerians. Clin Exp Hypertens. 2013;35:620–7 https://doi.org/10.3109/10641963.2013.776570.
Schulz KF, Altman DG, Moher D, CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. Trials. 2010;11:32 https://doi.org/10.1186/1745-6215-11-32.
Juszczak E, Altman DG, Hopewell S, Schulz K. Reporting of multi-arm parallel-group randomized trials: extension of the CONSORT 2010 statement. JAMA. 2019;321:1610–20 https://doi.org/10.1001/jama.2019.3087.
Piaggio G, Elbourne DR, Pocock SJ, Evans SJW, Altman DG, CONSORT Group. Reporting of noninferiority and equivalence randomized trials: extension of the CONSORT 2010 statement. JAMA. 2012;308:2594–604 https://doi.org/10.1001/jama.2012.87802.
Pocock SJ, Assmann SE, Enos LE, Kasten LE. Subgroup analysis, covariate adjustment and baseline comparisons in clinical trial reporting: current practice and problems. Stat Med. 2002;21:2917–30 https://doi.org/10.1002/sim.1296.
Norton EC, Miller MM, Kleinman LC. Computing adjusted risk ratios and risk differences in Stata. Stata J. 2013;13:492–509 https://doi.org/10.1177/1536867X1301300304.
Kahan BC, Morris TP. Reporting and analysis of trials using stratified randomisation in leading medical journals: review and reanalysis. BMJ. 2012;345:e5840 https://doi.org/10.1136/bmj.e5840.
Committee for Proprietary Medicinal Products. Points to consider on switching between superiority and non-inferiority. Br J Clin Pharmacol. 2001;52:223–8 https://doi.org/10.1046/j.0306-5251.2001.01397-3.x.
O’Brien E, Parati G, Stergiou G, Asmar R, Beilin L, Bilo G, et al. European Society of Hypertension position paper on ambulatory blood pressure monitoring. J Hypertens. 2013;31:1731–68 https://doi.org/10.1097/HJH.0b013e328363e964.
Myers MG, Valdivieso M, Chessman M, Kiss A. Can sphygmomanometers designed for self-measurement of blood pressure in the home be used in office practice? Blood Press Monit. 2010;15:300–4 https://doi.org/10.1097/MBP.0b013e328340d128.
SPRINT Research Group, Wright JT, Williamson JD, Whelton PK, Snyder JK, Sink KM, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103–16 https://doi.org/10.1056/NEJMoa1511939.
Kjeldsen SE, Mancia G. The un-observed automated office blood pressure measurement technique used in the SPRINT study points to a standard target office systolic blood pressure <140 mmHg. Curr Hypertens Rep. 2017;19:3 https://doi.org/10.1007/s11906-017-0700-y.
Sanders GD, Neumann PJ, Basu A, Brock DW, Feeny D, Krahn M, et al. Recommendations for conduct, methodological practices, and reporting of cost-effectiveness analyses: second panel on cost-effectiveness in health and medicine. JAMA. 2016;316:1093–103 https://doi.org/10.1001/jama.2016.12195.
Edejer TT-T, World Health Organization, editors. Making choices in health: WHO guide to cost-effectiveness analysis. Geneva: World Health Organization; 2003. https://www.who.int/choice/publications/p_2003_generalised_cea.pdf. (Accessed on 23 Dec 2020)
Salomon JA, Haagsma JA, Davis A, de Noordhout CM, Polinder S, Havelaar AH, et al. Disability weights for the Global Burden of Disease 2013 study. Lancet Glob Health. 2015;3:e712–23 https://doi.org/10.1016/S2214-109X(15)00069-8.
WHO | Noncommunicable diseases country profiles 2018. WHO n.d. http://www.who.int/nmh/countries/en/. (Accessed 26 Mar 2020).
Abegunde DO, Mathers CD, Adam T, Ortegon M, Strong K. The burden and costs of chronic diseases in low-income and middle-income countries. Lancet. 2007;370:1929–38 https://doi.org/10.1016/S0140-6736(07)61696-1.
Amuyunzu-Nyamongo M. Need for a multi-factorial, multi-sectorial and multi-disciplinary approach to NCD prevention and control in Africa. Glob Health Promot. 2010;17:31–2 https://doi.org/10.1177/1757975910363928.
Mapesi H, Paris DH. Non-communicable diseases on the rise in sub-Saharan Africa, the underappreciated threat of a dual disease burden. Praxis. 2019;108:997–1005 https://doi.org/10.1024/1661-8157/a003354.
Obel N, Thomsen HF, Kronborg G, Larsen CS, Hildebrandt PR, Sørensen HT, et al. Ischemic heart disease in HIV-infected and HIV-uninfected individuals: a population-based cohort study. Clin Infect Dis. 2007;44:1625–31 https://doi.org/10.1086/518285.
McDonald CL, Kaltman JR. Cardiovascular disease in adult and pediatric HIV/AIDS. J Am Coll Cardiol. 2009;54:1185–8 https://doi.org/10.1016/j.jacc.2009.05.055.
Islam FM, Wu J, Jansson J, Wilson DP. Relative risk of cardiovascular disease among people living with HIV: a systematic review and meta-analysis. HIV Med. 2012;13:453–68 https://doi.org/10.1111/j.1468-1293.2012.00996.x.
Data Collection on Adverse Events of Anti-HIV drugs (D:A:D) Study Group, Smith C, Sabin CA, Lundgren JD, Thiebaut R, Weber R, et al. Factors associated with specific causes of death amongst HIV-positive individuals in the D:A:D Study. AIDS. 2010;24:1537–48 https://doi.org/10.1097/QAD.0b013e32833a0918.
UNAIDS data 2019 | UNAIDS n.d. https://www.unaids.org/en/resources/documents/2019/2019-UNAIDS-data. (Accessed 20 Feb 2020).
Piper MA, Evans CV, Burda BU, Margolis KL, O’Connor E, Whitlock EP. Diagnostic and predictive accuracy of blood pressure screening methods with consideration of rescreening intervals: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2015;162:192–204 https://doi.org/10.7326/M14-1539.
Zeller A, Sigle J-P, Battegay E, Martina B. Value of a standard urinary dipstick test for detecting microalbuminuria in patients with newly diagnosed hypertension. Swiss Med Wkly. 2005;135:57–61 https://doi.org/2005/03/smw-10859.
Acknowledgements
The trial team would like to acknowledge Geofrey Isaya Mbunda, Jamali Bashiri Siru (Ifakara Health Institute), Elsie Ramapepe, and Ayesha Tarr (Solidarmed, Partnerships for Health) for their hard work and contribution. Additionally, we are grateful for IDMC members Dr. Pedro Pallangyo (Jakaya Kikwete Cardiac Institute, Tanzania), Dr. Joan Rwegamalila (Muhimbili National Hospital, Tanzania), Dr. Ally Olotu (Ifakara Health Institute, Tanzania), Dr. Tšepang Lekhela (Ministry of Health, Lesotho), and Dr. Tobias Erlanger (University of Basel, Switzerland).
Funding
This trial is funded by a grant from the Swiss National Science Foundation (32003B_185263) obtained by MW. HM receives his salary through the Swiss Government Excellence Scholarships for Foreign Scholars (ESKAS-Nr: 2018.0004). The funding sources had no role in the design of the study and will not be involved in data collection, data analysis, interpretation of the results and writing of the manuscript.
Author information
Authors and Affiliations
Contributions
MW is the responsible investigator of this trial, has conceived and designed the trial, and has received the grant. HM is the overall principal investigator. HM, TB, MR, NDL, DHP, and FV have contributed in conception and design of the trial and in protocol writing. RV, JM, AA, TG, AL, ML, GF, WG, MB, and DHP involved in critically revising the manuscript for important intellectual content. TG and FV perform statistical analysis. HW, ES, BL, MM, and MBR contribute to the data collection and management. All authors read, revised, and approved the final protocol and manuscript submitted.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This trial has been approved by Ethikkomission der Nordwest und Zentralschweiz, Switzerland (EKNZ, reference number: 2019-00817), the Institutional review board of Ifakara Health Institute (reference number: IHI/IRB/No: 29 - 2019), the National Institute for Medical Research (reference number: NIMR/HQ/R.8a/Vol. IX/3277), the Tanzania Medicines and Medical Devices Authority (reference number: TMDA A0020/CTR/0002/03), and the National Health Research and Ethics Committee, Ministry of Health of Lesotho (reference number: 224 - 2019).
The study investigator (or his/her designee) obtains written informed consent from all study participants before any study procedure and after explaining to the participant the nature of the study, its purpose, the procedures involved, the expected duration, the potential risks and benefits and any discomfort it may entail. Each participant is informed that participation in the study is voluntary, that he/she may withdraw from the study at any time, and that withdrawal of consent will not affect his/her subsequent medical assistance and treatment. The participant is given a copy of the document, dated and signed by him and the investigator (or his designee); the original is retained as part of the study records. If the participant is illiterate, the study information is being read to him/her in the presence of a valid witness and the signature of a witness and the fingerprint of the participant is obtained. Study participants do not receive any payment to be part of the study besides compensation for transport expenses for additional clinical visits. Non-consenting participants are not included in the study.
Locally, the results of this trial will be shared to the district officials during district management team meetings at the national research symposium of the Ministry of Health in both countries. Additionally, the results will be disseminated through international presentations at conferences and publication in peer-reviewed journals. We do not intend to use a professional medical writer.
Consent for publication
Consent is asked from study participants for publication of gathered study data without providing identifying information of the participant.
Competing interests
All authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mapesi, H., Gupta, R., Wilson, H.I. et al. The coArtHA trial—identifying the most effective treatment strategies to control arterial hypertension in sub-Saharan Africa: study protocol for a randomized controlled trial. Trials 22, 77 (2021). https://doi.org/10.1186/s13063-021-05023-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-021-05023-z