
Abstract
Background
Transforaminal epidural injections with steroids (TESI) are used increasingly for patients with sciatica. However, their safety, effectiveness, and cost-effectiveness are still a matter of debate. This a priori statistical analysis plan describes the methodology of the analysis for the STAR trial that assesses the (cost-)effectiveness of TESI during the acute stage of sciatica (< 8 weeks).
Methods
The STAR trial is a multicentre, randomized controlled, prospective trial (RCT) investigating the (cost-)effectiveness of TESI by making a three-group comparison among patients with acute sciatica due to a herniated lumbar disc (< 8 weeks): (1) TESI combined with levobupivacaine added to oral pain medication (intervention group 1) versus oral pain medication alone (control group), (2) intervention group 1 versus transforaminal epidural injection with levobupivacaine and saline solution added to oral pain medication (intervention group 2), and (3) intervention group 2 versus control group. Co-primary outcomes were physical functioning (Roland Morris Disability Questionnaire), pain intensity (10-point numerical rating scale), and global perceived recovery (7-point Likert scale, dichotomized into ‘recovered’ and ‘not recovered’). For all three comparisons, we defined the following minimal clinically relevant between-group differences: two points for pain intensity (range 0–10), four points for physical functioning (range 0–24) and a 20% difference in recovery rate. Secondary outcomes are health-related quality of life (EQ-5D-5L) and patient satisfaction (7-point Likert scale) and surgery rate. We also collected resource use data to perform an economic evaluation. Analyses will be conducted by intention-to-treat with p < 0.05 (two-tailed) for all three comparisons. Effects will be estimated using mixed models by maximum likelihood. For each comparison, mean differences, or difference in proportions, between groups will be tested per time point and an overall mean difference, or difference in proportions, between groups during the complete duration of follow-up (6 months) will be estimated. In the economic evaluation, Multivariate Imputation by Chained Equations will be used to handle missing data. Cost and effect differences will be estimated using seemingly unrelated regression, and uncertainty will be estimated using bootstrapping techniques.
Discussion
This statistical analysis plan provides detailed information on the intended analysis of the STAR trial, which aims to deliver evidence about the (cost-)effectiveness of TESI during the acute phase of sciatica (< 8 weeks).
Trial registration
Dutch National trial register NTR4457 (6 March 2014)
Similar content being viewed by others


Update
Sciatica or lumbar radicular syndrome is a disabling condition characterized by radiating leg pain, with or without low back pain [1]. Sciatica may be accompanied by neurological deficits, such as weakness of the leg muscles or sensory loss. About 85% of sciatica cases are caused by lumbar disc herniation [2]. During the first few weeks after onset, treatment primarily focusses on pain reduction and improvement of physical functioning (https://www.nice.org.uk/guidance/ng59/chapter/Recommendations). Pain medication and physiotherapy are usually initiated by the general practitioner. If patients are referred to a hospital in case of moderate to severe pain or neurological deficits, they are typically treated with epidural corticosteroid injections or surgery. However, the effectiveness, cost-effectiveness and safety [3,4,5,6] of epidural corticosteroid injections are still a matter of debate and therefore more high-quality RCTs are needed.
The STAR trial (STeroids Against Radiculopathy) assesses both the effectiveness and cost-effectiveness of transforaminal epidural injections with steroids (TESIs) in patients with acute sciatica (< 8 weeks post onset). This is done by making the following three comparisons: (1) TESI combined with levobupivacaine added to oral pain medication (intervention group 1) versus oral pain medication alone (control group), (2) intervention group 1 versus transforaminal epidural injection with levobupivacaine and saline solution added to oral pain medication (intervention group 2), and (3) intervention group 2 versus control group. Our hypothesis is that intervention-1 group will experience less pain and better physical functioning compared to both the control group and intervention group 2 and that intervention group 2 is more effective than the control group as well. Hence, these interventions will be assessed for superiority.
Participant recruitment commenced in January 2016 and was completed in November 2019. Data collection was completed in April 2020. This statistical analysis plan details the planned analyses for the STAR trial to facilitate transparency of our data analyses and was developed according to appropriate guidelines [7]. The initial statistical analysis plan was approved and signed by the study investigators on April 23th 2020 and was revised on September 1, 2020. All of the statistical analyses will be performed following data integrity checks and locking and will be commenced in October 2020.
Study overview
Trial design
The STAR trial is a multicentre, randomized controlled, prospective trial that investigates the effectiveness and cost-effectiveness of TESI by making a three-group comparison among patients with acute sciatica due to a herniated lumbar disc (< 8 weeks): (1) TESI combined with levobupivacaine added to oral pain medication (intervention group 1) versus oral pain medication alone (control group), (2) intervention group 1 versus transforaminal epidural injection with levobupivacaine and saline solution added to oral pain medication (intervention group 2), and (3) intervention group 2 versus control group. Follow-up is 6 months. On March 6, 2014, the protocol was registered at the Dutch Trial Register (number NTR 4457). On August 20, 2015, the design of the STAR trial was approved by the Medical research Ethics Committees United, Nieuwegein, The Netherlands (registration number NL 45805.100.15) and the study protocol has been published elsewhere [8].
Study population
Between January 13, 2016, and September 10, 2019, 141 eligible participants [8], who were seeking care for their back-related leg pain (sciatica), were recruited from two Dutch Neurology outpatient clinics (i.e. the Zaans Medisch Centrum, Zaandam and OLVG, Amsterdam, The Netherlands).
To be eligible for this study had to have < 8 weeks of sciatic symptoms and had to be seen by a neurologist in one of the two study centres upon referral by their general practitioners (GP). Additional inclusion criteria were (a) age between 18 and 75 years, (b) a magnetic resonance imaging (MRI) confirmed disc herniation with nerve root impingement causing clinical symptoms, (c) an average pain intensity of > 4 on a 10-point numerical rating scale (NRS) during the last week, (d) good understanding of the Dutch language, and (e) Internet access in order to be able to complete online questionnaires [8]. Exclusion criteria were (a) severe weakness of the legs (Medical Research Council [MRC] score < 3), (b) spinal surgery during the previous year at the symptomatic lumbar level, (c) lumbar spinal stenosis or spondylolisthesis as the cause of radicular pain diagnosed by MRI, (d) pregnancy, and (e) severe comorbidity (e.g. cancer) [8].
Sample size
We had initially aimed to include 264 patients (n = 88 per arm) [8]. This sample size was based on the three co-primary outcomes (i.e. pain, physical functioning, and global perceived effect), a 10% loss to follow-up, a power of 0.9, and a two-sided alpha of 0.05. We calculated that 48 patients would be needed per arm to detect a minimal clinical important difference of 20 points (SD = 30) for both leg and back pain on a 10-point NRS between intervention group 1 and control [9]. Moreover, 22 patients were estimated to be required per arm to detect a difference of 4 points (SD = 4) on the RDMQ-24 scale and 79 patients per arm to detect a difference on the dichotomized GPE of 20%. Unfortunately, this sample size was not reached, as the trial was stopped prematurely due to slow participant accrual. Stopping the trial was a decision by the research team only, meaning that there was no data monitoring board involved, and was based on prior evidence that very few trials with less than 50% of the required sample size at 1 to 2 years after launch ultimately attain sufficient accrual [10]. When trial inclusion stopped at September 10, 2019, 46, 50, and 45 patients were randomized to intervention group 1, intervention group 2, and control, respectively. Consequently, the analyses will likely to be slightly underpowered for pain intensity and global perceived effect, but not for physical functioning.
Randomization and treatment allocation
After providing informed consent and completing baseline questionnaires, eligible patients were randomized, stratified for treatment centre, by the study coordinator (BTM) using ALEA® software (NKI-AVL, The Netherlands). Alea® generated a random schedule of blocks with a maximum size of 6. Allocation across groups was at a 1:1:1 ratio.
Study conditions
A detailed description of the study conditions can be found in the design article [8].
In brief, the transforaminal epidural injection procedure was similar for intervention group 1 and intervention group 2. That is, the study participant was brought to a fluoroscopy room and placed in a prone position on the procedure table. Fluoroscopy was used for localization of MRI confirmed disc herniation. Target identification and needle entry into the targeted space was done following internationally accepted procedures [9]. In short, the skin was made sterile using chlorhexidine. The injections were given with 22 gauge 100 mm facet tipped needle (Pajunk RGN™). Right needle position was confirmed with the injection of 0.5–1.5 cc of Joversol 300 mg/ml contrast material (Optiray™ 300, Mallinckrodt). Once an image was obtained demonstrating contrast material spreading into the epidural space medial to a line connecting the ipsilateral lumbar vertebral pedicles, the injection was performed [8].
Patients in intervention group 1 received 1 ml of 5% levobupivacaine followed by 1 ml of 40 mg/ml methylprednisolone in an opaque syringe. Patients in intervention group 2 received 1 ml 5% levobupivacaine followed by 1 ml NaCl 0.9% [8].
All treatment groups were allowed to use oral pain medication and were permitted to go to a physiotherapist in case of kinesiophobia and/or an inactive lifestyle. All oral pain medication during the trial was registered by the participants themselves in Survalyzer, an online questionnaire (www.survalyzer.com). All patients participating in the trial underwent MR Imaging of the lumbar spine that was evaluated by a radiologist (see design article for scan protocol) [8].
Protocol deviations
Protocol deviations were defined as intervention group 1 and intervention group 2 patients who received no epidural injection, more epidural injections than prescribed by the study protocol, and/or a type of injection fluid other than the one prescribed by the study protocol. For the control group, protocol deviations were defined as patients who received one or more epidural injections in spite of being randomized to the oral pain medication alone condition. Protocol deviations will be confirmed prior to database lock for the final analysis. All protocol violators will be included in the main analysis and a per-protocol analysis will be performed to assess the impact of protocol deviations if more than 10% of the patients will be found to have deviated from the protocol.
Blinding
This pragmatic trial was partially blinded. Patients in intervention group 1 and intervention group 2 did not know which type of injection they received. However, the type of injection fluid was known to the anaesthesiologist performing the injections. Neurologists who performing the clinical follow-up of the patients were blinded for the type of injection (intervention group 1 versus intervention group 2). The same applied to research nurses.
Patient characteristics and study outcomes
Patients were asked to complete a web-based questionnaire, containing descriptive questions as well as questions on clinical outcomes and resource use, at baseline, 3 and 6 weeks, and 3 and 6 months after randomization using Survalyzer (www.survalyzer.com). The neurological examination at baseline, length and weight were registered in Openclinica for clinical data (https://www.openclinica.com/). Table 1 gives a schematic overview of the data collection process.
Baseline measurement
At baseline, all primary and secondary outcomes were measured and additional information was collected on:
-
Demographics: age, gender, length and weight, education level, work and marital status.
-
Episode details: back and leg pain duration.
-
Neurological examination: physical examination of the leg muscles using the Medical Research Council (MRC) scale for muscle strength; sensory examination: tests for perception of light touch, pin prick, and vibration sense of the lower extremities; reflex examination: tests for reflexes in the patellar (L4) and ankle (S1); straight leg raising (or Lasègue’s sign) and a finger-floor distance. Straight leg raising was considered positive if the patient experienced radicular pain when the leg is at an angle < 60°. A finger-floor distance of more than 25 cm was considered indicative for a herniated disc.
-
Magnetic resonance imagining: level and side of disc herniation.
Co-primary outcomes
Our co-primary outcomes included pain intensity (back and leg), physical functioning and global perceived recovery and were assessed at baseline, 3 and 6 weeks, and 3 and 6 months.
Pain intensity was assessed by asking patients about their average pain during the previous week, in both the back and the leg, and was rated using a 10-point NRS: 0 = no pain to 10 = worst imaginable pain [11].
Physical functioning was assessed using the Dutch version of the Roland Morris Disability Questionnaire (RDMQ) [12]. The RDMQ includes 24 items assessing normal daily activities. Each question has a ‘yes’ or ‘no’ option and the overall RDMQ-24 scale ranges from 0 to 24, with higher values indicating more physical limitations [13].
Global perceived recovery (GPR) was rated on a 7-point Likert scale, ranging from ‘completely recovered’ to ‘worse than ever’. The GPR was dichotomized into recovered (categories ‘completely’ and ‘much recovered’) and (categories ‘slightly recovered’, ‘no change’, ‘slightly worse’, ‘much worse’ and ‘worse than ever’) [14].
For the co-primary outcomes, we defined the following minimal clinically relevant between-group differences for all three comparisons: two points for pain intensity (range 0–10), four points for physical functioning (range 0–24) and a 20% difference in recovery rate across groups [9]. In accordance with the guidelines of the ‘European Medicines Agency’, we will only consider one intervention effective over another, if statistically significant and clinically relevant differences are found between them for all co-primary outcomes (https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-multiplicity-issues-clinical-trials_en.pdf) and therefore we will not adjust our analyses for multiplicity induced by having co-primary outcomes. We will also not adjust for multiplicity induced by having 3 comparators, because we will conduct various pairwise comparisons (i.e. intervention 1 versus control, intervention 2 versus control, intervention 1 versus control) with a clear hierarchy in anticipated effectiveness (i.e. intervention 1 > intervention 2 > control), instead of a global test of unordered groups [15].
Secondary outcomes
Secondary outcomes included health-related quality of life, patient satisfaction and surgery rate and were assessed at baseline, 3 and 6 weeks, and 3 and 6 months.
Health-related quality of life was assessed using the Euroqol-5 dimensions-5 levels (EQ-5D-5L) [16]. The EQ-5D-5L asks patients to rate the severity of their health problems (levels: no problems, slight problems, moderate problems, severe problems, unable to/extreme problems) on five health dimensions (health dimensions: mobility, self-care, usual activities, pain/discomfort, anxiety/depression). The patients’ resulting EQ-5D-5L health states will be converted to utility values ranging from 0 (equal to death) to 1 (equal to full health) using the Dutch tariff [17]. For the economic evaluation, quality-adjusted life years will be estimated by multiplying the time spent in a certain health state by its respective utility value.
Patient satisfaction was assessed using a 7-point Likert scale ranging from ‘not satisfied at all’ to ‘completely satisfied’ [18].
Surgery rate was assessed by keeping track of whether or not patients needed surgery in spite of conservative treatment (control group) and a possible epidural injection recorded in the case record form (CRF). Surgery rate was measured as dichotomous outcome, indicating whether patients received a surgery during follow-up (yes = 1/no = 0).
Confounding factors
Confounding factors were a priori selected based on evidence from existing studies in sciatica, and expertise within the study team [19, 20]. The factors were age, gender, body mass index (BMI) and severity of back and leg pain at baseline.
Societal costs
Intervention costs will be estimated using a micro-costing approach. That is, detailed information on the number of TESIs performed per patient and the cost per TESI were collected from hospital accounting records. Information regarding the use of all other kinds of resources was collected using online cost questionnaires administered at 3 weeks, 6 weeks, and 3 and 6 months. See supplemental file for the questionnaire. For assessing absenteeism and presenteeism, slightly adapted versions of the World Health Organization – Work Performance Questionnaire (WHO-HPQ) and the iMTA Productivity Cost Questionnaire (iPCQ) were used, respectively [21, 22]. Healthcare utilization, consisting of the use of primary care, secondary care, prescribed and over-the-counter medication, were valued using Dutch standard prices and unit prices derived from http://www.medicijnkosten.nl. If unavailable, prices according to professional organizations were used. Informal care and unpaid productivity losses were valued using a recommended Dutch shadow price [23]. Absenteeism was valued in accordance with the Friction Cost Approach, with a friction period of 12 weeks, and using gender-specific price weights. Presenteeism was valued using gender-specific price weights as well [24]. All costs were converted to Euros 2020 using consumer price indices.
Adverse events and safety issues
All adverse events (AEs) during the study were recorded on the case record form (CRF), whether or not caused by the study procedure. Registration included: the event, onset and end date, severity, relation to the study and action taken. AEs considered related to the study were judged by a medically qualified investigator and followed until resolution (or if the event was regarded stable). There were no AEs that resulted in withdrawal from the trial.
Registering and handling of data
A trial master file was established in Amsterdam by the coordinating investigator (BTM). The registering of data was done consecutively throughout the study. Data were registered in a case report form (CRF) for each patient. Throughout the study, the registering and handling of data followed the principles of good clinical practice (GCP). The data will be kept in a locked facility for 15 years after the study is finished. After this, it will be destroyed. The statistical master file is kept by the department of Data Management, at Amsterdam UMC (location VUMC) Amsterdam, the Netherlands.
Statistical analysis
All analyses described in this plan are considered a priori analyses in that they have been defined in the study protocol and/or this SAP. All post hoc analyses will be identified as such in the article. Analyses will be consistent with the intention-to-treat principle and will be performed using software package SPSS v26 and STATA v16.
Trial profile
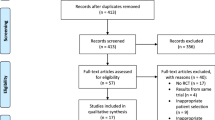
The following summaries will be presented in a flow diagram according to the CONSORT statement [25]: the number of patients with acute sciatica that were screened for eligibility at the Neurology outpatient clinics in Amsterdam and Zaanstad and the number of patients that was eventually randomized after providing informed consent, stratified for each treatment group. Additionally, the number and percentages of patients lost to follow-up will be reported per treatment arm, including information about the timing and reason(s) for loss to follow-up. See Fig. 1 for an overview.

STAR trial: enrollment and randomization
Data integrity
Prior to the analyses, the integrity of trial data will be checked by scrutinizing data files for omissions and errors. The source of any inconsistencies will be explored and resolved.
Methods for handling missing data
In the effect analyses, missing data will be handled using mixed models by maximum likelihood estimation [26]. In case more than 10% of patients have missing data, defined as having missing data on one or more variable, sensitivity analyses will be performed using mixed models with multiple imputation. For the economic evaluation, missing data will be multiply imputed, irrespective of the percentage of patients of missing data. This strategy is advised in economic evaluations, because total costs are the sum of numerous cost components, so total costs will already be missing if only one item is missing. Data will be multiply imputed using Multivariate Imputation by Chained Equations (MICE) and the number of imputed datasets will be determined using the loss of efficiency approach [27]. Imputation models will include all available cost and effect measure values as well as variables differing between groups at baseline, those variables related to the ‘missingness’ of data and variables related to the outcomes. Pooled estimates will be calculated using Rubin’s rules.
Evaluation of demographics and baseline patient characteristics
Demographic baseline characteristics will be described per treatment group (Table 2). Continuous variables will be summarized using standard measures of central tendency and dispersion, as either mean and standard error (data that with a normal distribution) or median and interquartile range (data with a skewed distribution). Dichotomous or categorical variables will be summarized by frequencies and percentages. In accordance with the CONSORT statement, we will not statistically test whether baseline differences across study groups [25, 28].
Primary analysis
All statistical tests of the primary and secondary analyses will be 2-sided and both 95% confidence intervals (95% CIs) and p values will be reported. Moreover, as indicated above, three pairwise comparisons will be conducted per outcome: (1) intervention 1 versus control, (2) intervention 2 versus control, and (3) intervention 1 versus control. The assumption of normal distribution will be checked by visual inspection and using a QQ-plot. Non-normally distributed data will be log-transformed. If normality will not be achieved after log-transformation, data will be dichotomized into either having a minimal clinically important improvement or not (e.g. ≥ 2 points for pain and ≥ 4 points for physical functioning [yes/no]). Pain intensity (back and leg) and physical functioning will be assessed using linear mixed models by maximum likelihood and global perceived recovery will be assessed using logistic mixed models by maximum likelihood. All models will have a 2-level structure, with time clustered within patients. Linear and logistic mixed models will be fitted using an ‘independent’ covariance matrix for the random effects, which allows for a distinct variance for each random effect within a random-effects equation and assumes that all covariances are 0. Linear mixed models will also use a large-sample approximation of the sampling distribution of the test statistic and will assume that all residuals are independent and identically distributed Gaussian with a common variance (www.stata.com). The necessity of having a random intercept and/or a random slope will be assessed using the likelihood-ratio test. For all co-primary outcomes, the main effect will be the pooled mean difference, or difference in proportions, across groups during the complete duration of follow-up. Additionally, mean differences, or differences in proportions, across groups will be tested per time point using time by treatment interactions. For all co-primary outcomes, adjusted (adjusted for the predefined confounders; see above) and unadjusted analyses will be performed and presented (Table 3).
Secondary analysis
Secondary outcomes health-related quality of life and satisfaction will be analysed using the same linear mixed models as the primary analysis. Surgery rate will be assessed using a logistic regression and both an adjusted and an unadjusted analysis will be performed and presented (Table 4).
Economic evaluation
The economic evaluation will focus on all three comparisons, i.e. intervention group 1 versus control group, intervention group 1 versus intervention group 2, and intervention group 2 versus control group.
The economic evaluation will be performed for all co-primary outcomes and QALYs. In the main analysis, the societal perspective will be applied, meaning that all costs will be included, irrespective of who pays or benefits from them. Unadjusted as well as adjusted cost differences across groups will be calculated for total and disaggregated costs. Ninety-five per cent CIs around those cost differences will be estimated using bias corrected and accelerated (BCA) bootstrapping, with 5000 replications. Cost and effect differences across groups will be estimated using seemingly unrelated regression (SUR) analyses, in which both are modelled simultaneously so that their possible correlation can be accounted for. Incremental cost-effectiveness ratios (ICERs) will be estimated by dividing the differences in costs by those in effects. The uncertainty surrounding the ICERs will be graphically illustrated by plotting bootstrapped cost-effect pairs on cost-effectiveness planes. Again, these bootstrapped cost-effect pairs will be estimated using the BCA bootstrap, with 5000 replications. An estimate of the joint uncertainty surrounding costs and effects will be provided by constructing cost-effectiveness acceptability curves (CEACs). These CEACs will provide an estimate of the probability of the interventions being cost-effective compared with each other. To assess the robustness of the results, three sensitivity analyses will be performed. In sensitivity analysis 1 (SA1), the healthcare perspective will be applied, meaning that only costs accruing to the formal Dutch healthcare system will be included in the analyses. In SA2, the human capital approach will be used instead of the friction cost approach for estimating absenteeism costs. In SA3, only data of patients with complete cost and effect measure values at all measurement points will be included.
Discussion
During the last decade, there has been extensive debate about the effectiveness of epidural corticosteroids for treating sciatica. A 2020 meta-analysis, as part of the Dutch multidisciplinary guideline on sciatica (https://richtlijnendatabase.nl/richtlijn/lumbosacraal_radiculair_syndroom/lumbosacraal_radiculair_syndroom_-_startpagina.html) (and based on 6 systematic reviews [4, 5, 29,30,31,32]), showed a small, but statistically significant, short-term (< 3 months) effect for leg pain of epidural corticosteroids versus placebo (mean difference (MD), 0.94 on a 10 point visual analogue scale (VAS) [95% CI, 0.14 to 1.73]). Moreover, for physical functioning, a small not clinically relevant standardized mean difference of 0.32 (95% BI − 0.58 to 1.22) was found in favour of epidural steroids. However, the level of evidence of these studies according to the GRADE approach [33] was regarded as low. Therefore, this two-centre, randomized controlled, prospective, single-blind trial (STeroids Against Radiculopathy [STAR]) will provide valuable information about the effectiveness, cost-effectiveness and safety of transforaminal epidural steroids in patients with sciatica shorter than 8 weeks, a subgroup that has hardly been addressed so far [34,35,36].
Unfortunately, however, we had to stop our trial prematurely, because of slow patient accrual, with only 53.4% of the required sample size being included in 2.5 years. Issues that affected slow patient accrual were the fact that (according to their guidelines) (https://richtlijnen.nhg.org/standaarden/lumbosacraal-radiculair-syndroom) Dutch general practitioners typically wait at least 6 weeks before referring patients with sciatica to a hospital; the fact that there were only 2 participating centres, and the fact that patients who believe in the superiority of epidural steroid injection over conservative treatment experience difficulty with being randomized and prefer active treatment with an epidural steroid injection. This is a well-known problem in back pain research [37, 38], but it will likely negatively affect the generalizability of our results to other (Dutch) sciatica patients and will result in the study being slightly underpowered for pain intensity and global perceived effect, but not for physical functioning.
Conclusion
The STAR trial aims to provide evidence about TESIs in the treatment of acute sciatica (< 8 weeks). This statistical analysis plan details the study’s planned analyses, to aid transparency of results, and may assist the design of studies in the future.
Trial status
Participant recruitment was completed in November 2019 and follow-up outcomes were collected till March 10, 2020. The data were available for analysis on May 15, 2020. The final analyses will commence after publication of the SAP.
Availability of data and materials
The datasets generated and/or analysed during the current study are not publicly available due to institution policy but are available from the corresponding author on reasonable request.
Abbreviations
- AE:
-
Adverse event
- AR:
-
Adverse reaction
- BCA:
-
Bias corrected and accelerated
- CEAC:
-
Cost-effectiveness acceptability curve
- CI:
-
Confidence interval
- CONSORT:
-
Consolidated Standards of Reporting Trials
- CRF:
-
Case record form
- EQ-5D-5 L:
-
Euroqol-5 dimensions-5 levels
- GCP:
-
Good clinical practice
- GPR:
-
Global perceived recovery
- GRADE:
-
Grades of recommendation, assessment, development and evaluation
- ICER:
-
Incremental cost-effectiveness ratios
- MCID:
-
Minimal clinically important difference
- MD:
-
Mean difference
- MICE:
-
Multivariate Imputation by Chained Equations
- MRC:
-
Medical Research Council
- MRI:
-
Magnetic resonance imaging
- NRS:
-
Numerical rating scale
- NTR:
-
Nederlands Trial Register
- PCQ:
-
Productivity Cost Questionnaire
- PRODISC:
-
Productivity and Disease Questionnaire
- QALY:
-
Quality-adjusted life year
- RCT:
-
Randomized controlled trial
- RDQ:
-
Roland-Morris Disability Questionnaire
- SA:
-
Sensitivity analysis
- SAP:
-
Statistical analysis plan
- SUR:
-
Seemingly unrelated regression
- TESI:
-
Transforaminal epidural injections with steroids
- TMF:
-
Trial master file
- VAS:
-
Visual analogue scale
- WHO:
-
World Health Organization
- WPQ:
-
Work Performance Questionnaire
References
Ropper AH, Zafonte RD. Sciatica. N Engl J Med. 2015;372(13):1240–8.
Porchet F, Wietlisbach V, Burnand B, Daeppen K, Villemure JG, Vader JP. Relationship between severity of lumbar disc disease and disability scores in sciatica patients. Neurosurgery. 2002;50:1253–9.
Van Dongen JM, Ketheswaran J, Tordrup D, Ostelo RWJG, Bertollini R, van Tulder MW. Health economic evidence gaps and methodological constraints in low back pain and neck pain: Results of the Research Agenda for Health Economic Evaluation (RAHEE) project. Best Pract Res Clin Rheumatol. 2016;30(6):981–93.
Lee JH, Kim DH, Kim DH, Shin KH, Park SJ, Lee GJ, Lee CH, Yang HS. Comparison of clinical efficacy of epidural injection with or without steroid in lumbosacral disc herniation: a systematic review and meta-analysis. Pain Physician. 2018;21(5):449–68.
Manchikanti L, Knezevic NN, Boswell MV, Kaye AD, Hirsch JA. Epidural injections for lumbar radiculopathy and spinal stenosis: a comparative systematic review and meta-analysis. Pain Physician. 2016;19(3):E365–410.
Benzon HT, Huntoon MA, Rathmell JP. Improving the safety of epidural steroid injections. JAMA. 2015;313(17):1713–4.
Gamble C, Krishan A, Stocken D, Lewis S, Juszczak E, Doré C, et al. Guidelines for the content of statistical analysis plans in clinical trials. JAMA. 2017;318(23):2337–43.
Ter Meulen BC, Maas ET, Vyas A, et al. Treatment of acute sciatica with transforaminal epidural corticosteroids and local anesthetic: design of a randomized controlled trial. BMC Musculoskelet Disord. 2017;18:215.
Ostelo RW, Deyo RA, Stratford P, Waddell G, Croft P, Von Korff M, Bouter LM, de Vet HC. Interpreting change scores for pain and functional status in low back pain: towards international consensus regarding minimal important change. Spine (Phila Pa 1976). 2008;33(1):90–4.
Bennette CS, Ramsey SD, McDermott CL, Carlson JJ, Basu A, Veenstra DL. Predicting low accrual in the National Cancer Institute’s cooperative group clinical trials. J Natl Cancer Inst. 2015;108(2):djv324.
Childs JD, Piva SR, Fritz JM. Responsiveness of the numeric pain rating scale in patients with low back pain. Spine (Phila Pa 1976). 2005;30(11):1331–4.
Gommans IHB, Koes BW, van Tulder MW. Validiteit en responsiviteit Nederlandstalige Roland Disability Questionnaire. Vragenlijst naar functionele status bij patiënten met lage rugpijn. Ned Tijdschr Fys. 1997;107:28–33.
Roland M, Fairbank J. The Roland-Morris questionnaire and the Oswestry disability questionnaire. Spine. 2000;25:3115–24.
Kamper SJ, Maher CG, Herbert RD, Hancock MJ, Hush JM, Smeets RJ. How little pain and disability do patients with low back pain have to experience to feel that they have recovered? Eur Spine J. 2010;19:1495–501.
Juszczak E, Altman DG, Hopewell S, Schulz K. Reporting of multi-arm parallel-group randomized trials: extension of the CONSORT 2010 statement. JAMA. 2019;321(16):1610–20.
Aaronson NK, Acquadro C, Alonso J, Apolone G, Bucquet D, Bullinger M, Bungay K, Fukuhara S, Gandek B, Keller S, et al. International quality of life assessment (IQO-LA) project. Qual Life Res. 1992;1:349–51.
Versteegh MM, Vermeulen KM, Evers SMAA, de Wit GA, Prenger R, Stolk EA. Dutch tariff for the five-level version of EQ-5D. Value Health. 2016;19(4):343–52.
Bombardier C. Outcome assessments in the evaluation of treatment of spinal disorders: summary and general recommendations. Spine (Phila Pa 1976). 2000;25(24):3100–3.
Konstantinou K, Dunn KM, Ogollah R, Lewis M, van der Windt D, Hay EM, ATLAS Study Team. Prognosis of sciatica and back-related leg pain in primary care: the ATLAS cohort. Spine J. 2018;18(6):1030–40.
De Schepper EI, Koes BW, Oei EH, Bierma-Zeinstra SM, Luijsterburg PA. The added prognostic value of MRI findings for recovery in patients with low back pain in primary care: a 1-year follow-up cohort study. Eur Spine J. 2016;25(4):1234–41.
Kessler RC, Barber C, Beck A, Berglund P, Cleary PD, McKenas D, Pronk N, Simon G, Stang P, Ustun TB, Wang P. The World Health Organization Health and Work Performance Questionnaire (HPQ). J Occup Environ Med. 2003;45(2):156–74.
Bouwmans C, Krol M, Severens H, Koopmanschap M, Brouwer W, Hakkaart-van RL. The iMTA productivity cost questionnaire: a standardized instrument for measuring and valuing health-related productivity losses. Value Health. 2015;18(6):753–8.
Kanters TA, Bouwmans CAM, van der Linden N, Tan SS, Hakkaart-van Roijen L. Update of the Dutch manual for costing studies in health care. PLoS One. 2017;12(11):e0187477.
Gold MR, Patrick DL, Torrance GW, Fryback DG, Hadorn DC, Kamlet MS, Daniels N, Weinstein MC. Identifying and valuing outcomes. In: Gold MRSJ, Russell LB, Weinstein MC, editors. In cost-effectiveness in health and medicine. New York: Oxford University Press; 1996. p. 82–134.
Schulz KF, Altman DG, Moher D, CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. BMC Med. 2010;8:18.
Twisk J, de Boer M, de Vente W, Heymans M. Multiple imputation of missing values was not necessary before performing a longitudinal mixed-model analysis. J Clin Epidemiol. 2013;66(9):1022–8.
White IR, Royston P, Wood AM. Multiple imputation using chained equations: issues and guidance for practice. Stat Med. 2011;30(4):377–99.
De Boer MR, Waterlander WE, Kuijper LD, Steenhuis IH, Twisk JW. Testing for baseline differences in randomized controlled trials: an unhealthy research behavior that is hard to eradicate. Int J Behav Nutr Phys Act. 2015;12:4.
Bhatia A, Flamer D, Shah PS, Cohen SP. Transforaminal epidural steroid injections for treating lumbosacral radicular pain from herniated intervertebral discs: a systematic review and meta-analysis. Anesth Analg. 2016;122(3):857–70.
Liu J, Zhou H, Lu L, Li X, Jia J, Shi Z, Yao X, Wu Q, Feng S. The effectiveness of transforaminal versus caudal routes for epidural steroid injections in managing lumbosacral radicular pain: a systematic review and meta-analysis. Medicine (Baltimore). 2016;95(18):e3373.
Lewis R, Williams N, Matar HE, Din N, Fitzsimmons D, Phillips C, Jones M, Sutton A, Burton K, Nafees S, Hendry M, Rickard I, Chakraverty R, Wilkinson C. The clinical effectiveness and cost-effectiveness of management strategies for sciatica: systematic review and economic model. Health Technol Assess. 2011;15(39):1–578. https://doi.org/10.3310/hta15390.
Pinto RZ, Maher CG, Ferreira ML, Hancock M, Oliveira VC, McLachlan AJ, Koes B, Ferreira PH. Epidural corticosteroid injections in the management of sciatica: a systematic review and meta-analysis. Ann Intern Med. 2012;157(12):865–77.
Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ (Clinical research ed). 2008;336(7650):924–6.
Spijker-Huiges A, Winters JC, van Wijhe M, Groenier K. Steroid injections added to the usual treatment of lumbar radicular syndrome: a pragmatic randomized controlled trial in general practice. BMC Musculoskelet Disord. 2014;15:341.
Bush K, Hillier S. A controlled study of caudal epidural injections of triamcinolone plus procaine for the management of intractable sciatica. Spine (Phila Pa 1976). 1991;16:572–5.
Mathews JA, Mills SB, Jenkins VM, Grimes SM, Morkel MJ, Mathews W, et al. Back pain and sciatica: controlled trials of manipulation, traction, sclerosant and epidural injections. Br J Rheumatol. 1987;26:416–23.
Mutubuki EN, van Helvoirt H, van Dongen JM, Vleggeert-Lankamp CLA, Huygen FJPM, van Tulder MW, Klopper-Kes HAHJ, Ostelo RWJG. Cost-effectiveness of combination therapy (mechanical diagnosis and treatment and transforaminal epidural steroid injections) among patients with an indication for a lumbar herniated disc surgery: protocol of a randomized controlled trial. Physiother Res Int. 2020;25(1):e1796.
Schreijenberg M, Luijsterburg PAJ, Van Trier YDM, Rizopoulos D, Koopmanschap MA, Voogt L, Maher CG, Koes BW. Discontinuation of the PACE plus trial: problems in patient recruitment in general practice. BMC Musculoskelet Disord. 2018;19(1):146.
Acknowledgements
The OLVG-hospital paid for the randomization software (ALEA), CRF software and web-based questionnaires. The authors acknowledge the help of doctors and trial nurses of the Zaans MC, Zaandam and OLVG Hospital Amsterdam for performing the study. The authors acknowledge W. Kraan from the data management group VU for his contribution to the study software.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
RO and HW principal investigators. RO, HW and BTM conceived the trial. RO, HW, MVDV and BTM contributed to the design of the trial. JVD oversaw the design of the statistical analyses. RO, JVD and BTM were involved in the analysis planning. BTM and JVD drafted the manuscript. RO, HW, MVDV, JVD and BTM contributed to the manuscript and approved the final version for publication.
Authors’ information
BTM MD, neurologist and PhD student, OLVG Teaching Hospital; JVD PhD Department of Epidemiology and Biostatistics Amsterdam Movement Sciences Research Institute; MVDV MD anaesthesiologist, Department of Anesthesiology and Pain Medicine at Zaans Medisch Centrum; HW MD PhD, neurologist at OLVG Teaching Hospital and professor of cognitive neurology at Vrije Universiteit Amsterdam; RO PhD, professor, Department of Health Sciences, Faculty of Science, Vrije Universiteit Amsterdam, Amsterdam Movement Sciences
Corresponding author
Ethics declarations
Ethics approval and consent to participate
On August 20, 2015, the RCT was evaluated positively by the Medical research Ethics Committees United, Nieuwegein, The Netherlands (registration number NL 45805.100.15) and the protocol was registered at the Dutch Trial Register (number NTR 4457). All participants gave their (written) informed consent before participation in the trial.
Consent for publication
Not applicable (anonymous data).
Competing interests
There are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Cost questionnaire.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
ter Meulen, B.C., van Dongen, J.M., van der Vegt, M. et al. STeroids Against Radiculopathy (STAR) trial: a statistical analysis plan. Trials 22, 80 (2021). https://doi.org/10.1186/s13063-020-05018-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-020-05018-2