
Abstract
Background
Tuberculosis (TB) in children is frequently paucibacillary and non-severe forms of pulmonary TB are common. Evidence for tuberculosis treatment in children is largely extrapolated from adult studies. Trials in adults with smear-negative tuberculosis suggest that treatment can be effectively shortened from 6 to 4 months. New paediatric, fixed-dose combination anti-tuberculosis treatments have recently been introduced in many countries, making the implementation of World Health Organisation (WHO)-revised dosing recommendations feasible. The safety and efficacy of these higher drug doses has not been systematically assessed in large studies in children, and the pharmacokinetics across children representing the range of weights and ages should be confirmed.
Methods/design
SHINE is a multicentre, open-label, parallel-group, non-inferiority, randomised controlled, two-arm trial comparing a 4-month vs the standard 6-month regimen using revised WHO paediatric anti-tuberculosis drug doses. We aim to recruit 1200 African and Indian children aged below 16 years with non-severe TB, with or without HIV infection. The primary efficacy and safety endpoints are TB disease-free survival 72 weeks post randomisation and grade 3 or 4 adverse events. Nested pharmacokinetic studies will evaluate anti-tuberculosis drug concentrations, providing model-based predictions for optimal dosing, and measure antiretroviral exposures in order to describe the drug-drug interactions in a subset of HIV-infected children. Socioeconomic analyses will evaluate the cost-effectiveness of the intervention and social science studies will further explore the acceptability and palatability of these new paediatric drug formulations.
Discussion
Although recent trials of TB treatment-shortening in adults with sputum-positivity have not been successful, the question has never been addressed in children, who have mainly paucibacillary, non-severe smear-negative disease. SHINE should inform whether treatment-shortening of drug-susceptible TB in children, regardless of HIV status, is efficacious and safe. The trial will also fill existing gaps in knowledge on dosing and acceptability of new anti-tuberculosis formulations and commonly used HIV drugs in settings with a high burden of TB. A positive result from this trial could simplify and shorten treatment, improve adherence and be cost-saving for many children with TB.
Recruitment to the SHINE trial begun in July 2016; results are expected in 2020.
Trial registration
International Standard Randomised Controlled Trials Number: ISRCTN63579542, 14 October 2014.
Pan African Clinical Trials Registry Number: PACTR201505001141379, 14 May 2015.
Clinical Trial Registry-India, registration number: CTRI/2017/07/009119, 27 July 2017.
Similar content being viewed by others

Background
Disease setting and context in the study
Of the estimated 10.4 million new tuberculosis (TB) cases globally per annum, approximately one million occur in children in Africa and South East Asia [1]. In Africa, where the TB and HIV epidemics are closely linked, children contribute approximately 30% of incident TB cases [2,3,4]. Furthermore, in countries with high HIV prevalence, the peak age-prevalence of TB has shifted towards younger adults. These adults are often parents of young children, increasing the exposure of children to TB. Among both HIV-infected children and adults, TB remains a leading cause of death and is the most common opportunistic infection, despite improved access to antiretroviral therapy (ART) [5,6,7].
Historically, studies of anti-tuberculosis treatment in children have lagged substantially behind adult studies and data regarding TB treatment extrapolated from adults. Children have also been seen to pose little risk of onward transmission. In combination with the challenges in confirming a TB diagnosis in children and the resource constraints, the focus has been on adult trials. The approaches used to treat childhood TB have relied on extrapolation to children of the evidence from clinical trials in adults who have a different spectrum of TB disease and lower bacillary burden [8].
The spectrum of TB disease in children
The TB disease spectrum observed in children is different compared to adults [9, 10]. Whereas a higher proportion of young children (below 3 years) have disseminated forms of TB, including miliary TB and TB meningitis, [9] primary childhood TB is typically more benign than adult TB. Three-quarters of children have intrathoracic TB, mainly with non-severe forms of intrathoracic lymph node TB [10,11,12]. Adult forms with pleural effusions and cavitation commonly emerge during adolescence [13].
TB diagnosis and measuring response to treatment in children
Microbiological confirmation of TB in younger children, who have the highest risk of TB disease, is challenging as they cannot spontaneously produce sputum. In addition, most children with pulmonary (intrathoracic) TB typically have paucibacillary disease. Of the 75% of children with intrathoracic disease, more than 85% have negative-smear microscopy on respiratory samples [11, 12]. Confirmation by culture (the diagnostic ‘gold standard’) is typically made only in approximately 30% of children [11, 14]. Of note, positive culture can also occasionally be obtained in children with normal chest radiographs and a positive tuberculin skin test or history of Mycobacterium tuberculosis exposure [15,16,17]. Therefore, the diagnosis of intrathoracic tuberculosis in children frequently relies on non-specific clinical symptoms and signs, supported by evidence of TB contact and/or TB infection and changes on chest radiography [18].
Data on objective measures of TB treatment response in children are limited. Response to anti-tuberculosis treatment can be evaluated by resolution of clinical symptoms, radiological features and smear and culture conversion in bacteriologically confirmed cases. Radiological resolution has limitations as some features may temporarily worsen and residual changes may persist at the end of treatment [19,20,21,22]. Repeat microbiological sampling during therapy may also not be feasible in the majority of children who stop coughing early in treatment. Although well-defined clinical case definitions for research are useful for TB diagnosis and evaluation of treatment response in paediatric studies, [8, 23] there are no published data on the gold standard of TB treatment response, i.e. long-term relapse-free survival.
Drug doses and fixed-dose formulations of anti-tuberculosis drugs in children
The doses of first-line anti-tuberculosis drugs recommended for children have been based on the same dose per kilogram of body weight that is recommended for adults [8]. In 2010, the WHO revised the doses of the first-line TB drugs in children, increasing daily doses of isoniazid (H) from 5 to 10 mg/kg, rifampicin (R) from 10 to 15 mg/kg, pyrazinamide (Z) from 25 to 35 mg/kg and recommending ethambutol (E) at a dose of 20 mg/kg/day [24]. The change was triggered by a review of the academic literature including pharmacokinetic (PK) studies showing that doses extrapolated from adults do not achieve comparable drug exposures in children [25,26,27,28,29]. With the revised doses it was thought to be possible to attain similar rifampicin, isoniazid and pyrazinamide concentrations to those in adults on standard doses [30,31,32].
WHO-recommended increased drug doses were difficult to implement in children using the original fixed-dose combination (FDC) RHZ 60/30/150 mg which, in particular, delivered low isoniazid doses [33]. New co-formulated RHZ 75/50/150 mg FDC tablets were launched in 2015 with rapid and wide uptake globally [34]. The new FDC, which we use in the SHINE trial, facilitates achieving the recommended doses [35].
Anti-TB and antiretroviral treatment interactions in HIV co-infected children
Rifampicin interacts with many antiretroviral drugs necessitating ART adjustment for children. For children ≥ 3 years, efavirenz-based ART is preferred during co-administration of anti-tuberculosis drugs as efavirenz has minimal drug interactions with rifampicin. For children < 3 years on lopinavir/ritonavir-based ART, two options are available: first, super boosting with additional ritonavir to overcome the rifampicin-inducing effect of liver enzymes, and second, changing to three nucleoside reverse transcriptase inhibitors (NRTIs) [36]. However, neither option is optimal. Super boosting with separate liquid ritonavir is frequently not feasible due to its unavailability, complexity of administration, short shelf-life and poor tolerability [37], and triple NRTI ART is less efficacious in viral suppression [38]. Alternative solutions for child-friendly dosing are urgently required for young children [36, 39].
TB treatment-shortening trials in adults
Early trials in HIV-negative adults with smear-negative TB in Hong Kong and Singapore showed that, in principle, reductions of treatment duration from 6 to 4 months have resulted in relapse rates of only 4% [40,41,42]. WHO guidelines subsequently recommended 4-month treatment duration for smear-negative TB in 1993, although this decision was soon reversed [43]. Recent trials of treatment-shortening in adults with smear positive TB have demonstrated poorer outcomes associated with shorter regimens [44,45,46]. However, there is no trial evidence about optimum regimen duration for children.
Rationale for the trial
The implementation of standard regimens for both adults and children is attractive for TB programmes. The needs of children with TB, who potentially benefit from shorter treatment and who form a considerable proportion of the global disease burden, should be addressed. The costs to children and their families, burden to health services and added toxicity associated with potentially overly long treatment regimens should be also considered. In addition, risks of suboptimal HIV control from drug-drug interactions, the increased pill-burden and its effect on adherence are significant issues for HIV-co-infected children [47] which could be ameliorated by shortening TB treatment.
Treatment outcomes appear considerably better in children than adults with multi-drug resistant (MDR) TB [48], likely because most children have paucibacillary disease. It therefore follows that TB outcomes for shorter first-line regimens could be as good as the longer regimens currently used in adults and children [49, 50]. In addition, there are similarities between minimal TB disease in children and smear-negative, paucibacillary TB in adults, in whom trials of shorter regimens have suggested success. This makes the implementation of trials of shorter treatment of standard first-line regimens conducted in children all the more important, and in different countries where the metabolism of anti-tuberculosis drugs may differ (India/Africa). Finally, revised WHO dosing guidelines for TB drugs aim to achieve substantially higher TB drug exposures in children which should be more efficacious, providing additional rationale to perform this treatment-shortening trial at the time that these dosing guidelines are being implemented globally.
Aims
The primary aim of the trial is to determine whether the standard 6-month regimen (8 weeks’ HRZE followed by 16 weeks’ HR) can be reduced with similar efficacy, to 4 months’ (8 weeks’ HRZE followed by 8 weeks’ HR), in African and Indian children with non-severe TB and regardless of HIV status, using revised WHO dosing guidelines and recently introduced new paediatric fixed-dose combination tablets.
The other objectives are:
-
1.
To determine whether the higher WHO-recommended doses of daily first-line anti-TB drugs, given as fixed-dose combination dispersible tablets, and prescribed according to weight bands, give appropriate drug exposures when compared with historical paediatric and adult pharmacokinetic (PK) data
-
2.
To determine, among HIV-infected children, whether currently recommended adjusted strategies and doses of antiretroviral drugs can appropriately overcome the effect of rifampicin at the new higher doses
Methods
Trial design
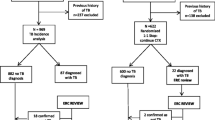
SHINE is an open-label, phase II/III, non-inferiority trial. Participants are randomised in a 1:1 ratio to the experimental 4-month or standard 6-month treatment arm. Both arms have an 8-week intensive phase with rifampicin, isoniazid, pyrazinamide and ethambutol followed by a continuation phase with rifampicin and isoniazid given for 8 weeks in the shorter arm vs 16 weeks in the standard arm (Fig. 1).

Trial schema
Setting and population
The study recruits children aged < 16 years diagnosed with minimal non-severe TB from diverse populations in three sites in Africa (Cape Town, South Africa; Kampala, Uganda; Lusaka Zambia) and two sites in India (Pune and Chennai). The trial sites were selected based on the expertise of treating children with TB, sufficient clinical trial experience and the potential for recruiting the required number of suitable participants within the agreed recruitment period.
The patient inclusion and exclusion criteria are detailed in Table 1.
Definition of minimal (non-severe) TB
Minimal TB in this protocol refers to a form of non-severe, symptomatic, smear-negative TB including extrathoracic lymph node TB, intrathoracic uncomplicated lymph node TB and other non-severe forms of pulmonary TB as per an established classification system [51].
Endpoints
Primary outcome
The main trial outcome for efficacy is categorised as follows:
-
1.
Favourable outcome; TB-free survival at week 72 post randomisation
-
2.
Unfavourable outcomes; a composite of death, reinfection or recurrence
-
3.
Not assessable; will include unknown outcomes at trial completion such as losses to follow-up after completing treatment and clinically well when last seen.
The safety endpoint will be grade 3 and 4 adverse events.
Secondary outcomes
-
1.
Mortality
-
2.
Adverse drug reactions within 30 days of completing treatment
-
3.
Relapse/re-infection-free survival including only cases adjudicated to be TB by an independent Endpoint Review Committee
-
4.
HIV viral suppression at weeks 24 and 48 in HIV-infected children
-
5.
Adherence and acceptability
-
6.
Bacterial infections requiring hospitalisation
Screening and enrolment procedures
The standard screening procedures include: a chest radiograph (CXR) and at least two consecutive respiratory samples including gastric aspirate, expectorated or induced sputum for smear microscopy, Xpert MTB/RIF© (Xpert; Cepheid, Sunnydale, CA, USA), culture and drug-susceptibility testing (DST). In children with extrathoracic cervical lymphadenopathy, lymph node needle aspiration will be undertaken. A Mantoux test or IGRA (interferon-gamma release assay) will be done as available.
At enrolment, a clinical history and physical examination is performed including measurements of weight, height, mid-upper arm circumference, paediatric WHO staging for HIV and signs/symptoms suggestive of TB.
Randomisation and allocation
Randomisation is undertaken at each site using a web-based system controlled through an authorised username and password. The method of minimisation with a random element is employed. To reduce potential predictability of the randomisation in this open-label study, the minimisation factors are not disclosed to the investigators.
Trial interventions
Children randomised to the 4-month arm receive rifampicin, isoniazid and pyrazinamide (plus ethambutol where given as part of standard care treatment) for an 8-week intensive phase followed by isoniazid and rifampicin for a continuation phase of a further 8 weeks. In the 6-month arm, children receive the same intensive phase as the other group followed by a continuation phase of 16 weeks.
TB treatment products, dosing schedule and treatment supervision
Anti-TB drugs are administered according the WHO dosing recommendations using new FDCs and new weight bands for children weighing < 25 kg (Table 2). Children weighing ≥ 25 kg are treated according to adult guidelines (Table 3).
Treatment is implemented in accordance with each site’s local practice. Typically this involves daily administration by the child’s carer(s) and documenting ingestion on TB treatment cards provided by the trial.
Adherence to trial medication is assessed by pill counts, specific adherence questions at each 4-weekly visit, as well as the number of doses recorded on the TB treatment card.
Protocol treatment discontinuation
Protocol treatment discontinuation is considered under any of the following circumstances:
1. Unacceptable toxicity or adverse event
2. Disease progression necessitating change of regimen (e.g. with drug-resistant TB or complication of disease)
3. Withdrawal of consent for treatment or participation by the participant’s carer/guardian
4. Participant’s DST-identifying drug resistance, including isoniazid mono-resistance
The children whose protocol treatment is discontinued remain in the trial for the purpose of follow-up and data analysis (unless the carer withdraws consent). Data are kept and included for participants who stop follow-up early, up to the point of exit.
Non-trial treatment
HIV-infected children enrolled in the trial are provided with ART in accordance with local country programmes including alternative ART drugs regimens indicated for concomitant use with rifampicin during the duration of the TB treatment. All HIV-infected children with TB receive co-trimoxazole prophylaxis as per local guidelines.
Trial assessments and follow-up
All participants are seen at screening, enrolment/randomisation and then at weeks 2, 4, 8, 12, 16, 20, 24, 28, 36, 48, 60 and finally at week 72, when they will exit the trial (Fig. 2).

SHINE trial assessment and follow-up schedules
A symptom checklist is performed at each visit to detect inter-current illnesses, new TB exposure including DR-TB, TB treatment failure/recurrence or relapse or adverse events to TB drugs (and ART if applicable). Respiratory samples for TB assessment and blood samples for biochemistry and haematology, as well as lymphocyte subsets and HIV1 viral load in HIV-infected children, are obtained as summarised in Fig. 2. Relevant samples are stored for pharmacogenomics, biomarkers and hair PK substudies at pre-specified time points (Fig. 2). A quality of life questionnaire, adapted for younger children from a standardised EQ-5D questionnaire [52], is administered at weeks 0, 16, 24, 48 and 72.
Participants are expected to attend all scheduled visits unless agreed in advance with the study team. Additional study visits are arranged when a participant misses a clinic visit or whenever clinical care is required.
Procedures for assessment of TB diagnosis and treatment response
TB diagnosis and treatment response will be adjudicated by an Endpoint Review Committee (ERC) with independent chair and membership. The ERC will review the endpoints (treatment failure or recurrence) and cause of death against standard criteria [23], blinded to treatment allocation, based on all available clinical, microbiological and molecular data obtained from the follow-up assessments and procedures. TB disease severity at diagnosis will be ascertained using Wiseman criteria [51].
CXRs are repeated at week 24, for suspected treatment failure or TB recurrence, and if clinically indicated. All CXRs will be prospectively read by the study site clinician to check eligibility for enrolment and retrospectively reviewed centrally by independent experts blind to treatment allocation using a standardised approach. Where available, Mantoux/IGRA is repeated at week 12 or 16 if the initial test was negative or missed at screening.
At each visit a clinical examination for TB signs and symptoms and new TB exposure is conducted to identify children with possible treatment failure or recurrence. Repeat specimens for smear microscopy and culture for M. tuberculosis are collected if clinically indicated. For children with culture-positive respiratory specimens, repeat TB microscopy and culture are attempted every 2 months until culture conversion. Lymph node needle aspiration is not routinely repeated during follow-up.
Procedures for assessing safety
Symptoms checklists and laboratory investigations are performed as part of safety monitoring at predetermined intervals (Fig. 2). In the case of adverse events, additional tests are done as clinically indicated.
To monitor for the potential ocular toxicity with ethambutol use, colour-vision testing is done at screening and at week 8 for children aged 3 years and older, with repeat testing at 16 weeks only in those patients with abnormal results. Children with obvious symptoms at any time within the intensive phase of treatment are referred to an ophthalmologist for further testing as appropriate. In the event of confirmed ocular toxicity, ethambutol is stopped.
For girls of reproductive age, or who have started menses, a pregnancy test is performed at screening. Any girl found to be pregnant is not eligible for the trial. Girls who become pregnant during the trial will be closely monitored and followed for treatment and pregnancy outcome.
Substudies
Pharmacokinetic (PK) studies
The trial includes two PK substudies. The first includes 65 children and describes the PK parameters of rifampicin, isoniazid and pyrazinamide when administered in the new FDC according to WHO weight-band doses in children weighing < 25 kg, or the adult formulation for children 25 to < 37 kg [18]. Ethambutol will be measured in the subset of children treated with this fourth drug.
The second substudy evaluates PK interactions between anti-TB and antiretroviral drugs (efavirenz, lopinavir, ritonavir, nevirapine, abacavir and zidovudine) in up to 60 HIV-infected children on ART. Children have cross-over intensive PK undertaken during the continuation phase and 4 weeks after stopping anti-TB treatment.
For both studies, seven blood samples (0.8 ml each) are collected before drug intake and then at 1, 2, 4, 6, 8 and 12 h after drug intake for intensive PK.
Hair PK sub-study
Novel assays to measure concentrations of two TB drugs, isoniazid and pyrazinamide, in small hair samples have been developed and validated. These assays have the potential to monitor adequate drug adherence and exposure and need further investigation. In the SHINE study, small hair samples are being collected in around 600 consenting participants to evaluate hair concentrations of isoniazid and pyrazinamide as surrogates of adherence and predictors of TB treatment outcomes (e.g. treatment success, recurrence and death) among HIV-infected and uninfected children.
Health economics
Economic evaluation of the alternative interventions will be undertaken. Resource use data are collected in the trial on health care visits, hospitalisations, TB and HIV medications, concomitant medications, and diagnostic tests. Estimates of the total costs of the two treatment strategies from a health sector perspective will be made using nationally representative unit costs combined with resource use data. Health outcomes will be estimated in terms of life years gained (LYGs), quality-adjusted life years (QALYs) and disability-adjusted life years (DALYs). The adapted quality of life questionnaire [52] will be combined with existing tariffs to estimate QALYs. Existing DALY weights for TB and TB-free survival will be used.
The nature of the economic analyses will largely depend upon the trial results. If the shortened regimen arm is non-inferior, and is not associated with any additional costs, this will indicate its cost-effectiveness. The cost savings from adopting this strategy at the national level will then be estimated based upon national estimates of childhood TB incidence. If the longer regimen arm is shown to be more effective, a cost-effectiveness analysis will be undertaken to investigate whether this represents ‘value for money’ to each of the health care systems in terms of the cost per QALY/DALY/LYG, and estimates of the opportunity costs of resources required to fund the intervention.
Social science
The substudy evaluates palatability and acceptability of the new FDC using acceptability questionnaires and semi-structured interviews in health facilities and at the homes of a subset of the South African trial population. Challenges with administration of the anti-TB drugs and differences between clinical advice and practice, as well as changes in administration and acceptability over the period of treatment, will be described.
Statistical considerations
Sample size and power
It is expected that TB recurrence (relapse or re-infection) and mortality rates will differ by HIV status and co-administration of ART. Based on estimates from the ARROW trial (Uganda and Zimbabwe) [53] and routine data from paediatric HIV clinics in South Africa, we estimate mortality in children HIV-negative, HIV-infected children on and not on ART to be 2%, 5% and 7%, respectively, over 18 months. Corresponding % estimates for TB recurrence are 3%, 4% and 6%, respectively, in these three groups. We assume that the proportion of HIV-infected children will be approximately 35% maximum (averaged across sites), of whom the majority will be on ART at the start of TB therapy. Using these estimates, the overall rate of TB recurrence or death was estimated at approximately 8%.
Non-inferiority margin
In discussion with study investigators, 6% was considered a reasonable margin of non-inferiority for the experimental arm for the endpoint of relapse or re-infection-free survival. This has been used in previous treatment-shortening phase III trials in adult smear-positive TB [45, 46].
Based on these assumptions, the total number of evaluable children required to demonstrate non-inferiority between the short-regimen arm and the control arm (90% power, one-sided 97.5% confidence intervals (CIs)), would be 860 across two arms. Based on experience from other phase III TB trials, 10% loss-to-follow-up was considered reasonable, resulting in a total of 956 children.
It is expected that a proportion of enrolled children treated for TB will not actually have TB, thus reducing the recurrence rate and increasing the chance of demonstrating non-inferiority. For this reason, although the primary analysis will include all randomised children (with assessable outcomes), a key secondary analysis will exclude children (assumed 20%) considered by the independent Blinded-endpoint Review Committee not to have TB at enrolment.
Due to the importance of this analysis the trial will be powered to demonstrate non-inferiority also in this sub-population, resulting in a total of 1200 children to be enrolled.
Analysis plan
There will be two main analysis populations: the modified intention-to-treat (MITT) and per-protocol (PP) analysis populations. While both of these will be considered primary in order to declare non-inferiority, consistent results should be obtained from key subgroup analyses (confirmed TB cases, both MITT and PP) and sensitivity analyses, although not all of these will necessarily need to satisfy the strict definition of non-inferiority.
The MITT analysis will include all randomised participants excluding those withdrawn from treatment because of a protocol violation at enrolment (late exclusions from the study, based on data collected prior to randomisation). The PP analysis will be restricted to participants who have received an adequate amount of study treatment. Any further exclusions in both MITT and PP populations will be detailed in the statistical analysis plan.
Definition of adequate treatment
The definition of adequate treatment sets limits both for the amount of treatment missed in each treatment phase and the amount of treatment missed overall. This definition of adequate treatment has been used in adult TB trials and is based on receiving at least 80% of the prescribed doses within a pre-specified time frame [45].
Primary efficacy analysis
The primary efficacy analysis will be the difference in proportions of the unfavourable outcomes (proportion with unfavourable outcome on the 4-month regimen arm less the proportion with unfavourable outcome on the 6-month regimen) adjusted for the minimisation factors.
Non-inferiority will be assessed using the upper bound of the one-sided 97.5% CI for this difference. If the upper bound of this CI is less than 6% (the margin of non-inferiority) the 4-month regimen will be considered non-inferior to the 6-month regimen.
PK substudies analysis plan
Non-linear mixed effects (NLME) models will be used to describe the population PK of the TB and ART drugs taking account of age, weight, race (African/Asian) and actual dose. In addition to describing the PK for each drug, these models would account for any non-linearity in the PK related to dose.
Differences in the Area Under the Curve (AUC) by age, HIV status, and between Asian vs African would be determined using model-based individual post-hoc Bayesian estimates. The geometric mean ratios of the post-hoc estimates of the AUCs for the TB drugs in the FDC compared to single drugs in regulatory approved products will be computed and evaluated against bioequivalence criteria.
Safety reporting
Adverse events and adverse reactions
The definitions of adverse events (AE) and adverse reactions (AR) outlined in the EU Directive 2001/20/EC Article 2 based on the principles of ICH GCP apply to this trial protocol (Table 4) [54].
The investigational medicinal products in this trial will be rifampicin, isoniazid, pyrazinamide and ethambutol. ARs include any untoward or unintended response to any of these drugs.
All AEs meeting the standard definitions above are reported, regardless of their relationship to TB. The seriousness of the events is assessed using the ICH-GCP definitions, and severity is determined in accordance with the 2014 Division of AIDS toxicity grading scale [55]. The causality and expectedness of all serious events or reactions in relation to the trial drugs is categorised as unrelated, unlikely, possibly, probably and definitely related, and for the latter three categories as expected and unexpected based on the Summary of Product Characteristics.
All SAEs, grade 3/4 adverse reactions and notable events (ocular toxicities, pregnancies and bacterial infections requiring hospitalisation) are notified to the Medical Research Council Clinical Trials Unit (MRC CTU) within one working day of the investigator becoming aware of the event. Other reportable events are reported within seven working days. The investigators at each site are also responsible for the onward reporting of adverse events to their ethics and regulatory bodies, as per their local reporting requirements. MRC CTU is responsible for annual onward reporting of a line listing of events to the trial sponsor, University College London (UCL). Investigators are responsible for reporting all SAEs, notable events and other adverse events meeting their reporting requirements that occur from the time of randomisation until 30 days after the last protocol treatment administration.
Quality assurance, data management and monitoring
UCL is responsible for trial oversight and ensuring that arrangements are in place for adequate management, monitoring, analysis and reporting of the trial.
Quality assurance and quality control are based on a formal risk assessment which leads to the development of the trial quality management plan implemented by the sponsor. On-site monitoring and data validation and assessment of adherence to International Conference on Harmonisation-Good Clinical Practice (ICH-GCP) principles is regularly conducted by independent local monitors at each site and periodically directly by MRC CTU on behalf of the sponsor. Each site is responsible for its own data entry and local trial management.
Trial governance
The trial is overseen by a number of committees each with distinct functions and membership. The Trial Steering Committee (TSC) consists of the chief investigator, site principal investigators and independent members. The TSC provides overall supervision for the trial and advice through its independent chair. The ultimate decision for the continuation of the trial lies with the TSC.
The Independent Data Monitoring Committee (IDMC) reviews confidential, accumulating data for the trial produced by the trial statisticians. The functions and frequency of its meeting are dictated by the DMC charter. The IDMC can recommend premature closure or reporting of the trial.
The Endpoint Review Committee (ERC), blinded to the trial arm, will determine the primary endpoint classification for all participants as favourable, unfavourable or unassessable. In addition, the ERC will examine evidence and adjudicate causes of death based on all available sources of data and determine the TB diagnosis at enrolment across sites for the key secondary analysis of those with definitive TB [23].
Discussion
SHINE is the first large-scale paediatric trial that will test whether 4-month therapy is as effective as the standard 6-month course in children with non-severe TB. Recently, three large multicentre trials to determine whether fluoroquinolone-containing regimens can shorten treatment from 6 to 4 months did not demonstrate non-inferiority of the 4-month regimens in adults with drug susceptible TB [44,45,46]. In SHINE, unlike in these adult trials, smear-negative children with non-severe paucibacillary disease will use revised doses of first-line anti-TB drugs. In addition, the trial will also demonstrate whether the PK parameters and safety profile of the new dosing recommendations for rifampicin, isoniazid, pyrazinamide and ethambutol across populations of Indian and African children are adequate. As TB often occurs alongside HIV infection, SHINE will provide answers for optimal anti-TB drug dosing in HIV co-infected children on concomitant ART. The nested economic and social science substudies enable SHINE to rapidly translate findings into budget relevant, patient-oriented, and context-sensitive policy guidance.
TB studies in children have long been neglected owing to the perception that paediatric TB is not a public health priority. Paediatric TB trials, in particular, face challenges because of difficulties in confirming TB bacteriological conversion as a study endpoint [8]. While acknowledging the limitations associated with the establishment of paediatric TB diagnosis and evaluating endpoints, both clinically diagnosed and bacteriologically confirmed cases of non-severe TB will be included, reflecting the pragmatic approaches utilised in routine clinical settings where children are diagnosed, treated and monitored predominantly using clinical and radiological parameters. The TB diagnoses and primary endpoints will be adjudicated by an independent ERC, blinded to the study arms, against the standard paediatric TB research consensus criteria [23, 51, 56]. The trial will utilise a centralised imaging review by independent paediatric TB radiology experts to assist with decisions on the endpoints. Although the main primary analysis will include all randomised children, it is anticipated that some children might not have TB, thus contributing to a non-inferiority outcome. To overcome this, the study was powered for demonstration of non-inferiority in a key subpopulation considered to have TB by the ERC.
The trial includes HIV-infected children as current evidence suggests no association between treatment duration and outcomes in patients on ART receiving rifampicin-containing regimens [57]. According to WHO guidance and existing programmatic approaches, HIV-infected and uninfected children should be treated for the same duration [18, 36]. Ethically, the risk profile of the trial will be similar to existing recommended approaches in routine clinical practice for TB treatment in children, regardless of HIV status.
Economic analyses will assess the cost-effectiveness of reducing treatment from 6 months to 4 months while the social science analyses will evaluate the palatability and acceptability of using the recently implemented paediatric anti-tuberculosis FDCs.
SHINE will confirm whether or not treatment-shortening of drug susceptible TB in children is efficacious and safe. It will also fill existing gaps in knowledge on dosing of the new anti-TB formulations and commonly used HIV drugs in settings with a high burden of childhood TB. A positive result from this trial (i.e. if 4 months is shown to be non-inferior to 6 months) will be to simplify treatment, to potentially improve adherence and to reduce treatment costs for a large proportion of children with TB (Additional file 1).
Trial status
The first child was enrolled to SHINE in July 2016 and recruitment is due to finish by the end of June 2018.
Abbreviations
- 3TC:
-
Lamivudine
- ABC:
-
Abacavir
- AE:
-
Adverse event
- ALT:
-
Alanine aminotransferase
- AR:
-
Adverse reaction
- ART:
-
Antiretroviral therapy
- ARV:
-
Antiretroviral
- AUC:
-
Area under the curve
- CTU:
-
Clinical trials unit
- CXR:
-
Chest x-ray
- DALY:
-
Disability-adjusted life years
- DST:
-
Drug-susceptibility test
- E:
-
Ethambutol
- EFV:
-
Efavirenz
- ERC:
-
Endpoint Review Committee
- FDC:
-
Fixed-dose combination
- GCP:
-
Good clinical practice
- H:
-
Isoniazid
- Hb:
-
Haemoglobin
- HIV:
-
Human immunodeficiency virus
- ICH:
-
International Conference on Harmonisation
- IDMC:
-
Independent Data Monitoring Committee
- IGRA:
-
Interferon-gamma release assay
- IRB:
-
Institutional review board
- ISRCTN:
-
International standard randomised controlled trial number
- LPV:
-
Lopinavir
- LPV/r:
-
Lopinavir with ritonavir
- LYG:
-
Life years gained
- MDR:
-
Multiple drug resistant tuberculosis
- MITT:
-
Modified intention-to-treat
- MRC:
-
Medical Research Council
- NNRTI:
-
Non-nucleoside reverse transcriptase inhibitor
- NRTI:
-
Nucleoside reverse transcriptase inhibitor
- NVP:
-
Nevirapine
- PK:
-
Pharmacokinetics
- PP:
-
Per protocol
- QALY:
-
Quality-adjusted life year
- R:
-
Rifampicin
- SAE:
-
Serious adverse event
- TB:
-
Tuberculosis
- TSC:
-
Trial Steering Committee
- UCL:
-
University College London
- WHO:
-
World Health Organisation
- ZDV:
-
Zidovudine
References
WHO. Global Tuberculosis Report 2017. Geneva: World Health Organisation; 2017.
Hesseling AC, Cotton MF, Jennings T, Whitelaw A, Johnson LF, Eley B, Roux P, Godfrey-Faussett P, Schaaf HS. High incidence of tuberculosis among HIV-infected infants: evidence from a South African population-based study highlights the need for improved tuberculosis control strategies. Clin Infect Dis. 2009;48(1):108–14.
Schaaf HS, Marais BJ, Hesseling AC, Brittle W, Donald PR. Surveillance of antituberculosis drug resistance among children from the Western Cape Province of South Africa—an upward trend. Am J Public Health. 2009;99(8):1486–90.
Schaaf HS, Marais BJ, Whitelaw A, Hesseling AC, Eley B, Hussey GD, Donald PR. Culture-confirmed childhood tuberculosis in Cape Town, South Africa: a review of 596 cases. BMC Infect Dis. 2007;7:140.
Dodd PJ, Yuen CM, Sismanidis C, Seddon JA, Jenkins HE. The global burden of tuberculosis mortality in children: a mathematical modelling study. Lancet Glob Health. 2017;5(9):e898–906.
Oliwa JN, Karumbi JM, Marais BJ, Madhi SA, Graham SM. Tuberculosis as a cause or comorbidity of childhood pneumonia in tuberculosis-endemic areas: a systematic review. Lancet Respir Med. 2015;3:235–43.
Venturini E, Turkova A, Chiappini E, Galli L, de Martino M, Thorne C. Tuberculosis and HIV co-infection in children. BMC Infect Dis. 2014;14(Suppl 1):S5.
Burman WJ, Cotton MF, Gibb DM, Walker AS, Vernon AA, Donald PR. Ensuring the involvement of children in the evaluation of new tuberculosis treatment regimens. PLoS Med. 2008;5(8):e176.
Marais BJ, Gie RP, Schaaf HS, Hesseling AC, Obihara CC, Starke JJ, Enarson DA, Donald PR, Beyers N. The natural history of childhood intra-thoracic tuberculosis: a critical review of literature from the pre-chemotherapy era. Int J Tubercul Lung Dis. 2004;8(4):392–402.
Lincoln EM, Harris LC, Bovornkitti S, Carretero RW. Endobronchial tuberculosis in children, a study of 156 patients. Am Rev Tubercul. 1958;77(1):39–61.
Marais BJ, Hesseling AC, Gie RP, Schaaf HS, Enarson DA, Beyers N. The bacteriologic yield in children with intrathoracic tuberculosis. Clin Infect Dis. 2006;42(8):e69–e71.
Marais BJ, Gie RP, Schaaf HS, Hesseling AC, Enarson DA, Beyers N. The spectrum of disease in children treated for tuberculosis in a highly endemic area. Int J Tubercul Lung Dis. 2006;10(7):732–8.
Weber HC, Beyers N, Gie RP, Schaaf HS, Fish T, Donald PR. The clinical and radiological features of tuberculosis in adolescents. Ann Trop Paediatr. 2000;20(1):5–10.
Zar HJ, Hanslo D, Apolles P, Swingler G, Hussey G. Induced sputum versus gastric lavage for microbiological confirmation of pulmonary tuberculosis in infants and young children: a prospective study. Lancet. 2005;365(9454):130–4.
Fox TG. Occult tuberculous infection in children. Tubercle. 1977;58(2):91–6.
Toppet M, Malfroot A, Hofman B, Casimir G, Cantraine F, Dab I. Tuberculosis in children: a 13-year follow up of 1714 patients in a Belgian home care centre. Eur J Pediatr. 1991;150(5):331–5.
Schaaf HS, Beyers N, Gie RP, Nel ED, Smuts NA, Scott FE, Donald PR, Fourie PB. Respiratory tuberculosis in childhood: the diagnostic value of clinical features and special investigations. Pediatr Infect Dis J. 1995;14(3):189–94.
WHO. Guidance for national tuberculosis programmes on the management of tuberculosis in children. 2nd ed. Geneva: World Health Organisation; 2014.
Abernathy RS, Dutt AK, Stead WW, Moers DJ. Short-course chemotherapy for tuberculosis in children. Pediatrics. 1983;72(6):801–6.
Al-Dossary FS, Ong LT, Correa AG, Starke JR. Treatment of childhood tuberculosis with a six month directly observed regimen of only two weeks of daily therapy. Pediatr Infect Dis J. 2002;21(2):91–7.
Ramachandran P, Kripasankar AS, Duraipandian M. Short-course chemotherapy for pulmonary tuberculosis in children. Indian J Tuber. 1998;45(2):83–87.
Swaminathan S, Raghavan A, Duraipandian M, Kripasankar AS, Ramachandran P. Short-course chemotherapy for paediatric respiratory tuberculosis: 5-year report. Int J Tubercul Lung Dis. 2005;9(6):693–6.
Graham SM, Cuevas LE, Jean-Philippe P, Browning R, Casenghi M, Detjen AK, Gnanashanmugam D, Hesseling AC, Kampmann B, Mandalakas A, et al. Clinical case definitions for classification of intrathoracic tuberculosis in children: an update. Clin Infect Dis. 2015;61(Suppl 3):S179–87.
WHO. Rapid Advice: Treatment of tuberculosis in children. Geneva: World Health Organisation; 2010.
McIlleron H, Willemse M, Werely CJ, Hussey GD, Schaaf HS, Smith PJ, Donald PR. Isoniazid plasma concentrations in a cohort of South African children with tuberculosis: implications for international pediatric dosing guidelines. Clin Infect Dis. 2009;48(11):1547–53.
Schaaf HS, Parkin DP, Seifart HI, Werely CJ, Hesseling PB, van Helden PD, Maritz JS, Donald PR. Isoniazid pharmacokinetics in children treated for respiratory tuberculosis. Arch Dis Child. 2005;90(6):614–8.
Graham SM, Bell DJ, Nyirongo S, Hartkoorn R, Ward SA, Molyneux EM. Low levels of pyrazinamide and ethambutol in children with tuberculosis and impact of age, nutritional status, and human immunodeficiency virus infection. Antimicrob Agents Chemother. 2006;50(2):407–13.
Thee S, Detjen A, Wahn U, Magdorf K. Rifampicin serum levels in childhood tuberculosis. Int J Tubercul Lung Dis. 2009;13(9):1106–11.
Thee S, Detjen A, Wahn U, Magdorf K. Pyrazinamide serum levels in childhood tuberculosis. Int J Tubercul Lung Dis. 2008;12(9):1099–101.
Thee S, Seddon JA, Donald PR, Seifart HI, Werely CJ, Hesseling AC, Rosenkranz B, Roll S, Magdorf K, Schaaf HS. Pharmacokinetics of isoniazid, rifampin, and pyrazinamide in children younger than two years of age with tuberculosis: evidence for implementation of revised World Health Organization recommendations. Antimicrob Agents Chemother. 2011;55(12):5560–7.
Bekker A, Schaaf HS, Draper HR, van der Laan L, Murray S, Wiesner L, Donald PR, McIlleron HM, Hesseling AC. Pharmacokinetics of rifampin, isoniazid, pyrazinamide, and ethambutol in infants dosed according to revised WHO-recommended treatment guidelines. Antimicrob Agents Chemother. 2016;60(4):2171–9.
McIlleron H, Hundt H, Smythe W, Bekker A, Winckler J, van der Laan L, Smith P, Zar HJ, Hesseling AC, Maartens G, et al. Bioavailability of two licensed paediatric rifampicin suspensions: implications for quality control programmes. Int J Tubercul Lung Dis. 2016;20(7):915–9.
Detjen A, Macé C, Perrin C, Graham SM, Grzemska M. Adoption of revised dosage recommendations for childhood tuberculosis in countries with different childhood tuberculosis burdens. Public Health Action. 2012; https://doi.org/10.5588/pha.12.0052. [Accessed 28 Dec 2017]
TB-Alliance-and-Unitaid. New pathways for childhood TB treatment—Lessons from the STEP TB Project. In: TB Alliance, Unitaid; 2017.
New fixed-dose-combinations for the treatment of TB in children. http://www.who.int/tb/FDC_Factsheet.pdf. [Accessed 28 Dec 2017]
WHO. Consolidated guidelines on the use of antiretroviral drugs for treating and preventing hiv infection—recommendation for a public health approach. 2nd ed. Geneva: World Health Organisation; 2016.
Ren Y, Nuttall JJ, Egbers C, Eley BS, Meyers TM, Smith PJ, Maartens G, McIlleron HM. Effect of rifampicin on lopinavir pharmacokinetics in HIV-infected children with tuberculosis. J Acquir Immune Defic Syndr. 2008;47(5):566–9.
Bwakura-Dangarembizi M, Kendall L, Bakeera-Kitaka S, Nahirya-Ntege P, Keishanyu R, Nathoo K, Spyer MJ, Kekitiinwa A, Lutaakome J, Mhute T, et al. A randomized trial of prolonged co-trimoxazole in HIV-infected children in Africa. N Engl J Med. 2014;370(1):41–53.
Oudijk JM, McIlleron H, Mulenga V, Chintu C, Merry C, Walker AS, Cook A, Gibb DM, Burger DM. Pharmacokinetics of nevirapine in HIV-infected children under 3 years on rifampicin-based antituberculosis treatment. AIDS. 2012;26(12):1523–8.
Hong Kong Chest Service/Madras Tuberculaosis Rearsrch Centre/British MRC. A controlled trial of 2-month, 3-month, and 12-month regimens of chemotherapy for sputum-smear-negative pulmonary tuberculosis. Results at 60 months. Am Rev Respir Dis. 1984;130(1):23–8.
Hong Kong Chest Service/Madras Tuberculaosis Rearsrch Centre/British MRC. A controlled trial of 3-month, 4-month, and 6-month regimens of chemotherapy for sputum-smear-negative pulmonary tuberculosis. Results at 5 years. Hong Kong Chest Service/Tuberculosis Research Centre, Madras/British Medical Research Council. Am Rev Respir Dis. 1989;139(4):871–6.
Teo SK, Tan KK, Khoo TK. Four-month chemotherapy in the treatment of smear-negative pulmonary tuberculosis: results at 30 to 60 months. Ann Acad Med Singap. 2002;31(2):175–81.
WHO. Tuberculosis control workskop report and revision of ‘Treatment of tuberculosis: guidelines for national programs’. Geneva: World Health Organisation; 1995.
Merle CS, Fielding K, Sow OB, Gninafon M, Lo MB, Mthiyane T, Odhiambo J, Amukoye E, Bah B, Kassa F, et al. A four-month gatifloxacin-containing regimen for treating tuberculosis. N Engl J Med. 2014;371(17):1588–98.
Gillespie SH, Crook AM, McHugh TD, Mendel CM, Meredith SK, Murray SR, Pappas F, Phillips PP, Nunn AJ. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N Engl J Med. 2014;371(17):1577–87.
Jindani A, Harrison TS, Nunn AJ, Phillips PP, Churchyard GJ, Charalambous S, Hatherill M, Geldenhuys H, McIlleron HM, Zvada SP, et al. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N Engl J Med. 2014;371(17):1599–608.
Violari A, Cotton MF, Gibb DM, Babiker AG, Steyn J, Madhi SA, Jean-Philippe P, McIntyre JA. Early antiretroviral therapy and mortality among HIV-infected infants. N Engl J Med. 2008;359(21):2233–44.
Seddon JA, Hesseling AC, Willemse M, Donald PR, Schaaf HS. Culture-confirmed multidrug-resistant tuberculosis in children: clinical features, treatment, and outcome. Clin Infect Dis. 2012;54(2):157–66.
Donald PR, Maritz JS, Diacon AH. The pharmacokinetics and pharmacodynamics of rifampicin in adults and children in relation to the dosage recommended for children. Tuberculosis (Edinb). 2011;91(3):196–207.
Schaaf HS, Cilliers K, Willemse M, Labadarios D, Kidd M, Donald PR. Nutritional status and its response to treatment of children, with and without HIV infection, hospitalized for the management of tuberculosis. Paediatr Int Child Health. 2012;32(2):74–81.
Wiseman CA, Gie RP, Starke JR, Schaaf HS, Donald PR, Cotton MF, Hesseling AC. A proposed comprehensive classification of tuberculosis disease severity in children. Pediatr Infect Dis J. 2012;31(4):347–52.
EuroQuol: EQ-5D™ a standardised instrument for use as a measure of health outcome. 2015. https://euroqol.org/eq-5d-instruments/. Accessed 6 Apr 2018.
Kekitiinwa A, Cook A, Nathoo K, Mugyenyi P, Nahirya-Ntege P, Bakeera-Kitaka S, Thomason M, Bwakura-Dangarembizi M, Musiime V, Munderi P, et al. Routine versus clinically driven laboratory monitoring and first-line antiretroviral therapy strategies in African children with HIV (ARROW): a 5-year open-label randomised factorial trial. Lancet. 2013;381(9875):1391–403.
European-Commission. EU Directive 2001/20/EC Article 2. In: European Commission; 2001.
U.S. Department of Health and Human Services, National Institutes of Health, National Institute of Allergy and Infectious Diseases, Division of AIDS. Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.0. [November 2014]. Available from:http://rsc.tech-res.com/docs/default-source/safety/daids_ae_grading_table_v2_nov2014.pdf. [Accessed 6 Apr 2018]
Graham SM, Ahmed T, Amanullah F, Browning R, Cardenas V, Casenghi M, Cuevas LE, Gale M, Gie RP, Grzemska M, et al. Evaluation of tuberculosis diagnostics in children: 1. Proposed clinical case definitions for classification of intrathoracic tuberculosis disease. Consensus from an expert panel. J Infect Dis. 2012;205(Suppl 2):S199–208.
Ahmad Khan F, Minion J, Al-Motairi A, Benedetti A, Harries AD, Menzies D. An updated systematic review and meta-analysis on the treatment of active tuberculosis in patients with HIV infection. Clin Infect Dis. 2012;55(8):1154–63.
Acknowledgements
The SHINE team would like to thank all the staff in all the participating sites and the children and families that have participated in the trial to date.
Funding
SHINE is funded by the Joint Global Health Trials Scheme of the Department for International Development, UK (DFID), the Wellcome Trust and the Medical Research Council (MRC UK), Grant number MR/L004445/1; and TB Alliance. This work was supported by core support to the UK Medical Research Council [MC_UU_12023/26].
Trial drugs are manufactured by Macleods Pharmaceuticals Ltd.
The funders, the sponsor (University College London) and the drug manufacturer had no role in the study design or report writing.
Author information
Authors and Affiliations
Consortia
Contributions
CC drafted the first version of this manuscript. DG, MC, AC and AT conceived and designed the overall study with contributions from MT, AH, EW, CC, VMa, SH, MvdZ, PM, VMu, AK, PKB, RA, HM, AMD and MP. HM, RA and VMa led the design and implementation of the pharmacokinetic substudies and GH of the social science studies. AMD leads the TB laboratory components of the trial and MP is responsible for clinical radiology aspects. AT, EW, PAR, MvdZ, MK, SB, PKB and AK are responsible for patient recruitment, care and follow-up. MT and EOP are responsible for the study management, co-ordination and governance. AC developed the statistical methods of the study. All authors reviewed the manuscript for intellectual content and approved the final version of the report.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The trial has been approved by the Ethics Committees of the all participating sites in South Africa (Stellenbosch University and University of Cape Town Research Ethics Committees), Uganda (Joint Clinical Research Centre Institutional Review Board), Zambia (University of Zambia Biomedical Research Ethics Committee and National Health Research Ethics Committee), India (BJMC Ethics Committee-Pune, National Institute of Research in Tuberculosis Ethics Committee-Chennai and Health Ministry Screening Committee) and by University College London (UCL Research Ethics Committee). The study was also approved by the regulatory authorities in all the participating countries.
Written informed consent (parents/caregivers) and assent (children whose understanding is considered adequate) will be obtained before any trial procedures are carried out and participants’ confidentiality will be maintained throughout the trial in line with the standard ICH-GCP principles.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
SPIRIT 2013 Checklist: recommended items to address in a clinical trial protocol and related documents. (DOC 125 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chabala, C., Turkova, A., Thomason, M.J. et al. Shorter treatment for minimal tuberculosis (TB) in children (SHINE): a study protocol for a randomised controlled trial. Trials 19, 237 (2018). https://doi.org/10.1186/s13063-018-2608-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-018-2608-5