
Abstract
Background
There is an unprecedented rise in the prevalence of stroke in sub-Saharan Africa (SSA). Secondary prevention guidelines recommend that antihypertensive, statin and antiplatelet therapy be initiated promptly after ischemic stroke and adhered to in a persistent fashion to achieve optimal vascular-risk reduction. However, these goals are seldom realized in routine clinical care settings in SSA due to logistical challenges.
We seek to assess whether a polypill containing fixed doses of three antihypertensive agents, a statin and antiplatelet therapy taken once daily per os for 12 months among recent stroke survivors would result in carotid intimal thickness regression compared with usual care (UC).
Methods
The Stroke Minimization through Additive Anti-atherosclerotic Agents in Routine Treatment (SMAART) trial is a phase 2, open-label, evaluator-blinded trial involving 120 Ghanaian recent-ischemic-stroke survivors. Using a computer-generated sequence, patients will be randomly allocated 1:1 into either the intervention arm or UC. Patients in the intervention arm will receive Polycap DS® (containing aspirin, 100 mg; atenolol, 50 mg; ramipril, 5 mg; thiazide, 12.5 mg; simvastatin, 20 mg) taken as two capsules once daily. Patients in the UC will receive separate, individual secondary preventive medications prescribed at the physician’s discretion. Both groups will be followed for 12 months to assess changes in carotid intimal thickness regression – a surrogate marker of atherosclerosis – as primary outcome measure. Secondary outcome measures include adherence to therapy, safety and tolerability, health-related quality of life, patient satisfaction, functional status, depression and cognitive dysfunction.
Discussion
An efficacy-suggesting SMAART trial could inform the future design of a multi-center, double-blinded, placebo-controlled, parallel-group, randomized controlled trial comparing the clinical efficacy of the polypill strategy for vascular risk moderation among stroke survivors in SSA.
Trial registration
ClinicalTrials.gov, ID: NCT03329599. Registered on 11 February 2017.
Similar content being viewed by others


Background
There has been an astronomical rise in the prevalence of stroke in sub-Saharan Africa (SSA) which, when compared to stroke profiles in high-income countries (HIC) is characterized by a younger age of onset, higher early and long-term fatality rates, and more severe cognitive deficits, emotional and social isolation and disability among survivors [1,2,3,4,5,6,7,8,9,10]. Stroke survivors in SSA (vs. HIC) are especially at high risk for recurrent vascular events or death due to several contextural factors including uncoordinated health systems, under-controlled vascular risk factors [11, 12], and lack of care affordability.
Among stroke survivors, a major source of subsequent mortality and functional decline is from recurrent stroke and myocardial infarction (MI) [13,14,15,16,17]. Prevention of future vascular events is critical to reducing the morbidity/mortality of patients with stroke, since the risk is highest within 1 year of the index stroke [18, 19]. Antihypertensive, antithrombotic, anticoagulant, antidiabetic and lipid-lowering therapies form the foundation of modern secondary preventive strategies for strokes and other types of cardiovascular disease (CVD) [20]. Combination therapy using a statin, aspirin and antihypertensive agents has been associated with reductions in stroke, myocardial infarction (MI) and mortality risk compared to monotherapy [21], and an 80% reduction in overall cardiovascular event risk when aspirin, beta-blockers, lipid-lowering drugs and angiotensin-converting enzyme inhibitors (ACEIs) are used simultaneously [22]. Hence, most guidelines recommend that secondary prevention interventions for stroke (comprising antihypertensive, statin and antiplatelet therapy) should be initiated promptly after stroke and adhered to in a persistent fashion to achieve the goals of risk reduction for vascular events [23, 24]. However, these goals are seldom realized in routine clinical care settings, especially in Africa, due to logistical and affordability challenges [25,26,27,28,29,30,31,32,33,34,35]. Of note, the neurologist:population ratio in SSA ranges from 1 per 162,885 persons to none in 11 countries (vs. 1 per 29,200 persons in the US) [36], underscoring the need to identify relatively simple strategies that can be applied broadly for stroke survivors in resource-constrained settings.
The use of evidence-based therapies for vascular risk reduction among stroke patients receiving conventional care in low- and middle-income countries (LMIC) is extremely low [37]. Usually, an individual recovering from stroke is prescribed multiple medications to treat various risk factors indefinitely, and this often engenders poor adherence and non-persistence with these efficacious, evidence-based, preventive therapies [25,26,27,28,29,30,31,32,33,34,35]. Indeed, reports emanating from HIC suggest that continual utilization of secondary prevention medications is challenged, with short-to-medium-term persistence rates ranging between 60 to 90% [29,30,31,32,33,34]. Thus, non-adherence, treatment complexity, pill burden and limited expert input are the principal antagonists militating against widespread compliance to life-saving cardiovascular-disease-prevention medication interventions, leading the World Health Organization (WHO) to advocate for interventions that will address the factors contributing to non-adherence to CVD treatments as a priority issue [38].
Fixed-dose combination pills, also known as “polypills,” containing generic drugs: aspirin, a statin and blood pressure (BP)-lowering medication(s) may be a viable low-cost avenue to broadly improve medication adherence and consequently reduce further disability or death on a large scale among stroke survivors in SSA [39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54]. The principal objectives for the polypill strategy are improving drug adherence by reducing pill burden and thereby improving risk factor control and potentially reducing vascular event risk as a cost-effective intervention [44]. Feasibility studies have been conducted using polypills vs. “usual care,” (UC) with strong evidence for improved adherence [55,56,57], superior, or at least non-inferior, efficacy in systolic BP (SBP) and low-density lipoprotein cholesterol (LDL-C) control [40, 41, 46, 50, 55,56,57], better acceptability [55,56,57] and comparable safety profile and better cost-effectiveness in both LMIC and HIC [58,59,60,61]. Although these benefits are yet to translate into significantly higher reductions in hard outcomes vs. UC, the majority of the available studies have had a relatively short duration of follow-up (on average 8 weeks to 6 months) whereas it has been previously established that there is at least a 12–24-month lag phase before the maximum benefits of SBP and LDL-C reduction by these medications are typically observed [62, 63].
Hence, the overarching objective of the Stroke Minimization through Additive Anti-atherosclerotic Agents in Routine Treatment (SMAART) trial is to assess whether a polypill containing fixed doses of three antihypertensive agents, a statin and antiplatelet therapy taken once daily per os for 12 months would result in carotid intimal thickness regression, improved adherence, and tolerability compared with the UC group on separate individual secondary preventive medications among Ghanaian first-time stroke survivors.
Methods
Trial design
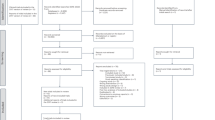
SMAART is a phase II, randomized, open-label, blinded-endpoint clinical trial to evaluate the effect of a polypill taken once daily per os in improving CIMT regression and adherence to secondary risk reduction drug intake vs. a UC group with separate individual secondary preventive medications among Ghanaians with recent-onset stroke or transient ischemic attack (TIA) in a single-center study. See Fig. 1 for the study algorithm.

Stroke Minimization through Additive Anti-atherosclerotic Agents in Routine Treatment (SMAART) trial algorithm
Study settings
SMAART will be conducted at the Komfo Anokye Teaching Hospital (KATH), a tertiary-referral center and teaching hospital with 1000 beds. There is a stroke unit and a neurology outpatient service [64]. Stroke survivors encountered at the Family Medicine, Internal Medicine and Neurology Outpatient Clinics will be enrolled into the trial to capture UC experiences of stroke patients as well as the use of the polypill in these diverse settings. Each year, on average 450 stroke survivors are discharged from the hospital and approximately 300 stroke survivors are seen at the Family Medicine, Internal Medicine, Neurology Outpatient Clinics at KATH. Of note, the site has built up experience in stroke epidemiological studies as well as clinical trials involving stroke survivors [65,66,67].
Study participants
The participants will include 120 adult Ghanaian recent stroke/TIA patients (within 2 months of stroke onset) meeting the inclusion/exclusion criteria who will be randomly assigned to either the intervention or the UC arm.
Inclusion criteria
-
1.
Above the age of 18 years; male or female
-
2.
Stroke/TIA diagnosis no longer than 2 months before enrollment. Only ischemic strokes including lacunar and large-vessel atherosclerotic subtypes are eligible
-
3.
Legally competent to sign informed consent
Exclusion criteria
-
1.
Unable to sign informed consent and having no proxy
-
2.
Contraindications to any of the components of the polypill
-
3.
Severe cognitive impairment/dementia or severe global disability limiting the capacity of self-care
-
4.
Severe congestive cardiac failure (NYHA grades III–IV)
-
5.
Severe renal disease, estimated glomerular filtration rate (eGFR) < 30 ml/min/1.73 m2), renal dialysis; awaiting renal transplant, or transplant recipient
-
6.
Cancer diagnosis or treatment in the past 2 years
-
7.
Nursing/pregnant mothers
-
8.
Participants not agreeing to the filing, forwarding or use of their pseudonymized data
Case definition
The diagnosis of stroke will be defined as an acute episode of focal cerebral, spinal or retinal dysfunction caused by infarction of central nervous system tissue, not resulting in death. Patients meeting the eligibility criteria will be allocated to the experimental or active comparator arm. Subjects with stroke may also present with at least one of the following additional conditions: documented diabetes mellitus or previous treatment with an orally administered hypoglycemic agent or insulin; documented hypertension (HT) > 140/90 mmHg or previous treatment with antihypertensive medications; mild to moderate renal dysfunction (eGFR 60–30 ml/min/1.73 m2); or prior MI.
Intervention
Patients allocated to the experimental arm will receive two Polycap DS® (containing aspirin, 100 mg; atenolol, 50 mg; ramipril, 5 mg; thiazide, 12.5 mg; simvastatin, 20 mg) taken per os once a day. Polycap DS® is produced by Cadila Pharmaceuticals, Ahmedabad, India. Patients assigned to the polypill will have their antihypertensive agents, lipid modifiers and antithrombotic agents withdrawn and replaced with the polypill if they are already receiving such treatments before enrollment. Since our focus is to isolate the effect of the polypill strategy itself and create equipoise, at study inception providers for patients in both study arms will receive a brief one-off training and a one-off email synopsis on guideline-recommended biomarker targets after stroke [23]. To maximize participant’s adherence to the study medications, the following procedures will be implemented:
-
Participant education during all visits on the importance of taking study medication, including timing, storage and what to do in the event of a missed dose
-
Participants will be instructed to return unused capsules at each follow-up visit, and returned capsules will be counted and recorded by the study team
-
Supervised dosing during face-to-face visits, when appropriate
-
Application of the Morisky Green Questionnaire (MAQ) to participants at each yearly follow-up visit
-
Participants will be provided with contact details of the responsible researcher so that they can make contact if for any reason they are unable to continue their study medication or have missed multiple doses and are unsure whether to continue
Usual care group
Patients allocated to the UC arm will receive standard-of-care therapies for secondary prevention with drugs and doses left to the discretion of the treating physicians. Measures outlined to ensure adherence to study medications will be also be deployed in the UC group.
Management of possible treatment-related side effects
The following strategies will be employed for patients with side effects deemed due to study medication and sufficiently severe to warrant a change of treatment:
-
If there are clear contraindications to one or more of the antihypertensive agents – ACEI, beta-blocker or thiazide; antithrombotic agent or lipid modifiers due to side effects, the polypill will be stopped and an open-label treatment with other components of the polypill excluding the class(es) for which the contraindication has developed
-
If there are symptoms of severe hypotension, one approach will be to stop the polypill, introduce the individual components gradually at reduced doses and consider re-starting at a later date
-
Indication for a high dose of a particular agent – the agent will be added as open label without the need to unblind
Discontinuation of study treatment
Participants who have had the study drug discontinued for any reason other than the above should be encouraged to restart the study drug as soon as practically and medically appropriate at the discretion of the investigator.
The investigator must not deviate from the protocol except when there is a contraindication due to the patient’s condition or the patient is intolerant of the drugs. In these cases the study treatment should be discontinued. Study treatment should also be discontinued if any of the following occurs:
-
1.
Serious adverse events (SAEs) which are, in the opinion of the investigator, related to the study treatment
-
2.
The investigator feels it is in the subject’s best interest
In the event of discontinuation of treatment, the patient should still remain in follow-up and attend study visits as scheduled. Physicians would then prescribe other medications for risk factor control and reasons for discontinuation documented.
Relevant concomitant care and interventions permitted or prohibited during the trial
Other medications deemed necessary for the management of other comorbidities, such as diabetes mellitus, will be permitted.
Study procedures
Recruitment of study subjects
A total number of 120 patients will be randomized (1:1) to treatment arms. Patients with stroke meeting the inclusion/exclusion criteria will be recruited from the Family Medicine, Internal Medicine and Neurology Outpatient Clinics at KATH. Randomization will take place within 8 weeks of the index event (ischemic stroke) in a 1:1 ratio to one of the two arms: polypill versus UC.
Allocation and concealment
Randomization of subjects in blocks of 4 will be conducted by a statistician using a computer-generated random sequence of numbers. Consented participants will be assigned to either study arm at the baseline visit using the computer-generated randomization sequence. The randomization sequence uses a minimization algorithm to ensure balance of prognostic factors such as stroke severity, an average of three recorded baseline SBP measurements, and whether or not they are taking background BP-lowering agents, lipid modifiers and antithrombotic agents. Each sequence generated will be kept concealed in an envelope which will be opened by the research coordinator in the presence of the consenting study participant at enrollment.
Blinding
Physicians and sonographers who will be assessing primary outcomes and research assistants assessing feasibility and secondary outcomes will remain blinded as to patients’ group status throughout the study.
Screening evaluation
All potentially eligible participants referred to, or who contact the study team directly, will undergo screening to determine their eligibility using the pre-specified criteria.
Enrollment evaluation
Information on stroke type from a cranial computed tomography (CT) scan performed within 10 days of stroke symptom onset will be reviewed by the principal investigator (PI) (FSS); stroke subtype information where available will be sought to classify ischemic stroke using the TOAST classification [68] into cardio-embolic, large-vessel and lacunar ischemic stroke. Stroke severity and functional status at enrollment will be assessed using the modified NIHSS [69] and Modified Rankin Score [70], followed by a detailed assessment of vascular risk factors, namely HT, diabetes mellitus, dyslipidemia, cigarette smoking, cardiac diseases (atrial fibrillation, ischemic heart disease, cardiomyopathies), from history and physical examination. Blood samples for baseline assessments of renal and liver function tests, lipid profile and HbA1C will be collected and contraindications for study medications assessed. All concomitant medications will be recorded in the Case Report Form.
Follow-up and outcome evaluations
Participants will be followed for 12 months with scheduled visits at months 1, 3, 6, 9 and 12 for clinical assessments and primary, secondary and tertiary/feasibility study outcome evaluations as shown in Table 1 and the Additional file 1: Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) Checklist and SPIRIT Figure (Fig. 2). Prior to scheduled visits, patients will be reminded of their appointment 1 week and a day before the visit using telephone calls. Arrangements will be made for home visits for participants unable to attend visits or visits will be re-scheduled within 2 weeks where home visits are not feasible.

Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) Figure: SMAART trial protocol – schedule of enrollment, intervention and assessments
Primary outcome measure
The primary endpoint will be CIMT burden. Each study participant will undergo carotid Doppler ultrasonic evaluation at baseline and month 12 for evidence of clinical or sub-clinical carotid artery disease – a validated surrogate marker of atherosclerosis [71]. To achieve reliable ultrasonic measurements of the common carotid artery IMT a standardized protocol and strict quality control procedures will be followed by two local experienced sonographers who will be blinded to the participants group status and risk factor levels to ensure that we achieve unbiased results [72, 73]. CIMT will be measured at 1-cm portions of the distal left and right common carotid artery far walls with a linear transducer (transducer frequency of 7.5 MHz) with axial resolution of 0.10 mm, and calculated automatically over three cardiac cycles following the Mannheim consensus [73]. The average thickness of the left and right carotid arteries will be used as the outcome measure.
Secondary outcome measures
These will include (1) medication adherence indicators assessed using the self-reported Morisky-Green Questionnaire, pill count and medication possession ratio; (2) safety and tolerability indicators such as changes in renal and liver function tests, side effects and treatment discontinuations; (3) health-related quality of life; (4) patient satisfaction; (5) changes in cognitive function; (6) depression and (7) functional status.
Tertiary outcome measures
These will include CVD risk-factor-control indices, such as change in mean SBP between baseline and month 12, as well as changes in mean LDL-C from baseline to month 12. Major adverse cardiovascular events, such as recurrent stroke, MI, CVD-related deaths and all-cause mortality, are included as tertiary outcome measures.
Management of possible treatment-related side effects
Participants who experience side effects will be reviewed by their physicians to assess severity and appropriate measures instituted.
Sample size justification
The end-of-study CIMT value will be subtracted from the baseline CIMT value and divided by the length of follow-up. The rate of change in CIMT (mm/year) between treatment arms will be tested with a two-sided t test. The rate of change in common CIMT in treated patients is around 0.085 mm/year with a standard deviation of 0.035 [74]. We assume that polypill improvement leads to a halting of CIMT progression with an assumed rate of change of 0.0825 mm/year. With a two-sided alpha of 0.05 and a 90% power we need 82 patients (104 patients with 20% dropout rate).
Data management
All data entry will be collected onto a Case Record Form (CRF) and doubly entered into a password-protected, validated, encrypted study electronic CRF on REDCap. This web-based data management system will allow for real-time data query generation for values entered outside of pre-set valid ranges and consistency checking. This system will speed up data reporting and assist overall trial management. The Internet-based data management system is managed at the Kwame Nkrumah University of Science and Technology (KNUST) and hosted by the Medical University of South Carolina server, which has extensive experience in clinical trial data capture and security. Data entry will be performed at the KNUST/KATH using a password-protected, encrypted HTTPS connection. Only staff listed in the delegation log will be given the unique individual password to access the Internet-based data management system. The database will undergo logic checks to ensure that data are entered in mandatory fields and undergo value-range checks to ensure accuracy and to reduce the chance of missing data. Reports and data query management will also be included in the system to assist with centralized online monitoring by the data manager and statistician.
Statistical analysis plan
Prior to addressing each hypothesis, univariate descriptive statistics and frequency distributions will be calculated as appropriate for all biological variables (including gender, age) comparing individuals by treatment arms (polypill arm vs. UC arm). Briefly, box plots will be used to examine the relative distribution of variables stratified by treatment arm. Non-parametric and equivalent parametric statistics will be utilized to compare groups. Appropriate regression models (linear regression for continuous outcomes such as carotid-media thickness; Cox proportional hazards regression for time-to-event measures such as recurrent strokes, CVD events, deaths and defaults) will be used to estimate the association of covariates with each outcome. When building models for each specific aim, the first stage of the model building algorithm will involve testing whether the individual covariates are correlated with the main outcome variables. A liberal alpha = 0.20 will be used for these unadjusted analyses [75]. Once the initial pool of candidate predictors has been identified, regression models consisting of multiple covariates will be fitted to identify potential confounders and effect modifiers. To achieve unbiased and robust results, optimal combinations of predictors, including interaction effects, will be identified and used for further analysis based upon whether or not they are a confounder, by whether they did not improve the model fit, or increased the standard error of the parameter estimate of the primary covariates. Finally, each model will be rigorously assessed for collinearity and goodness-of-fit using residual analysis. Model diagnostics will be performed using tools (in SAS or STATA) that detect outliers and influential data points. We will use diagnostic measures, such as residual deviance, the hat matrix diagonal and residual chi-squared deviance and the difference between chi-square goodness-of-fit, when an observation is deleted [76]. Plots of these against predicted values will be used to investigate the influence of each data point on the model. We will handle missing data using several techniques including multiple imputation and propensity score methods [77, 78].
Data Safety Monitoring Board (DSMB)
A DSMB, comprised of three experienced external experts, will meet twice per year to review the safety, ethics and outcomes of the study. The responsibilities of the DSMB will include monitoring blinded response variables and safety outcomes for early dramatic benefits or potential harmful effects and provide reports on recommendations to continue or temporarily halt recruitment to the study. The DSMB will be governed by a charter that will outline their responsibilities, procedures and confidentiality, and will review unblinded data from the study at regular intervals during follow-up and monitor BP differences between the two groups, dropout rates and event rates. The first meeting will be held within 3–6 months after the start of the study recruitment. One or two formal interim analysis will be planned to review data relating to patient safety and quality of trial conduct.
Harms
All SAEs and adverse events of special interest (AESI) experienced by a participant after the informed consent document is signed and until the end of the study will be collected and reported to the Institutional Review Board and applicable regulatory guidelines. If an SAE is unresolved at the conclusion of the study, a clinical assessment will be made by the medical monitor as to whether continued follow-up of the SAE is warranted.
Discussion
The overarching objective of the proposed SMAART trial is to assess whether a polypill containing fixed doses of three antihypertensive agents, a statin and antiplatelet therapy taken once daily per os would result in carotid intimal thickness regression, improved adherence and tolerability among first-time stroke survivors in SSA. The ultimate objective would be the design of a future multi-center, double-blinded, placebo-controlled, parallel-group randomized controlled trial (RCT) comparing the clinical efficacy of the polypill strategy vs. UC in the African context to derive locally relevant, high-quality evidence for routine deployment of polypill for CVD risk moderation among stroke survivors in LMIC. There are six major RCTs on polypills currently on-going or planned, three of these RCTs – TIPS-3 [79], HOPE-3 [80] and HOPE-4 [81] – are primary prevention studies, two of them – PROPS [82] and SECURE [83] – are secondary prevention trials and PolyIran study [84] is aimed at both primary and secondary CVD prevention. As far as we are aware, a potential SMAART trial could be the first study to evaluate the feasibility of a polypill on secondary risk reduction among stroke survivors in SSA with the possibility of generating results that will inform the future research agenda and policy on the utility of the polypill for comprehensive vascular risk reduction in the region.
We anticipate the following potential challenges:
-
1.
Subject accrual and retention: multiple tactics will be used to facilitate high patient retention including obtaining full contact information for patients, caregivers and relatives at time of enrollment and updating every 3 months. Transportation support will be provided when needed and follow-up visits will be scheduled, whenever possible, on days that subjects already have scheduled clinic visits
-
2.
Meeting sample size: there is a high case load of stroke patients with approximately 700 cases per year. At a recruitment rate of five cases per week, recruitment of 120 stroke patients meeting eligibility criteria is a realistic objective
-
3.
Missing data: we anticipate that there may be missing data due to loss to follow-up but we will minimize this by home visits where geographically feasible, providing transportation stipends and employing statistical methodology to account for missing data
Trial status
Recruitment is anticipated to begin in April 2018.
Abbreviations
- ACEI:
-
Angiotensin-converting enzyme inhibitor
- ALT:
-
Alanine transaminase
- AST:
-
Aspartate transaminase
- BP:
-
Blood pressure
- CIMT:
-
Carotid intima-media thickness
- CT:
-
Computerized tomography
- CVD:
-
Cardiovascular disease
- DBP:
-
Diastolic blood pressure
- eGFR:
-
Estimated glomerular filtration rate
- EQ-5D:
-
European Quality of Life Questionnaire-5 Dimensional
- HIC:
-
High-income countries
- HOPE-3:
-
Heart Outcomes Prevention Evaluation-3
- HOPE-4:
-
Heart Outcomes Prevention Evaluation-4
- HTN:
-
Hypertension
- KATH:
-
Komfo Anokye Teaching Hospital
- LDL-C:
-
Low-density lipoprotein cholesterol
- LMIC:
-
Low- and middle-income countries
- MOCA:
-
Montreal Cognitive Assessment
- NIHSS:
-
National Institute of Health Stroke Scale
- NSTEMI:
-
Non-ST segment elevated myocardial infarction
- NYHA:
-
New York Heart Association
- PROPS:
-
Preventative Role Of a fixed dose combination Pill in Stroke
- RCT:
-
Randomized controlled trial
- SBP:
-
Systolic blood pressure
- SECURE:
-
Secondary Prevention of Cardiovascular Disease in the Elderly Trial
- SMAART:
-
Stroke Minimization through Additive Anti-atherosclerotic Agents in Routine Treatment
- SSA:
-
Sub-Saharan Africa
- STEMI:
-
ST segment elevated myocardial infarction
- TIPS-3:
-
The International Polycap Study 3
- UC:
-
Usual care
- WHO:
-
World Health Organization
References
Feigin VL, Lawes CM, Bennett DA, Barker-Collo SL, Parag V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: a systematic review. Lancet Neurol. 2009;8(4):355–69.
Feigin VL. Stroke epidemiology in the developing world. Lancet. 2005;365:2160–1.
Sarfo FS, Acheampong JW, Tetteh LA, Oparebea E, Akpalu A, Bedu-Addo G. The profile of risk factors and in-patient outcomes of stroke in Kumasi, Ghana. Ghana Med J. 2014;48(3):127–34.
Sarfo FS, Awuah DO, Nkyi C, Akassi J, Opare-Sem OK, Ovbiagele B. Recent patterns and predictors of neurological mortality among hospitalized patients in Central Ghana. J Neurol Sci. 2016;363:217–24.
Sarfo FS, Akassi J, Kyem G, Adamu S, Awuah D, Kantanka OS, et al. Long-term outcomes of stroke in a Ghanaian outpatient clinic. J Stroke Cerebrovasc Dis. 2018;27(4):1090–9.
Sarfo FS, Jenkins C, Singh A, Owolabi M, Ojagbemi A, Adusei N, Saulson R, Ovbiagele B. Post-stroke depression in Ghana: characteristics and correlates. J Neurol Sci. 2017;379:261–5.
Ojagbemi A, Owolabi M, Atalabi M, Baiyewu O. Stroke lesions and post-stroke depression among survivors in Ibadan, Nigeria. Afr J Med Sci. 2013;42(2):245–51.
Sarfo FS, Nichols M, Qanungo S, Teklehaimanot A, Singh A, Mensah N, et al. Stroke-related stigma among West Africans: patterns and predictors. J Neurol Sci. 2017;375:270–4.
Sarfo FS, Akassi J, Adamu S, Obese V, Ovbiagele B. Burden and predictors of vascular cognitive impairment among long-term Ghanaian stroke survivors. J Stroke Cerebrovasc Dis. 2017;26(11):2553–62.
Owolabi MO. Consistent determinants of post-stroke health-related quality of life across diverse cultures: Berlin-Ibadan study. Acta Neurol Scand. 2013;128(5):311–20.
Mensah G, Roth G, Sampson U, Moran A, Feigin V, Forouzanfar MH, et al. Mortality from cardiovascular diseases in sub-Saharan Africa, 1990–2013: a systematic analysis of data from the Global Burden of Disease Study 2013. Cardiovasc J Afr. 2015;26:S6–S10.
Owolabi MO, Karolo-Anthony S, Akinyemi R, Arnett D, Gebregziabher M, Jenkins C, et al. The burden of stroke in Africa: a glance at the present and a glimpse into the future. Cardiovasc J Afr. 2015;26(2 Suppl 1):S27–38.
Bronnum-Hansen H, Davidsen M, Thorvaldsen P, for the Danish MONICA Study Group. Long-term survival and cause of death after stroke. Stroke. 2001;32:2131–6.
Kernan WN, Viscoli CM, Brass LM, Makuch RW, Sarrel PM, Roberts RS, et al. The Stroke Prognosis Instrument II (SPI-II): a clinical prediction instrument for patients with transient ischemic and non-disabling ischemic stroke. Stroke. 2000;31:456–62.
Petty GW, Brown RD, Whisnant JP, Sicks JD, O’Fallon WM, Wiebers DO. Survival and recurrence after first cerebral infarction. A population-based study in Rochester, Minnesota 1975–1989. Neurology. 1998;50:208–16.
Viscoli CM, Brass LM, Kernan WN, Sarrel PM, Suissa S, Horwitz RI. Estrogen replacement after ischemic stroke: report of the Women’s Estrogen for Stroke Trial (WEST). N Engl J Med. 2001;345:1243–9.
Vernino S, Brown RD, Sejvar JJ, Sicks JD, Petty GW, O’Fallon M. Cause-specific mortality after cerebral infarction. A population-based study. Stroke. 2003;34:1828–32.
Coull A, Lovett JK, Rothwell PM. Population based study of early risk of stroke after transient ischaemic attack or minor stroke: implications for public education and organisation of services. BMJ. 2004;328:326.
Kleindorfer D, Panagos P, Pancioli A, Khoury J, Kissela B, Woo D, et al. Incidence and short-term prognosis of transient ischemic attack in a population-based study. Stroke. 2005;36:720–3.
Sacco RL, Boden-Albala B, Gan R, Chen X, Kargman DE, Shea S, et al. Stroke incidence among white, black, and Hispanic residents of an urban community: the Northern Manhattan Stroke Study. Am J Epidemiol. 1998;147:259–68.
Lafeber M, Spiering W, van der Graaf Y, Nathoe H, Bots ML, Grobbee DE, et al. The combined use of aspirin, a statin, and blood pressure-lowering agents (polypill components) and the risk of vascular morbidity and mortality in patients with coronary artery disease. Am Heart J. 2013;166:282–9.
Yusuf S. Two decades of progress in preventing vascular disease. Lancet. 2002;360:2–3.
Kernan WN, Ovbiagele B, Black HR, Bravata DM, Chimowitz MI, Ezekowitz MD, et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(7):2160–236.
The European Stroke Organization (ESO) Executive Committee and the ESO Writing Committee. Guidelines for management of ischemic stroke and transient ischemic attack 2008. Cerebrovasc Dis. 2008;25:457–507.
Ho PM, Bryson CL, Rumsfeld JS. Medication adherence: its importance in cardiovascular outcomes. Circulation. 2009;119:3028–35.
Naderi SH, Bestwick JP, Wald DS. Adherence to drugs that prevent cardiovascular disease: meta-analysis on 376,162 patients. Am J Med. 2012;125:882–7.
Chowdhury R, Khan H, Heydon E, Shroufi A, Fahimi S, Moore C, et al. Adherence to cardiovascular therapy: a meta-analysis of prevalence and clinical consequences. Eur Heart J. 2013;34:2940–8.
Hamann GF, Weimar C, Glahn J, Busse O, Diener HC. Adherence to secondary stroke prevention strategies – results from the German Stroke Data Bank. Cerebrovasc Dis. 2003;15:282–8.
Glader EL, Sjolander M, Eriksson M, Lundberg M. Persistent use of secondary preventive drugs declines rapidly during the first 2 years after stroke. Stroke. 2010;41(2):397–401.
Bushnell CD, Olson DM, Zhao X, Pan W, Zimmer LO, Goldstein LB, et al. Secondary preventive medication persistence and adherence 1 year after stroke. Neurology. 2011;77(12):1182–90.
Ovbiagele B, Kidwell C, Selco S, Razinia T, Saver J. Treatment adherence rates one year after initiation of a systematic hospital-based stroke prevention program. Cerebrovasc Dis. 2005;20:280–2.
Lummis H, Sketris I, Gubitz G, Joffres M, Flowerdew G. Medication persistence rates and factors associated with persistence in patients following stroke: a cohort study. BMC Neurol. 2008;10:25. https://doi.org/10.1186/1471-2377-8-25. Accessed 19 Oct 2016.
Xu J, Ju Y, Wang C, Wang Y, Liu L, Zhao X, et al. Patterns and predictors of antihypertensive medication used 1 year after ischemic stroke or TIA in urban China. Patient Prefer Adher. 2013;7:71–9.
Ji R, Liu G, Shen H, Wang Y, Li H, Peterson E, et al. Persistence of secondary prevention medications after acute ischemic stroke or transient ischemic attack in Chinese population: data from China National Stroke Registry. Neurol Res. 2013;35(1):29–36.
Sarfo FS, Ovbiagele B, Akassi J, Kyem G. Baseline prescription and one-year persistence of secondary prevention drugs after an index stroke in central Ghana. eNeurologicalSci. 2017;6:68–73.
Owolabi MO, Bower JH, Ogunniyi A. Mapping Africa’s way into prominence in the field of neurology. Arch Neurol. 2007;64:1696–700.
Furie KL, Kasner SE, Adams RJ, et al. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(1):227–76.
World Health Organization. Adherence to long-term therapies: evidence for action. 2003.
Wiley B, Fuster V. The concept of the polypill in the prevention of cardiovascular disease. Ann Global Health. 2014;80:24–34.
Truelove M, Patel A, Bompoint S, Brown A, Cass A, Hills GS, et al. The effect of a cardiovascular polypill strategy on pill burden. Cardiovasc Ther. 2015;33:347–52.
Castellano JM, Sanz G, Penalvo JL, Bansilal S, Fernandez-Ortiz A, Alvarez L, et al. A polypill strategy to improve adherence: results from the FOCUS project. J Am Coll Cardiol. 2014;64:2071–82.
Castellano JM, Copeland-Halperin R, Fuster V. Aiming at strategies for a complex problem of medical nonadherence. Glob Heart. 2013;8:263–71.
Becerra V, Gracia A, Desai K, Abogunrin S, Brand S, Chapman R, et al. Cost-effectiveness and public health benefit of secondary cardiovascular disease prevention from improved adherence using a polypill in the UK. BMJ Open. 2015;5:e007111.
Wald DS, Morris JK, Wald NJ. Randomized polypill crossover trial in people aged 50 and over. PLoS One. 2012;7:e41297.
Group PC, Rodgers A, Patel A, Berwanger O, Bots M, Grimm R, et al. An international randomized placebo-controlled trial of a four-component combination pill (“polypill”) in people with raised cardiovascular risk. PLoS One. 2011;6:e19857.
Patel A, Cass A, Peiris D, Usherwood T, Brown A, Jan S, et al. A pragmatic randomized trial of a polypill-based strategy to improve use of indicated preventive treatments in people at high cardiovascular disease risk. Eur J Prev Cardiol. 2015;22:920–30.
Webster R, Patel A, Selak V, Billot L, Bots ML, Brown A, et al. Effectiveness of fixed dose combination medication (‘polypills’) compared with usual care in patients with cardiovascular disease or at high risk: a prospective, individual patient data meta-analysis of 3140 patients in six countries. Int J Cardiol. 2016;205:147–56.
Malekzadeh F, Marshall T, Pourshams A, Gharravi M, Aslani A, Nateghi A, et al. A pilot double-blind randomised placebo-controlled trial of the effects of fixed-dose combination therapy (“polypill”) on cardiovascular risk factors. Int J Clin Pract. 2010;64(9):1220-7.
Selak V, Elley CR, Crengle S, Harwood M, Doughty R, Arroll B, et al. IMProving Adherence using Combination Therapy (IMPACT) design and protocol of a randomized controlled trial in primary care. Contemp Clin Trials. 2011;32:909–15.
Soliman EZ, Mendis S, Dissanayake WP, Somasundaram NP, Gunaratne PS, Jayasingne IK, et al. A polypill for primary prevention of cardiovascular disease: a feasibility study of the World Health Organization. Trials. 2011;12:3.
Elley CR, Gupta AK, Webster R, Selak V, Jun M, Patel A, et al. The efficacy and tolerability of “polypills”: meta-analysis of randomised controlled trials. PLoS One. 2012;7(12):e52145.
Yusuf S, Pais P, Sigamani A, Xavier D, Afzal R, Gao P, et al. Comparison of risk factor reduction and tolerability of a full-dose polypill (with potassium) versus low-dose polypill (polycap) in individuals at high risk of cardiovascular diseases: the Second Indian Polycap Study (TIPS-2) investigators. Circ Cardiovasc Qual Outcomes. 2012;5:463–71.
Sarfo FS, Kyem G, Ovbiagele B, Akassi J, Sarfo-Kantanka O, Agyei M, et al. One-year rates and determinants of poststroke systolic blood pressure control among Ghanaians. J Stroke Cerebrovasc Dis. 2017;26(1):78-86.
Yusuf S, Pais P, Afzal R, Xavier D, Teo K, Eikelboom J, et al. Effects of a polypill (Polycap) on risk factors in middle-aged individuals without cardiovascular disease (TIPS): a phase II, double-blind, randomized trial. Lancet. 2009;373:1341–51.
Lafeber M, Grobbee DE, Schrover IM, Thom S, Webster R, Rodgers A, et al. Comparison of a morning polypill, evening polypill and individual pills on LDL-cholesterol, ambulatory blood pressure and adherence in high-risk patients; a randomized crossover trial. Int J Cardiol. 2015;181:193–9.
Thom S, Poulter N, Field J, Patel A, Prabhakaran D, Stanton A, et al. Effects of a fixed-dose combination strategy on adherence and risk factors in patients with or at high risk of CVD: the UMPIRE randomized clinical trial. JAMA. 2013;310:918–29.
Selak V, Elley CR, Bullen C, Crengle S, Wadham A, Rafter N, et al. Effect of fixed dose combination treatment on adherence and risk factor control among patients at high risk of cardiovascular disease: randomized controlled trial in primary care. BMJ. 2014;348:G3318.
Bautista LE, Vera-Cala LM, Ferrante D, Herrera VM, Miranda JJ, Pichardo R, et al. A “polypill” aimed at preventing cardiovascular disease could prove highly cost-effective for use in Latin America. Health Aff. 2013;32:155–64.
Ong KS, Carter R, Vos T, Kelaher M, Anderson I. Cost-effectiveness of interventions to prevent cardiovascular disease in Australia’s indigenous population. Heart Lung Circ. 2014;23:414–21.
Khonputsa P, Veerman LJ, Bertram M, et al. Generalized cost-effectiveness analysis of pharmaceutical interventions for primary prevention of cardiovascular disease in Thailand. Value Health Reg Issues. 2012;1:15–22.
Ito K, Shrank WH, Avorn J, Patrick AR, Brennan TA, Antman EM, et al. Comparative cost-effectiveness of interventions to improve medication adherence after myocardial infarction. Health Serv Res. 2012;47:2097–117.
Collins R, Peto R, MacMahon S, Hebert P, Fiebach NH, Eberlein KA, et al. Blood pressure, stroke, and coronary heart disease. Part 2, Short-term reductions in blood pressure: overview of randomized drug trials in their epidemiological context. Lancet. 1990;335:827–38.
Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomized trials of statins. Lancet. 2005;366:1267–78.
Sarfo FS, Akassi J, Badu E, Okorozo A, Ovbiagele B, Akpalu A. Profile of neurological disorders in an adult neurology clinic in Kumasi, Ghana. eNeurologicalSci. 2016;3:69–74.
Akpalu A, Sarfo FS, Ovbiagele B, Akinyemi R, Gebregziabher M, Obiako R, et al. Phenotyping stroke in sub-Saharan Africa: Stroke Investigative Research and Education Network (SIREN) phenomics protocol. Neuroepidemiology. 2015;45(2):73–82.
Sarfo FS, Treiber F, Jenkins C, Patel S, Gebregziabher M, Singh A, et al. Phone-based Intervention under Nurse Guidance after Stroke (PINGS): study protocol for a randomized controlled trial. Trials. 2016;17(1):436.
Sarfo FS, Treiber F, Gebregziabher M, Adamu S, Patel S, Nichols M, et al. PINGS (Phone-based Intervention under Nurse Guidance after Stroke): interim results of a pilot randomized controlled trial. Stroke. 2018;49(1):236–9.
Kolominsky-Rabas PL, Weber M, Gefeller O, Neundoerfer B, Heuschmann PU. Epidemiology of ischaemic stroke subtypes according to TOAST criteria: incidence, recurrence, and long-term survival in ischaemic stroke subtypes: a population-based study. Stroke. 2001;32:2735–40.
Lyden PD, Lu M, Levine S, Brott TG, Broderick J. A Modified National Institutes of Health Stroke Scale for use in stroke clinical trials. Preliminary Reliability and Validity. Stroke. 2001;32:1310–7.
Wilson JT, Hareendran A, Hendry A, Potter J, Bone I, Muir KW. Reliability of the modified Rankin Scale across multiple raters: benefits of a structured interview. Stroke. 2005;36(4):777–81.
Chambless LE, Heiss G, Folsom AR, Rosamond W, Szklo M, Sharrett AR, et al. Association of coronary heart disease incidence with carotid arterial thickness and major risk factors: the Atherosclerosis Risk in Communities (ARIC) Study, 1987–1993. Am J Epidemiol. 1997;146(6):483–94.
De Groot E, Hovingh GK, Wiegman A, Duriez P, Smit AJ, Fruchart JC, et al. Measurement of arterial wall thickness as a surrogate marker for atherosclerosis. Circulation. 2004;109(23 Suppl 1):III33–8.
Touboul PJ, Hennerici MG, Meairs S, Adams H, Amarenco P, Desvarieux M, et al. Mannheim intima-media thickness consensus. Cerebro Vasc Dis. 2004;18(4):346–9.
Naqvi TZ, Lee MS. Carotid intima-media thickness and plaque in cardiovascular risk assessment. JACC Cardiovasc Imaging. 2014;7(10):1025–38.
Lehmann E. Non-parametics: statistical methods based on ranks. Englewood Cliffs: Prentice Hall; 1998.
Hosmer DW, Lemeshow S. Applied logistic regression. 2nd ed. New York: Wiley; 2000.
Agresti A. Categorical data analysis. New York; Wiley; 2002.
Little RJA, Rubin DB. Statistical analysis with missing data. New York: Wiley; 2002.
Yusuf S. The International Polycap Study 3 (TIPS). 2015. ClinicalTrials.gov Identifier: NCT0164637. www.clinicaltrials.gov. Accessed 18 Oct 2016.
Heart Outcomes Prevention Evaluation-3 (HOPE-3). 2015. https://clinicaltrials.gov/ct2/show/NCT00468923, NCT00468923. Accessed 18 Oct 2016.
Heart Outcomes Prevention and Evaluation-4 (HOPE-4). 2016. https://clinicaltrials.gov/ct2/show/NCT01826019, NCT01826019. Accessed 19 Oct 2016.
Mant J. PROPS-preventative role of a fixed dose combination pill in stroke: a multi-centre open label randomized controlled trial of a fixed dose combination pill versus standard care for secondary prevention of stroke in a primary care setting (ISRCTN58452386). ISRCTN Registry, 2015.
Secondary Prevention of Cardiovascular Disease in the Elderly Trial (SECURE). 2015. https://clinicaltrials.gov/ct2/show/NCT02596126, NCT02596126. Accessed 19 Oct 2016.
Ostovaneh MR, Poustchi H, Hemmeing K, Marjani H, Pourshams A, Nateghi A, et al. Polypill for the prevention of cardiovascular disease (PolyIran): study design and rationale for a pragmatic cluster randomized controlled trial. Eur J Prev Cardiol. 2015;22:1609–17.
Morisky DE, Green LW, Levine DM. Concurrent and predictive validity of a self-reported measure of medication adherence. Med Care. 1986;24(1):67–74.
Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van LF, Greene T, Coresh J. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–12.
U.S. Department of Health and Human Services, National Institutes of Health, National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE). Version 4.0, 2010.
EuroQol Group. EuroQol—a new facility for the measurement of health-related quality of life. Health Policy (Amsterdam, Netherlands). 1990;16(3):199–208.
Atkinson MJ, Sinha A, Hass SL, Colman SS, Kumar RN, Brod M, et al. Validation of a general measure of treatment satisfaction, the Treatment Satisfaction Questionnaire for Medication (TSQM), using a national panel study of chronic disease. Health Qual Life Outcomes. 2004;2:12.
Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, et al. The Montreal Cognitive Assessment, MOCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 53(4):695–9.
Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–71.
Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62.
Acknowledgements
Not applicable.
Trial sponsor
Department of Neurology, Medical University of South Carolina, Charleston, SC, USA.
Funding
National Institute of Health-National Institute of Neurological Disorders and Stroke; R21 NS103752–01. The study sponsor and funders will play no active role in study design; collection, management, analysis and interpretation of data; writing of the report; and the decision to submit reports for publication and will have no ultimate authority over any of these activities.
Availability of data and materials
Not applicable.
Confidentiality
Data will be kept confidential and anonymized for analyses.
Dissemination plans
Study findings will be presented in conference abstracts, poster presentations and scientific publications in medical journals. The principal investigator will work with co-investigators to generate manuscripts for publications.
Author information
Authors and Affiliations
Contributions
Conception of the study: BO and FSS. Drafting the manuscript: FSS. Critical review of the manuscript: OSK, SA, VO, JV, RT, VS and BO. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval for the study was been obtained from The Committee of Human Research Publication and Ethics, KNUST, Ghana (reference number: CHRPE/AP/524/17) and the IRB at the Medical University of South Carolina, Charleston, SC, USA. Important protocol modifications will be sought from both ethics committees.
Informed consent: written informed consent will be obtained from all patients before enrollment by a trained research coordinator. Patients are free to withdraw at any time.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
SPIRIT 2013 Checklist: recommended items to address in a clinical trial protocol and related documents. (DOC 121 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sarfo, F.S., Sarfo-Kantanka, O., Adamu, S. et al. Stroke Minimization through Additive Anti-atherosclerotic Agents in Routine Treatment (SMAART): study protocol for a randomized controlled trial. Trials 19, 181 (2018). https://doi.org/10.1186/s13063-018-2564-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-018-2564-0