
Abstract
Background
Subacromial impingement syndrome (SIS) is a painful, and often long lasting, shoulder condition affecting patient function and quality of life. In a recent study, we observed major strength impairments in shoulder external rotation and abduction (~30%) in a population of patients with pronounced and long-lasting SIS. However, the current rehabilitation of such strength impairments may be inadequate, with novel rehabilitation programmes including exercise therapy only improving external rotation strength by 4–13%.
As these previous studies are the basis of current practice, this suggests that the strengthening component could be inadequate in the rehabilitation of these patients, and it seems likely that more emphasis should be placed on intensifying this part of the rehabilitation.
The purpose of this study is to investigate the effectiveness of a programme consisting of progressive home-based resistance training using an elastic band, aimed at improving shoulder external rotation and abduction strength, added to usual care and initiated shortly after diagnosis has been established.
Methods
A pragmatic randomised controlled superiority trial will be conducted, including 200 patients with pronounced and long-lasting SIS, diagnosed using predefined criteria. Participants will be randomised to receive either an add-on intervention of progressive home-based resistance training using an elastic band in addition to usual care or usual care alone in a 1:1 allocation ratio. The randomisation sequence is computer generated, with permuted blocks of random sizes. The primary outcome will be change in Shoulder Pain And Disability Index (SPADI) score from baseline to 16 weeks follow-up. Outcome assessors are blinded to group allocation. Intervention receivers will be kept blind to treatment allocation through minimal information about the content of the add-on intervention and control condition until group allocation is final. Analyses are performed by blinded data analysts.
Discussion
If effective, the simple shoulder strengthening exercise programme investigated in this trial could easily be added to usual care. The usefulness of the trial is further supported by the magnitude of the problem, the information gained from the study and the pragmatism, patient centeredness and transparency of the trial.
Trial registration
The trial is pre-registered at ClinicalTrials.gov with the ID NCT02747251 on April 19, 2016.
Similar content being viewed by others


Background
Shoulder disorders are the second most frequent musculoskeletal reason for contacting a general practitioner [1]. Nearly half of these incidents are categorised as subacromial impingement syndrome (SIS) [2], a painful, and often long lasting [3], condition affecting patient function and quality of life [4]. In Denmark, the incidence of SIS reported in primary care is approximately 8 per 1000 inhabitants per year [5]. Further, with an average yearly cost per incident case of shoulder disorder of €4000 [6], as reported in Sweden, the societal costs related to SIS are noteworthy. However, most of the costs (74%) are related to a sub-group of patients (12%) with high pain intensity and more pronounced and long-lasting disability [7]. In this sub-group, sick leave from paid work accounts for more than half (61%) of the costs [7].
The latest systematic review on the efficacy of exercise therapy in the treatment of SIS suggests that an exercise intervention improves patient-reported function and pain in patients with long-lasting SIS (≥ 3 months), to a degree equivalent to surgery followed by post-operative rehabilitation [8]. In addition, an intervention including specific exercises significantly reduced the amount of operations in patients on the waiting list for surgery for SIS [9]. Consistent with these studies, the Danish national clinical guidelines for treatment of SIS recommends an exercise intervention lasting at least 3 months [10]. However, it is unknown which specific components of the heterogeneous exercise interventions are associated with a better outcome [11] and, though not demonstrated in patients with long-lasting SIS, patient adherence to the prescribed exercise interventions is also likely to moderate the effects of such interventions.
Mechanisms
Resistance training, aimed at strengthening the rotator cuff muscles and scapula stabilising muscles, is an important component of most novel exercise interventions for long-lasting SIS [9, 12, 13]. This seems relevant as patients with SIS have significant force impairments in both the glenohumeral and the scapulothoracic joint [4, 14, 15]. However, these force impairments are most pronounced in the glenohumeral joint, with a 33% deficit in external rotation force and a 29% deficit in abduction force compared to only 8% and 18% force deficit in protraction and retraction of the scapula, respectively [15]. This suggests that specific training of the glenohumeral muscles is especially relevant. Such specific resistance training of the, often degenerated [16], rotator cuff muscles and tendons also seems relevant as this training modality is known to improve muscle and tendon health through various pathways [17].
Need for a trial
It is uncertain if the novel exercise intervention programmes, aimed at patients with long-lasting SIS, are focusing sufficiently on strengthening exercises. Accordingly, changes in muscle force are only tested in a few randomised controlled trials (RCTs), including resistance training in the rehabilitation of patients with SIS [12, 13, 18, 19]. In these studies, maximum force in external rotation increased by only 4–15% in average from baseline to follow-up in patients doing strengthening exercises [12, 13, 18, 19], which is far from restoring the 33% impairment in external rotation force previously reported [15]. Though strength gain may not be the primary aim of the intervention, the sparse effect on this objective outcome points towards a possible gap in the rehabilitation of patients with long-lasting SIS. This suggests that the exercise dose received by patients might have been too small, either because the prescribed resistance training programmes were too mild, or simply due to lack of adherence to the prescribed programmes. The trial described in this protocol will aim to investigate the effectiveness of adding a simple shoulder strengthening exercise programme to usual care in patients with long-lasting SIS.
Existing knowledge
To further ensure that we do not initiate a redundant trial, we have performed a systematic literature review. On January 26, 2016, we performed a systematic literature search on controlled trials investigating the effect of resistance training in patients with SIS. We searched Medline via PubMed (86 hits) and Embase via OVID (38 hits) using search strings for condition (shoulder impingement) and intervention (resistance training). We also searched www.clinicaltrials.gov for registered (finalised, ongoing or planned) trials. We only identified one trial [18] in which the effect of resistance training was isolated as the active part of the intervention. Additionally, in a recent systematic review, only one study [20] investigating the effect of an exercise intervention in patients with persistent SIS was identified, and in that study the exercise intervention was compared to surgery. In conclusion, the existing evidence regarding the effect of resistance training in patients with persistent SIS is very limited and we therefore consider this to provide a clear-cut ethical, scientific and economic justification for the trial described in this protocol.
Dose selection
As previously stated, the exercise dose received by patients might have been too small in previous studies, either because the prescribed resistance training programmes were too mild or simply due to lack of adherence to the prescribed programmes. In the study by Lombardi et al. [18], patients allocated to an intervention consisting of supervised strengthening exercises only increased 9% in external rotation strength (derived from Lombardi et al. [18]), which was not significantly different from the waiting list control group. As exercise sessions were supervised, this absence of effect is likely a consequence of the prescribed dose being too small, with exercises for flexion, extension, internal and external rotation performed twice a week in 8 weeks with 2 × 8 repetitions at 50% and 70% of 6RM, respectively. One way to efficiently increase the exercise dose in rehabilitation is to include home-based exercises, as was done in a study by Bennell et al. [12]. In that study, an intervention consisting of manual therapy and home exercises was found superior to placebo in improving abduction strength (significant 15% increase), but not external rotation strength (non-significant 4% increase), pain scores or patient-reported function at the end of treatment. The limited gains in shoulder strength observed in the study by Bennell et al. [12] might also be a consequence of an insufficient exercise dose received by the patients. However, it is difficult to determine whether the sparse improvements in shoulder strength are due to the programme being too mild or because of low adherence, as the amount of external resistance applied to exercises is not described. Nevertheless, exclusion of non-adherent patients from the analyses did not alter the results, indicating that the sparse improvements in shoulder strength were not only due to low adherence.
In the current study, we aim to increase the received dose of strengthening exercises aimed at the rotator cuff muscles in patients with long-lasting SIS without aggravating their symptoms and with minimal risk of adverse events. To achieve this, the intervention will start with exercises using low relative resistance but high volume (number of sets per session and frequency of sessions). This approach is also relevant, as resistance training with low intensity/resistance, high frequency and approximately 3–4 sets per muscle group is associated with the highest gains in muscle strength in untrained individuals [21]. All sets are to be continued to failure, as this maximises the gain in local muscle endurance [22]. Furthermore, the low resistance will minimise pain during exercises and protect the muscle from overload injuries, ensuring that all patients are able to complete the programme.
The focus of exercises is on isometric holds and slow dynamic strengthening with a long time under tension (TUT). A high TUT is achieved through exercises with a low contraction velocity and the inclusion of an isometric component to increase the exercise stimulus without increasing external resistance; this in turn reduces the peak load on the involved, and possibly damaged [16], muscles and tendons. Thus, exercises become safer and less painful, which again will allow patients to actually adhere to the exercise intervention. Furthermore, the isometric part will be performed with the shoulder in approximately 30–45 degrees of scaption, the position in which the compression forces on the supraspinatus muscle and tendon are lowest [23, 24]. Exercises will mainly focus on external rotation and abduction, functions in which both supraspinatus and infraspinatus muscles are highly active [25].
Possible effect modifiers
Patients with long-lasting SIS have lower mechanical pressure pain thresholds (PPT), both at the shoulder region and in other anatomical regions (such as the tibialis anterior), which may reflect an increased manifestation of peripheral and central pain sensitisation [26]. Pain sensitisation modulates the patients’ pain experience and, furthermore, the presence of hyperalgesia or referred pain before decompression surgery significantly worsens the outcome after 3 months [26], while more pain catastrophizing is related to persistence of symptoms in patients with long-lasting SIS [27]. It therefore seems likely that manifestations of peripheral and central pain sensitisation, and peripheral adaptations to long-lasting pain, could also affect the outcome of an exercise intervention. This could be either through an alteration in adherence to the intervention due to pain sensitisation, or through other mechanisms related to central pain sensitisation and peripheral adaptations.
The effect of scapula function on treatment outcome in patients with SIS is often debated, and one could argue that the increased emphasis on rotator cuff muscles, as is proposed in the current trial, would neglect an important aspect of the rehabilitation. Accordingly, it has been suggested that rotator cuff emphasis should only be after scapula control is achieved, as insufficient dynamic stability of the scapula could cause abnormal shoulder kinematics and impingement symptoms [28]. Assuming this, the effect of the add-on intervention in the current study, focusing mainly on strengthening exercises for the rotator cuff, could be modified be the presence of scapula dyskinesis. However, Mulligan et al. [29] recently found that treatment outcome was not significantly different between patients randomised to scapula stabilisation first, or rotator cuff strengthening first, questioning the importance of maintaining a scapula focus in the beginning of rehabilitation.
Explanation for choice of comparators
This study aims to investigate the effectiveness of adding a simple shoulder strengthening exercise programme to usual care in patients with long-lasting SIS. Therefore, the relevant control condition is usual care, which is to be compared to the intervention condition involving an add-on exercise programme and usual care.
This is relevant because usual care for patients that are covered by the Danish National Guidelines for Treatment of SIS [10], and hence have a medically justified need for general rehabilitation, is referral to general rehabilitation under the Danish health act § 140. Such rehabilitation is likely based on the most novel evidence from previously mentioned RCTs [12, 13, 18], where only limited gains in muscle strength are found. It therefore seems unlikely that the current treatment addresses the issue of glenohumeral muscle strength to a sufficient degree.
Furthermore, direct monitoring of adherence to the add-on intervention makes it possible to conduct secondary analysis, investigating the relationship between exercise adherence and changes in both strength and patient-reported function.
Methods
Objectives
Primary research question
Based on the literature reviewed in the “Background” section, we asked the following research question:
Is a 16-week simple home-based shoulder strengthening exercise programme, in addition to usual care, superior to usual care alone for improving patient-reported shoulder function 16 weeks after baseline, measured using the Shoulder Pain And Disability Index (SPADI), in patients with persistent (> 3 months) subacromial impingement syndrome referred to further examination at a hospital?
The research question was framed using the PICOT model [30] with the following options for each element:
-
Population: Patients with persistent SIS (> 3 months)
-
Intervention: 16-week simple home-based shoulder strengthening exercise programme added to usual care
-
Control: Usual care
-
Outcome: Patient-reported shoulder function
-
Time frame: 16 weeks after baseline
Primary objective
The primary objective is therefore to investigate the effectiveness of adding a 16-week simple home-based shoulder strengthening exercise programme to usual care, compared to usual care alone, on changes in patient-reported shoulder function (SPADI score) from baseline to end of treatment (16 weeks) in patients with SIS.
Primary research hypothesis
Our hypothesis is that a 16-week simple home-based shoulder strengthening exercise programme, in addition to usual care, is superior to usual care alone for improving patient-reported shoulder function 16 weeks after baseline, measured using the SPADI, in patients with persistent (> 3 months) subacromial impingement syndrome referred to further examination at a hospital.
Secondary objectives
-
1.
To investigate the effectiveness of adding a 16-week simple home-based shoulder strengthening exercise programme to usual care, compared to usual care alone, on changes in shoulder abduction and external rotation strength from baseline to end of treatment (16 weeks) in patients with SIS.
-
2.
To investigate the modifying effects of pain sensitisation, pain catastrophizing and scapula function on the effectiveness of adding a 16-week simple home-based shoulder strengthening exercise programme to usual care, compared to usual care alone, on changes in patient-reported shoulder function and shoulder abduction and external rotation strength from baseline to end of treatment (16 weeks) in patients with SIS.
-
3.
To investigate the dose-response relationship between objectively monitored adherence to the add-on intervention and change in patient-reported shoulder function, shoulder abduction strength and external rotation strength.
-
4.
To investigate the relationship between central and peripheral pain sensitisation and change in patient-reported shoulder function, shoulder abduction strength and external rotation strength, and to what degree this is mediated through adherence to the intervention.
Trial design
The SExSI-Trial is a pragmatic, assessor blinded, randomised, controlled superiority trial, with a two-group parallel design. Patients will be randomised to either usual care or a home-based intervention consisting of progressive high volume resistance training in addition to usual care with a 1:1 allocation. The primary end-point will be change in patient-reported shoulder function 16 weeks after baseline.
This clinical trial protocol is based on the PREPARE Trial guide [31] and the SPIRIT checklist [32] (Additional file 1). The trial report will adhere to the CONSORT guidelines for reporting parallel group randomised trials, the CONSORT extension for pragmatic trials http://www.consort-statement.org, and the TIDieR template for intervention description and replication [33].
Study setting and eligibility criteria
Participants will be recruited from the secondary care orthopaedic outpatient clinic at Hvidovre Hospital. Consecutive sampling will be used to ensure generalisability of the results. Accordingly, all patients who are referred for the first time to the clinic for examination of their current shoulder disorder lasting at least 3 months, who are living in the Capitol Region of Copenhagen, Denmark, who are not pregnant, do not permanently use strong pain medication (defined as morphine analgesic or similar), are aged 18–65 and are considered able to understand spoken and written Danish will be evaluated for eligibility by an orthopaedic specialist. All eligible patients will be provided with written information about the study.
Patients will be invited to participate if they meet the following inclusion criteria: (1) At least three positive of the five diagnostic tests for subacromial impingement syndrome (Hawkins-Kennedy test, Neer’s test, pain-full arc, Resisted External Rotation test and Jobe’s test) [34]; (2) Have been provided with or offered a rehabilitation plan due to a medically justified need for general rehabilitation after discharge from the hospital under the Danish Health Act § 140; and (3) hand in a completed SPADI questionnaire on the day of the medical examination.
Patients will be excluded if they fulfil any of the exclusion criteria, namely (1) have a radiologically verified new or previous fracture related to the shoulder joint, including the scapula; (2) have radiologically verified glenohumeral osteoarthritis; (3) have a clinically suspected luxation or sub-luxation of the glenohumeral, acromioclavicular or sternoclavicular joint; or (4) have a clinically suspected labral lesion, complete traumatic tear of the rotator cuff, frozen shoulder or other competing diagnoses (i.e. rheumatoid arthritis, cancer, neurological disorders, fibromyalgia, psychiatric illness).
Interventions
Both groups will receive usual care consisting of an offer of referral to general rehabilitation in the municipal clinic under the Danish health Act § 140, sometimes with the option to instead choose a private physiotherapy clinic, partly at their own expense. The referral is standard procedure when a patient with SIS is considered to have a medically justified need for general rehabilitation, as identified during the examination at the orthopaedic outpatient department (secondary care unit). This procedure is based on the Danish National clinical guideline on diagnostics and treatment of patients with selected shoulder disorders [10], as exercise therapy is recommended as first line of treatment for SIS.
In the context of this study, usual care includes all treatment received by a patient during the time between baseline and follow-up, except that included in ‘Strengthen your Shoulder’. Therefore, usual care might include a range of treatment modalities including advice, stretching, exercises, manual therapy, massage, acupuncture and electrotherapy at the discretion of the treating physiotherapist and doctor. A description of usual care following the TIDieR guidelines [33] is included in Additional file 2.
Patients in the intervention group will, in addition to usual care, receive instructions regarding a home-based, progressive, high volume resistance-training programme immediately after randomisation. Instructions in the exercises are provided at baseline (week 0) and after 2, 5, 10 and 16 weeks. The exercise programme has been developed in cooperation with a team of experts with extensive knowledge of both exercise physiology and rehabilitation and training of patients with shoulder disorders (MC, TB, KT and MR). The design of the exercises, and their progression, is based on the relevant literature regarding exercise physiology, shoulder biomechanics and strength training principles for both healthy people and patients with shoulder disorders. A standard protocol for individual adaptation of the programme based on pain response is part of the intervention.
A thorough description of the intervention, and modifications of the intervention, following the TIDieR guidelines [33], is included in Additional file 3, and the full set of strength training descriptors, as suggested by Toigo and Boutellier [35], are presented in Table 1. An online instruction video of the exercises will be made available upon publication of the main results of the trial, and a link to these is provided in Additional file 3.
Interventions – adherence
Lack of adherence to an exercise intervention is a major problem when aiming at investigating the effect of an intervention. In the previously described studies that include unsupervised resistance training in rehabilitation of SIS patients, monitoring adherence is either done using a log-book filled in by the patients [9, 12] or not described at all [13]. Using only patient-reported measures of adherence may limit the possibility of detecting which exercise dose is actually received by the patient. Accordingly, in a recent systematic review, the authors did not identify a single measure of patient-reported adherence to unsupervised home-based rehabilitation exercises to be sufficiently investigated [36]. Furthermore, findings from one previous study revealed that the dose of exercises reported in exercise diaries were 2.3 times higher than that collected through a system which monitored the exact TUT, being the total time a muscle resists weight during each set [37]. This clearly underlines the relevance of collecting objective data on exercise adherence. In addition, it seems unlikely that patients are always able to ‘take’ precisely the exercise dose prescribed. This issue was unfolded in a recent study, revealing that, just 2 weeks after initial instructions, only 7 out of 29 young healthy individuals were able to use the correct TUT, when performing shoulder exercises with an elastic band [38].
In the SExSI-Trial, adherence to the add-on progressive high volume resistance training intervention will be captured using the BandCizer©. The BandCizer© is a small device which is mounted on the elastic band during exercises to measure the TUT, number of repetitions and total work load for all exercises performed. Exactly TUT is a promising objective measure of exercise adherence, for which the BandCizer© is specifically developed and validated [39,40,41]. By directly monitoring the actual TUT for the prescribed exercises a more precise and objective distinction between adherent and less adherent patients will be possible.
In both the intervention group and the control group, the time spent on exercises related to usual care will be monitored by self-report, using a text-message based system (SMS-track©). Each week all participants will receive a text-message question regarding the time spent on exercises performed for their shoulder disorder. Participants in the intervention group will be instructed not to include the time spent on the add-on intervention in this report.
In the intervention group, face-to-face adherence reminders will be given by the investigator administering the intervention at the initial intervention instruction and at each subsequent intervention appointment (week 0, 2, 5, 10 and 16). The reminders will focus on (1) the importance of performing all prescribed exercises and on correct execution of exercises; (2) emphasising that the intervention is an add-on to usual care, and should not be a substitution for this; and (3) counteracting important barriers to ongoing engagement with home exercises, as identified by Littlewood et al. [42], namely the simplicity of the intervention (lack of potential effectiveness), lack of an early appreciable symptom response, when symptoms are reduced to a certain point, and a lack of self-efficacy.
Intervention adherence will further be enhanced by supplying a leaflet with the programme to stick on the fridge, and via the proactive follow-up intervention appointments, aspects known to improve patients adherence to a home-based exercise programme [42]. For an English version of the leaflets, see Additional files 4, 5 and 6.
Interventions – concomitant care
Concomitant use of corticosteroid injections will be based on the treating orthopaedic specialist’s evaluation, which is in accordance to the departments standard procedure and in line with the Danish national guidelines for treatment of SIS [10]. For pain relief, concomitant use of orally suspended NSAIDs will be permitted at patients’ own discretion. Corticosteroid injections and use of pain medication will be recorded.
Outcomes
Primary outcome
The primary outcome will be the change in the patient-reported outcome (PRO) score SPADI [43]. SPADI is a widely used shoulder-specific PRO [44], considered one of the most responsive shoulder PROs [45,46,47,48], with a minimal clinical important difference (MCID) of 8 to 13 points [45, 48] and a high standardised response mean of 1.38. The SPADI is easy and fast to complete [48] and, in a recent systematic review [49], it was highlighted as one of three PROs for patients with rotator cuff disease for which the psychometric properties are supported by most strong or moderate evidence.
Baseline values for the primary outcome will be collected from patient records, and passed on by the treating physician to the primary investigator, as completion of the SPADI questionnaire is an integral part of the medical examination. Primary outcome data will be further collected at follow-up weeks 5, 10 and 16, with the main analysis conducted for changes from baseline to 16 weeks follow-up, reported as the difference in mean change between groups.
Secondary outcomes
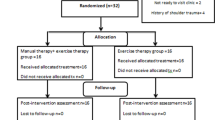
Secondary outcome data will be collected at one or more of the time-points (1) baseline assessment, (2) 5 weeks follow-up, (3) 10 weeks follow-up and (4) 16 weeks follow-up. See participant timeline in the SPIRIT diagram (Fig. 1) for further details.

Summary of measures to be collected (SPIRIT figure)
For the following secondary outcomes, the main analysis will be conducted for changes from baseline to 16 weeks follow-up, reported as the difference in mean change between groups.
-
Maximum isometric voluntary contraction (MVC) in abduction and external rotation measured using a handheld dynamometer in neutral position (torque in Newton meter per kilo body weight, Nm/kg). Measures of strength relate directly to the intervention.
-
Range of movement (ROM) in active shoulder abduction (0 to 180°), measured using a digital inclinometer, a widely used measure of function in patients with SIS.
-
An average of least pain and average pain experienced during the last week (Numeric Rating Scale: 0–10), a valid and reliable measure of pain [50]. A measure of pain is important, as pain is one of the cardinal complaints associated with SIS [51].
-
Pain catastrophizing (score from 0 to 52), being exaggerated negative thoughts related to experienced or anticipated pain, which might be a risk factor for chronicity [52], measured using the Danish version of the Pain Catastrophizing Scale.
-
Health-related Quality of Life index (EQ-5D-index, range −0.167 to 1.00) measured with the EQ-5D-3 L [53].
-
Health-related Quality of Life Visual Analogue Scale (EQ-5D visual analogue scale, score 0–100, 0 = lowest health-related quality of life) measured with the EQ-5D-3 L [53].
For the following secondary outcomes, the main analysis will be conducted for the outcome at 16 weeks follow-up, reported as the difference between groups at 16 weeks follow-up.
-
Temporal Summation of Pain (TSP), a measure of central excitability (range 0 to 10 cm on electronic Visual analogue scale). A higher degree of TSP has previously been found in patients with unilateral SIS compared to healthy controls [54], and the decrease in TSP after surgical intervention is reported to be associated with the outcome of surgery [26].
-
Conditioned Pain Modulation (CPM), a measure of the attenuated pain response (increase in tolerated pressure) to a painful pressure stimulus during application of another painful pressure stimulus [55]. CPM is a proxy measure of the function of the endogenous analgesia system, and patients with SIS have a lower CPM compared to healthy controls [54].
-
Pressure Pain Threshold (PPT) is a measure of local mechanical hyperalgesia. It measures how much mechanical pressure is needed to elicit the first onset of pain. PPT is measured in kPa as the threshold for first detection of pain. Patients with SIS have lower PPTs in the shoulder region when compared to matched controls, possibly indicating peripheral sensitisation of the nervous system [26].
-
Scapular Dysfunction (yes/no), measured using the modified Scapula Assistance Test, assessing if scapula muscle dysfunction influences the shoulder disorder. The result of the modified Scapula Assistance Test is a judgement of either positive or negative.
-
Scapula Dyskinesia (yes/no), measured using the Scapula Dyskinesis Test, the result of which is a judgment of either positive or negative test.
-
Global Impression of Change, measured on a 7-point Likert scale ranging from ‘Much better, a very important improvement’ to ‘Much worse, an important aggravation’.
-
Patient Acceptable Symptom State, measured as the dichotomous answer (yes/no) to a standardised question regarding the acceptability of the current state of the shoulder symptoms.
Other outcomes of interest will be pain during testing of external rotation MVC, abduction MVC and active abduction ROM, as reported by the participant on a numeric pain rating scale (0 to 10, 0 = no pain), using standardised verbal anchors for the instruction [50].
Sample size
The sample size estimation is based on the primary outcome SPADI. Studies on patients with SIS have shown standard deviations (SD) for SPADI change score after 11–12 weeks of between 17 [12] and 18.5 [56], while longer durations seem to entail larger SDs, with a SD of 22 reported for SPADI change from 0 to 22 weeks [12]. The latter is similar to our own unpublished data from SIS patients, showing a SD of 22.5 for SPADI change from 0 to 6 months. Based on this previous data, we expect a common SD of 19.5 for SPADI change values from week 0 to 16, the primary outcome. For the purpose of this study, the MCID for SPADI will be considered to be 10 points, based on previous studies [45, 48]. The negative effect that any dropouts will have on the statistical power will, to some degree, be reduced by the use of multiple imputation. However, the imputation of data will not fully redeem this, as multiple imputation can cause an underestimation of the effect and a larger variation in outcomes. Therefore, we aim at having a high power of 95% to verify an effect equal to or higher than the MCID of 10 points on SPADI, at a 5% significance level. To obtain this, a total of 200 patients will be required (100 in each group). This corresponds to a power of 89.7% in case of a 20% dropout and a power of 85.4% in case of a 40% dropout.
Recruitment strategies
All participants will be recruited consecutively from the arthroscopic centre at the orthopaedic department, Hvidovre Hospital immediately after undergoing clinical examination performed by an orthopaedic specialist, who will also make the initial eligibility screening and provide written information about the trial for eligible patients. In order to achieve adequate enrolment, the orthopaedic specialists, who are at the first line of recruitment, will be informed about the progress of the trial at regular formal and informal meetings.
Procedures and data collection methods
At baseline, after written consent is signed and before randomisation, all baseline assessments (Fig. 1) will be conducted by clinical physiotherapists who are trained in all assessment procedures and mutually aligned in order to improve the quality of data. All post-allocation assessments (week 5, 10 and 16) will be performed by the same group of testers in order to secure consensus regarding the instructions in questionnaires and conduction of tests. Participants will be instructed not to take any pain medication during the 8 hours prior to any assessment. Outcome assessors will also be trained in counselling for adherence to follow-up testing (to facilitate retention) and avoidance of disclosing allocation in a uniform manner. The study instruments are all described in detail in Additional file 7.
In general, once a participant is enrolled, every reasonable effort will be made to collect all outcomes for that participant, regardless of any deviations from the intervention protocol. Participants will receive text-message reminders for all scheduled follow-up appointments. The weekly text-messages with standardised questions regarding exercise time will also ensure that participants keep in mind their participation in the study. If participants do not attend their scheduled follow-up assessment, they will be contacted and offered to re-schedule to another date. If a participant should withdraw consent to follow-up assessments, the participant is offered the option to continue with the assessment of PROs.
Reasons for non-adherence to add-on intervention will be recorded by the investigator providing the intervention, either at the intervention visits or by phone if the participant does not come to the scheduled appointment. Reasons for non-retention will be recorded by the outcome assessor if this participant withdraws during a follow-up session, or by the primary investigator in a telephone interview.
Data management
All patient-reported questionnaires (SPADI, Pain Catastrophizing Scale, EQ-5D-5 L, Baseline and End of treatment) will be filled in by the patient in a paper format, and all data from the physiological measurements and clinical tests will be entered in a paper Case Report Form. Subsequently, all data will be entered in EpiData (version 3.1 or newer) by study personnel, using blinded double data entry to ensure data quality. The data entry form will support valid values and range checks where applicable. The original forms will be kept on file at a secure location on the study site for a period of 3 years after completion of the study. Data collected through SMS-track is entered directly by the participant as they send the answer in a text-message. The validity of data will be secured through answer validation, where answers that do not fit the specified requirements will be rejected. Reminder messages will be automatically generated in case of missing answers.
All data from the BandCizer units will be stored on the unit and transferred the BandCizer Backend Software (BandCizer®, Denmark) via Bluetooth connection to a dedicated repeater unit with internet access. All electronically entered data will be stored on a secure drive at the study site.
No data monitoring committee will be composed and no formal stopping guidelines and corresponding interim analyses are planned. No other interim analyses are planned.
Allocation
Participants will be randomly assigned to either the control or intervention group (CG or IG) at a 1:1 allocation ratio, using a computer generated randomisation schedule of permuted blocks of random sizes ranging from 4 to 10.
Participants will be randomised using sequentially numbered, opaque, sealed envelopes. Investigators taking part in allocation and data collection will be blinded to block sizes and randomisation sequence at all times during the study period. Allocation concealment will be ensured, as the envelopes will not be opened before the participant has been irreversibly included in the study.
The creation of the randomisation schedule of permuted blocks of random sizes, and subsequent packaging of sequentially numbered, opaque, sealed envelopes will be performed by persons not else involved in the trial.
The final enrolment and subsequent allocation of participants will be conducted by investigators not taking part in any outcome assessment, who will be blinded to the randomisation sequence at all times during the intervention period. Outcome assessors will not take part in any of the processes related to allocation.
Blinding
Outcome assessors performing the outcome assessment at baseline and follow-up weeks 5, 10 and 16 will be blinded to group allocation. Given the nature of the intervention, which requires the treating therapist to know the intervention, blinding of intervention providers is not deemed feasible, and therefore will not be performed. In order to obtain valid results from this trial, intervention receivers will be kept blind to treatment allocation. This will be attained through minimal information about the content of the add-on intervention and control condition until group allocation is final. Accordingly, participants will be informed (written and orally) that the study compares two different treatment regimens, and that both include the treatment elements normally offered, and adhere to the clinical guideline. Additionally, study participant will be strongly inculcated not to enclose or discuss treatment allocation with the outcome assessors, medical doctors or physiotherapists providing usual care.
All pre-defined analyses will be performed by a data analyst blinded to allocation.
Emergency unblinding
Investigators and caregivers will be encouraged to consult with the Medical Advisor (PH) in a case where unblinding seems relevant. As the intervention is only an add-on to usual care, and it will be modified with respect to the participants pain response, the probability of circumstances occurring where unblinding could be relevant seem very slim.
Statistical methods – outcomes
For the primary end-point, namely SPADI score after 16 weeks, and for all continuous secondary outcomes listed in Table 2, a constrained linear mixed model (cLMM) will be applied in order to compare the change from baseline to 16 weeks in the IG to that in the CG. The model will contain the outcome at 16 weeks as the dependent variable, treatment group (IG or CG) as the main effect and both baseline score and any additional follow-up measurements as repeated measurements, to estimate the differences in mean change between groups and corresponding 95% confidence intervals (95% CI) (Table 3). The covariance structures will be selected based on the MAICE procedure [57].
Mean scores and corresponding 95% CIs will be reported for all outcome time-points (t0, t1, t2 and t3) (Table 3). Within group changes between all outcomes assessment time-points (t0-t1, t1-t2 and t2-t3), and between baseline and last assessment (t0 and t3), and corresponding 95% CI, will be reported (Table 4). Finally, differences in within group changes between all outcomes assessment time-points (t0-t1, t1-t2 and t2-t3) and the corresponding 95% CIs will be reported (Table 5). Results from the repeated measures analysis with SPADI score as outcome will be visualised as illustrated in Fig. 2.

Visualisation of changes in SPADI score in the intervention and control group, respectively (example, not based on data)
For comparison of binary outcomes, proportions will be compared using a χ2 test, and odds ratio estimates and corresponding 95% CIs will be reported.
Results from analyses comparing IG and CG for all primary and secondary outcomes will be reported in the second paper outlined in the ‘Dissemination policy’ section, except for the results on secondary outcomes regarding pain sensitisation and scapula dyskinesia, which will be reported in the third and fourth paper outlined in the ‘Dissemination policy’ section, respectively.
P values will be reported to the fourth decimal. For all tests, a two-sided significance level of ≤ 0.05 will be applied. All analyses will be conducted using up-to-date versions of SPSS and SAS.
Statistical methods – additional analyses
Subgroup analyses
For the outcomes SPADI score, abduction MVC and external rotation MVC, subgroup analyses investigating the modifying effect of scapula dysfunction, scapula dyskinesia, temporal summation of pain, conditioned pain modulation, pain pressure threshold (site 1) and pain catastrophizing, respectively, will be conducted. These will be performed using a cLMM, similar to the corresponding main analyses, but including a dichotomised value for the first measurement of the relevant variable as the interaction term and reporting the effect estimates for each subgroup.
The following sensitivity analyses will be performed for the primary outcome and reported together with the primary analyses in the primary trial report:
-
A per protocol analysis, similar to the primary analysis, will be performed. From the IG, only patients who attended intervention appointments on weeks 0, 5 and 10 will be included in this analysis, while all patients in the CG will be included.
-
Adjusted cLMM to adjust the effect of treatment allocation on SPADI score for any baseline differences between groups. Baseline variables will be included as covariates if (1) the difference between groups is more than 0.3 of the common SD for continuous outcomes, or (2) proportions are significantly different (P < 0.05) between groups, for binary outcomes.
-
Sensitivity analyses to investigate the importance of the clinometric properties of the SPADI score may be relevant. This is based on a very recent study [58], which evaluates the SPADI score using the Rasch model. The study was published after initiation of this trial.
Three exploratory dose-response analyses will be performed. Only participants in the IG will be included in these analyses. First, the interacting effect of adherence to the intervention, TSP and CPM, on change in external rotation MVC, abduction MVC and SPADI score (outcomes), will be investigated. Secondly, whether the effect of TSP and CPM is mediated by adherence to the intervention will also be assessed. These analyses will be conducted as available-case analyses.
For all included variables, data from each time-period (t0-t1, t1-t2 and t2-t3) will be included as repeated measurements in these analyses. The outcome variable is the change between first and last measurement in a time-period. ‘Adherence’ to the intervention is the total TUT in a time-period. ‘Central Pain Sensitization’ score (TSP or CPM) is the score from the last time-point in each time-period.
Additionally, results will also be presented for similar analyses adjusted for Usual Care, calculated for each time-period as the average of time spent on usual care, as reported by text-message, in all weeks included in the relevant time-period.
Statistical methods – analysis population and missing data
All main analyses will be conducted as intention-to-treat analyses, including all randomised participants, regardless of protocol adherence. Participants will be analysed as randomised. To create a full analysis dataset for the intention-to-treat analyses, missing outcome data will be imputed using multiple imputations based on the variables of all previous scores in the relevant outcome, age, sex and allocation.
Harms
Adverse events will be defined in this context as any unintended, unfavourable findings, symptom or illnesses that occur during the assessment or the add-on intervention, whether it can be attributed to the assessment or not. Adverse events will be recorded in part by the patient as a spontaneous recording during assessment and by open questioning.
Acute exacerbations of shoulder symptoms will be recorded by the primary investigator (MC) and, as a safety precaution, in case a medical evaluation is required, the participant will be referred to the Medical Advisor (PH). Patients in the intervention group experiencing exacerbations lasting more than 1 week will be referred to the Medical Advisor (PH) and evaluated if further medical care is needed.
Serious unexpected side effects or serious adverse events will be reported to the Capital Regional Ethics Committee in Denmark within 7 days after sponsor or the primary investigator has become aware of the incident. Serious adverse events will be categorised according to the definitions established by the United States Food and Drug Administration [59] and will be assessed by the primary investigator for possible relations with the assessment and/or intervention to consider whether there is a reasonable possibility that the adverse event can be caused by either.
No audits are planned.
Dissemination policy
All results from the study, be they positive, negative or inconclusive, will be published in international scientific journals. The project leader will enforce publication. Furthermore, the results will be presented at national and international conferences. Working titles for scientific publications are listed below. In the primary trial report, all collected outcomes will be defined and referenced to the dissemination plan below if data or analyses are not reported in the primary trial report.
-
1.
The Strengthening Exercises in Shoulder Impingement trial (The SExSI-Trial): Protocol for a pragmatic, assessor blinded, parallel-group, randomised, controlled, superiority trial investigating the effectiveness of adding a simple shoulder strengthening exercise programme to usual care, in patients with long-lasting subacromial impingement syndrome.
-
2.
The effectiveness of adding to usual care a simple programme of Strengthening Exercises In Subacromial Impingement patients (The SExSI-Trial): A randomised controlled trial (Primary trial report focusing on the primary outcome and analysis, as well as supportive secondary analyses for the primary outcome).
-
3.
The dose-response relationship between adherence to the intervention and changes in shoulder strength and patient-reported shoulder function: Pre-defined secondary analyses from the SExSI-Trial.
-
4.
The effectiveness of the SExSI-Trial add-on intervention on pain sensitisation, the modifying effect pain sensitisation on treatment outcomes and mediating effects of pain sensitisation on the dose-response relationship between intervention adherence and changes in outcomes: Pre-defined secondary analyses from the SExSI-Trial.
-
5.
The modifying effect of scapula dysfunction at baseline on shoulder strength and function outcomes and the effectiveness of the add-on intervention on scapula dysfunction: Pre-defined secondary analyses from the SExSI-Trial.
Additionally, the results of this study will be communicated directly to the participants, who will be encouraged to comment on disseminated trial results in order to improve their further public dissemination. Furthermore, trial results will be disseminated to the public in general through the daily press.
Dissemination policy – authorship
Decisions on authorship eligibility will adhere to the Harvard author guideline statement as endorsed by the Faculty Council of Harvard Medical School (https://hms.harvard.edu/sites/default/files/assets/Sites/Ombuds/files/AUTHORSHIP%20GUIDELINES.pdf).
Topics suggested for presentation and/or publication, including suggestion and justification for authors to be reviewed for the Writing Committee, will be presented to the members of the Steering Committee. The Steering Committee will form the Writing Committee and decide on author sequence. Disputes regarding authorship will be settled by the Primary investigator after consulting with the Supervising investigator.
Discussion
This trial (the SExSI-Trial) will investigate the effectiveness of adding a simple shoulder strengthening exercise programme to usual care in patients with long-lasting SIS. If the intervention is found effective in improving patient-reported shoulder function, the intervention will be easily implemented as an addition to usual care.
The current trial is planned and designed to maximise the usefulness of the trial results by focusing on features that have recently been suggested as important when considering the usefulness of clinical research [60]. We further believe that the research question raised in this trial satisfies the FINER-criteria [61], as it is considered both Feasible, Interesting, Novel, Ethical and Relevant (Table 6).
Firstly, the ‘problem base’ and ‘context placement’ have been described in the Background section. Accordingly, the treatment of shoulder impingement syndrome is a problem that is worthwhile to address, based on the extent of the problem and the substantial societal costs related to the disorder. Furthermore, the systematic literature review, as is presented in the Background section, clearly indicates that knowledge is lacking with regards to the effect of strengthening exercises in the rehabilitation of patients with long-lasting SIS. The SExSI-Trial will provide relevant insight into this area, contributing to fill this gap.
Secondly, a focus on information gain, pragmatism and patient centeredness is secured through methodological considerations. The trial has been accordingly designed to optimise the information gained from the study by implementing multiple follow-up time-points and a close monitoring of intervention adherence. This makes it possible to conduct relevant dose-response analyses, which in turn will guide future research. The pragmatism of the trial is improved by the use of broad eligibility criteria, a consecutive sampling strategy and usual care as the comparator, aspects that will improve the generalisability of the trial results. Moreover, the choice of SPADI as the primary outcome is in line with the fact that pain and loss of function are the main complaints associated with SIS [51]. This improves the patient centeredness of the trial, as the goal of the intervention is meaningful to the patients.
Finally, the pre-registration at clinicaltrials.gov and publication of this trial protocol, including intervention descriptions based on the TIDieR framework [33], greatly improves the transparency of the trial conduct and results. Collectively, all of these efforts are likely to improve the usefulness of the trial results, and hence the relevance of the trial itself.
Strengths and limitations
Aside from the above, this trial has some methodological strengths and limitations which must be taken into account when interpreting the study findings.
Strengths
The blinding of participants, data analysts, outcome assessor and usual care providers greatly decreases the risk of bias in the current trial, supporting the internal validity of study findings. The external validity is further improved by the use of consecutive sampling, thereby improving the generalisability of the results.
Limitations
Given the nature of the intervention, the intervention providers could not be blinded, which increases the risk of bias in the trial. Furthermore, the use of a single centre study design, and the fact that the intervention is provided in a setting outside the normal usual care, decreases the external validity of the study, as study findings might not be generalisable to other settings. It is not within the scope of this study to conduct cost-effectiveness analyses or to conduct qualitative work to evaluate patient acceptability and experience of the home-based exercise programme. While the objective monitoring of adherence will show to what degree patients have accepted and managed to perform the intervention as prescribed, qualitative work could be conducted in order to investigate patient experiences with the intervention and reasons for adherence and non-adherence in order to improve implementation of the intervention. Cost-effectiveness analyses could also be performed in order to inform policy-makers and support implementation. The current study will, nevertheless, provide level-one evidence regarding the effectiveness of the add-on intervention.
Trial status
Protocol version
Issue date: 10 May 2017, Protocol amendment 01, Author MC.
Revision chronology
Version 1, 25.02.2016 Original.
Version 2, 10.05.2017 Amendment 1. Primary reason for amendment: EQ-5D-3 L was added as outcome at 1-year follow-up.
Recruitment
Recruitment was initiated May 1, 2016, and is expected to be finalised April 1, 2018.
Abbreviations
- CG:
-
control group
- cLMM:
-
constrained linear mixed model
- CPM:
-
conditioned pain modulation
- IG:
-
intervention group
- MCID:
-
minimal clinical important difference
- MVC:
-
maximum isometric voluntary contraction
- PPT:
-
pressure pain threshold
- PRO:
-
patient-reported outcome
- ROM:
-
range of movement
- SExSI:
-
Strengthening Exercises in Shoulder Impingement
- SIS:
-
subacromial impingement syndrome
- SPADI:
-
Shoulder Pain And Disability Index
- TSP:
-
temporal summation of pain
- TUT:
-
time under tension
References
Steinfeld R, Valente RM, Stuart MJ. A commonsense approach to shoulder problems. Mayo Clin Proc. 1999;74:785–94.
van der Windt DA, Koes BW, de Jong BA, Bouter LM. Shoulder disorders in general practice: incidence, patient characteristics, and management. Ann Rheum Dis. 1995;54:959–64.
van der Windt DA, Koes BW, Boeke AJ, Devillé W, De Jong BA, Bouter LM. Shoulder disorders in general practice: prognostic indicators of outcome. Br J Gen Pract J R Coll Gen Pract. 1996;46:519–23.
MacDermid JC, Ramos J, Drosdowech D, Faber K, Patterson S. The impact of rotator cuff pathology on isometric and isokinetic strength, function, and quality of life. J Shoulder Elb Surg. 2004;13:593–8.
Sundhedsstyrelsen. Impingementsyndrom/rotator cuff-syndrom og traumatisk rotator cuff-ruptur Del 2: Faglige visitationsretningslinjer. København: Sundhedsstyrelsen; 2011.
Virta L, Joranger P, Brox JI, Eriksson R. Costs of shoulder pain and resource use in primary health care: a cost-of-illness study in Sweden. BMC Musculoskelet Disord. 2012;13:17.
Kuijpers T, van Tulder MW, van der Heijden GJMG, Bouter LM, van der Windt DAWN. Costs of shoulder pain in primary care consulters: a prospective cohort study in The Netherlands. BMC Musculoskelet Disord. 2006;7:83.
Abdulla SY, Southerst D, Côté P, Shearer HM, Sutton D, Randhawa K, et al. Is exercise effective for the management of subacromial impingement syndrome and other soft tissue injuries of the shoulder? A systematic review by the Ontario Protocol for Traffic Injury Management (OPTIMa) Collaboration. Man Ther. 2015;20(5):646–56.
Holmgren T, Björnsson Hallgren H, Öberg B, Adolfsson L, Johansson K. Effect of specific exercise strategy on need for surgery in patients with subacromial impingement syndrome: randomised controlled study. BMJ. 2012;344:e787.
Sundhedsstyrelsen. National klinisk retningslinje for diagnostik og behandling af patienter med udvalgte skulderlidelser. Sundhedsstyrelsen. 2013.
Hanratty CE, McVeigh JG, Kerr DP, Basford JR, Finch MB, Pendleton A, et al. The effectiveness of physiotherapy exercises in subacromial impingement syndrome: a systematic review and meta-analysis. Semin Arthritis Rheum. 2012;42:297–316.
Bennell K, Wee E, Coburn S, Green S, Harris A, Staples M, et al. Efficacy of standardised manual therapy and home exercise programme for chronic rotator cuff disease: randomised placebo controlled trial. BMJ. 2010;340:c2756.
Maenhout AG, Mahieu NN, De Muynck M, De Wilde LF, Cools AM. Does adding heavy load eccentric training to rehabilitation of patients with unilateral subacromial impingement result in better outcome? A randomized, clinical trial. Knee Surg Sports Traumatol Arthrosc. 2013;21:1158–67.
Celik D, Sirmen B, Demirhan M. The relationship of muscle strength and pain in subacromial impingement syndrome. Acta Orthop Traumatol Turc. 2011;45:79–84.
Clausen MB, Witten A, Holm K, Christensen KB, Attrup ML, Hölmich P, et al. Glenohumeral and scapulothoracic strength impairments exists in patients with subacromial impingement, but these are not reflected in the shoulder pain and disability index. BMC Musculoskelet Disord. 2017;18:302.
Dean BJF, Franklin SL, Carr AJ. A systematic review of the histological and molecular changes in rotator cuff disease. Bone Joint Res. 2012;1:158–66.
Folland JP, Williams AG. The adaptations to strength training: morphological and neurological contributions to increased strength. Sports Med Auckl NZ. 2007;37:145–68.
Lombardi I, Magri AG, Fleury AM, Da Silva AC, Natour J. Progressive resistance training in patients with shoulder impingement syndrome: a randomized controlled trial. Arthritis Rheum. 2008;59:615–22.
Ingwersen KG, Jensen SL, Sørensen L, Jørgensen HR, Christensen R, Søgaard K, et al. Three months of progressive high-load versus traditional low-load strength training among patients with rotator cuff tendinopathy: primary results from the double-blind randomized controlled RoCTEx Trial. Orthop J Sports Med. 2017;5:2325967117723292.
Ketola S, Lehtinen J, Arnala I, Nissinen M, Westenius H, Sintonen H, et al. Does arthroscopic acromioplasty provide any additional value in the treatment of shoulder impingement syndrome?: a two-year randomised controlled trial. J Bone Joint Surg (Br). 2009;91:1326–34.
Peterson MD, Rhea MR, Alvar BA. Applications of the dose-response for muscular strength development: a review of meta-analytic efficacy and reliability for designing training prescription. J Strength Cond Res. 2005;19:950–8.
Izquierdo M, Ibañez J, González-Badillo JJ, Häkkinen K, Ratamess NA, Kraemer WJ, et al. Differential effects of strength training leading to failure versus not to failure on hormonal responses, strength, and muscle power gains. J Appl Physiol. 2006;100:1647–56.
Andarawis-Puri N, Kuntz AF, Ramsey ML, Soslowsky LJ. Effect of glenohumeral abduction angle on the mechanical interaction between the supraspinatus and infraspinatus tendons for the intact, partial-thickness torn, and repaired supraspinatus tendon conditions. J Orthop Res. 2010;28:846–51.
Bey MJ, Song HK, Wehrli FW, Soslowsky LJ. Intratendinous strain fields of the intact supraspinatus tendon: the effect of glenohumeral joint position and tendon region. J Orthop Res. 2002;20:869–74.
Boettcher CE, Cathers I, Ginn KA. The role of shoulder muscles is task specific. J Sci Med Sport. 2010;13:651–6.
Sanchis MN, Lluch E, Nijs J, Struyf F, Kangasperko M. The role of central sensitization in shoulder pain: A systematic literature review. Semin Arthritis Rheum. 2015;44:710–6.
van der Windt DAWM, Kuijpers T, Jellema P, van der Heijden GJMG, Bouter LM. Do psychological factors predict outcome in both low-back pain and shoulder pain? Ann Rheum Dis. 2007;66:313–9.
Kibler WB, Sciascia A. Current concepts: scapular dyskinesis. Br J Sports Med. 2010;44:300–5.
Mulligan EP, Huang M, Dickson T, Khazzam M. The effect of axioscapular and rotator cuff exercise training sequence in patients with subacromial impingement syndrome: a randomized crossover trial. Int J Sports Phys Ther. 2016;11:94–107.
Thabane L, Thomas T, Ye C, Paul J. Posing the research question: not so simple. Can J Anaesth J Can Anesth. 2009;56:71–9.
Bandholm T, Christensen R, Thorborg K, Treweek S, Henriksen M. Preparing for what the reporting checklists will not tell you: the PREPARE Trial guide for planning clinical research to avoid research waste. Br J Sports Med. 2017;51:1494–501.
Chan A-W, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža-Jerić K, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158:200–7.
Hoffmann TC, Glasziou PP, Boutron I, Milne R, Perera R, Moher D, et al. Better reporting of interventions: template for intervention description and replication (TIDieR) checklist and guide. BMJ. 2014;348:g1687.
Michener LA, Walsworth MK, Doukas WC, Murphy KP. Reliability and diagnostic accuracy of 5 physical examination tests and combination of tests for subacromial impingement. Arch Phys Med Rehabil. 2009;90:1898–903.
Toigo M, Boutellier U. New fundamental resistance exercise determinants of molecular and cellular muscle adaptations. Eur J Appl Physiol. 2006;97:643–63.
Bollen JC, Dean SG, Siegert RJ, Howe TE, Goodwin VA. A systematic review of measures of self-reported adherence to unsupervised home-based rehabilitation exercise programmes, and their psychometric properties. BMJ Open. 2014;4:e005044.
Rathleff MS, Bandholm T, KA MG, Harring SI, Sørensen AS, Thorborg K. New exercise-integrated technology can monitor the dosage and quality of exercise performed against an elastic resistance band by adolescents with patellofemoral pain: an observational study. J Physiother. 2016;62:159–63.
Faber M, Andersen MH, Sevel C, Thorborg K, Bandholm T, Rathleff M. The majority are not performing home-exercises correctly two weeks after their initial instruction-an assessor-blinded study. PeerJ. 2015;3:e1102.
Rathleff MS, Bandholm T, Ahrendt P, Olesen JL, Thorborg K. Novel stretch-sensor technology allows quantification of adherence and quality of home-exercises: a validation study. Br J Sports Med. 2014;48:724–8.
Rathleff MS, Thorborg K, Rode LA, McGirr K, Sørensen AS, Bøgild A, et al. Adherence to commonly prescribed, home-based strength training exercises for the lower extremity can be objectively monitored using the Bandcizer. J Strength Cond Res. 2015;29(3):627–36.
Skovdal Rathleff M, Thorborg K, Bandholm T. Concentric and eccentric time-under-tension during strengthening exercises: validity and reliability of stretch-sensor recordings from an elastic exercise-band. PLoS One. 2013;8:e68172.
Littlewood C, Malliaras P, Mawson S, May S, Walters S. Patients with rotator cuff tendinopathy can successfully self-manage, but with certain caveats: a qualitative study. Physiotherapy. 2014;100:80–5.
Christiansen DH, Andersen JH, Haahr JP. Cross-cultural adaption and measurement properties of the Danish version of the Shoulder Pain and Disability Index. Clin Rehabil. 2013;27:355–60.
Roe Y, Soberg HL, Bautz-Holter E, Ostensjo S. A systematic review of measures of shoulder pain and functioning using the International classification of functioning, disability and health (ICF). BMC Musculoskelet Disord. 2013;14:73.
Angst F, Goldhahn J, Drerup S, Aeschlimann A, Schwyzer H-K, Simmen BR. Responsiveness of six outcome assessment instruments in total shoulder arthroplasty. Arthritis Rheum. 2008;59:391–8.
Angst F, Schwyzer H-K, Aeschlimann A, Simmen BR, Goldhahn J. Measures of adult shoulder function: Disabilities of the Arm, Shoulder, and Hand Questionnaire (DASH) and its short version (QuickDASH), Shoulder Pain and Disability Index (SPADI), American Shoulder and Elbow Surgeons (ASES) Society standardized shoulder assessment form, Constant (Murley) Score (CS), Simple Shoulder Test (SST), Oxford Shoulder Score (OSS), Shoulder Disability Questionnaire (SDQ), and Western Ontario Shoulder Instability Index (WOSI). Arthritis Care Res. 2011;63(Suppl 11):S174–88.
Beaton D, Richards RR. Assessing the reliability and responsiveness of 5 shoulder questionnaires. J Shoulder Elb Surg. 1998;7:565–72.
Paul A, Lewis M, Shadforth MF, Croft PR, Van Der Windt DA, Hay EM. A comparison of four shoulder-specific questionnaires in primary care. Ann Rheum Dis. 2004;63:1293–9.
Huang H, Grant JA, Miller BS, Mirza FM, Gagnier JJ. A systematic review of the psychometric properties of patient-reported outcome instruments for use in patients with rotator cuff disease. Am J Sports Med. 2015;43:2572–82.
Jensen MP, Turner JA, Romano JM, Fisher LD. Comparative reliability and validity of chronic pain intensity measures. Pain. 1999;83:157–62.
Roach KE, Budiman-Mak E, Songsiridej N, Lertratanakul Y. Development of a shoulder pain and disability index. Arthritis Care Res. 1991;4:143–9.
Sullivan MJL, Bishop SR, Pivik J. The Pain Catastrophizing Scale: development and validation. Psychol Assess. 1995;7:524–32.
Rabin R, de Charro F. EQ-5D: a measure of health status from the EuroQol Group. Ann Med. 2001;33:337–43.
Valencia C, Kindler LL, Fillingim RB, George SZ. Investigation of central pain processing in shoulder pain: converging results from 2 musculoskeletal pain models. J Pain. 2012;13:81–9.
Yarnitsky D. Conditioned pain modulation (the diffuse noxious inhibitory control-like effect): its relevance for acute and chronic pain states. Curr Opin Anaesthesiol. 2010;23:611–5.
Kromer TO, de Bie RA, Bastiaenen CHG. Physiotherapy in patients with clinical signs of shoulder impingement syndrome: a randomized controlled trial. J Rehabil Med. 2013;45:488–97.
Akaike H. A new look at the statistical model identification. IEEE Trans Autom Control. 1974;19:716–23.
Jerosch-Herold C, Chester R, Shepstone L, Vincent JI, MacDermid JC. An evaluation of the structural validity of the shoulder pain and disability index (SPADI) using the Rasch model. Qual Life Res. 2017. Ahead of print. doi:https://doi.org/10.1007/s11136-017-1746-7.
Reporting Serious Problems to FDA - What is a Serious Adverse Event? http://www.fda.gov/Safety/MedWatch/HowToReport/ucm053087.htm. Accessed 27 Jan 2016.
Ioannidis JPA. Why most clinical research is not useful. PLoS Med. 2016;13:e1002049.
Hulley SB, editor. Designing Clinical Research. 4th ed. Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins; 2013.
Acknowledgements
The authors would like to acknowledge all orthopaedic specialists and nurses at Sports Orthopaedic Research Center (SORC-C), Orthopaedic Department, Hvidovre Hospital, for their valuable inputs during the planning of this study. We also thank Kasper Krommes and Laura Krohn for their valuable contributions related to the preparation of outcome assessment.
Funding
Metropolitan University College Copenhagen, as the work place of the primary investigators, is partly funding the salary and tuition fees for MC (DKR 900,000), the salary of other study personnel (DKR 120,000) and SMS-track expenses (DKR 30,000). Praksisfonden is partly funding the salary and tuition fees for MC, the salary of other study personnel and testing equipment (DKR 805,000). The Danish Rheumatism Association is funding testing equipment (DKR 50,000). Further external funding will be applied for. The Center for Neuroplasticity and Pain (CNAP) is supported by the Danish National Research Foundation (DNRF121).
The funders (Metropolitan University College, Praksisfonden and the Danish Rheumatism Association) had no role in the design of this study and will not have any role during its execution, analyses, interpretation of the data or decision to submit results. The trial sponsor is the Sports Orthopaedic Research Center – Copenhagen (SORC-C) at the Department of Orthopaedic Surgery, Copenhagen University Hospital, Hvidovre, the department where the project is being conducted. The head of SORC-C, Professor Per Hölmich, took part in designing the trial and forms part of the Steering Committee.
Availability of data and materials
Upon publication of the trial results, a fully anonymised participant-level dataset and corresponding statistical code will be made publicly available if required by the scientific journal in which the results are published. If the scientific journal does not require a full dataset, a fully anonymised participant-level dataset will be made available from the corresponding author upon reasonable request. There are no contractual agreements that limit the investigators’ access to the dataset.
Author information
Authors and Affiliations
Contributions
The study design and completion is conducted in collaboration between the Sports Orthopaedic Research Center – Copenhagen (SORC-C), Arthroscopic Center, Department of Orthopaedic Surgery Hvidovre Hospital; Physical Medicine & Rehabilitation Research – Copenhagen (PMR-C), Amager-Hvidovre Hospital; and the School of Physiotherapy, Department of Physiotherapy and Occupational Therapy, Faculty of Health and Technology, Metropolitan University College. KT and MC conceived the study idea. MC, KT, TB and MR initiated the study design. MC, KT, TB, MR, MZ, KC and PH contributed to study design and are members of the steering committee. PH and KT helped with implementation. KC provided statistical expertise in the trial design and planning of the primary and secondary statistical analyses. MC drafted the study protocol and all authors contributed to protocol refinement and approved the final protocol. MC and KT will together ensure execution and completion of the project. MC is the grant holder and primary investigator, and takes overall responsibility for patient inclusion and data collection. MC and KT will conduct training of study personnel in testing procedures. MR will instruct the primary investigator and other study personnel on the use of BandCizer technology, and contribute specifically in managing the data collected through this. MC will draft the manuscripts for publications with contributions and approval of final versions from all authors. MC, KT, TB and MR will contribute to the design of the study intervention. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The trial protocol, the informed consent forms and other requested documents have been reviewed and approved by the Capitol Regional Ethics Committee in Denmark (H-16016763) with respect to the scientific content and the compliance to the applicable health science regulations. Safety reports, containing a list of all serious side effects and serious events, will be sent to the ethics committee. Within 90 days after completion of the study, the sponsor and the primary investigator will inform the ethics committee that the study is closed.
Consent or assent
At the end of the medical examination, the treating orthopaedic specialist or the cooperating nurse will introduce the trial to patients fulfilling the eligibility criteria. Patients will be informed about the request for participation in the trial, and will be provided with participant information and a brochure on test subject’s rights. As soon as possible thereafter, patients will be contacted by one of the investigators. A final eligibility screening will be conducted and eligible patients will be offered a meeting to provide oral information about the trial, their rights as test subjects and the possibility to ask clarifying questions. Patients are offered deliberation time before being asked to sign written consent forms regarding willingness to participate.
Consent or assent – ancillary studies
In the oral information and in the participant information sheets, patients will be informed that a 1 year follow-up is planned for all participants, and that this is covered by the written consent forms. Data for the 1 year follow-up will, in part, be collected through text messages, using the SMS-track© (data hosted at a BFIH certified data centre that meets the ISO 27002 standards). Through answers to auto-generated standardized SMS questions, patients will be asked to report sick leave due to their shoulder disorder (weekly). Questions regarding surgery, if any, for SIS (yearly), the EQ-5D-3 L questionnaire (yearly), and all questions from the SPADI questionnaire (after 6 months and 1 year) will be administered using the REDCap® system.
Confidentiality
All study-related information on participants will be stored securely on the study site. All completed paper forms will be stored in locked file cabinets before and after data is entered to an electronic database. All electronic participants’ information will be stored on a secured study-specific drive at the study site. Access to the study-specific drive will be limited to users currently working on the project and therefore need access to the secured drive in order to store and/or analyse data. Access will be limited by individual person-specific login and passwords. A list of persons with access to the study-specific drive will be kept up-to-date by the primary investigator. Appointment schedules and any other listings that link participant to other identifying information will be stored separately in the hospital’s secured intranet system or in separate locked file cabinets in an area with limited access. Study information will not be released outside of the study without the permission of the relevant participant. The study will be reported to and approved by the Danish Data Protection Agency before start of inclusion, and will adhere to the “Act on Processing Personal Data”.
Ancillary and post-trial care
In general, no ancillary or post-trial care is provided to participants in the trial. However, the details of the intervention will be made public shortly after the trial is over, and the intervention could therefore be replicated by physiotherapists in ordinary settings.
Consent for publication
Written informed consent for publication of images in the intervention leaflet was obtained from the person appearing on the pictures. A copy of the consent form is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Populated SPIRIT checklist (PDF 172 kb)
Additional file 2:
TIDieR control, usual care. (PDF 113 kb)
Additional file 3:
TIDieR intervention, strengthen your shoulder. (PDF 207 kb)
Additional file 4:
Strengthen your shoulder, intervention leaflet 1. (PDF 411 kb)
Additional file 5:
Strengthen your shoulder, intervention leaflet 2. (PDF 557 kb)
Additional file 6:
Strengthen your shoulder, intervention leaflet 3. (PDF 588 kb)
Additional file 7:
Data collection methods – study instruments. (PDF 235 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Clausen, M.B., Bandholm, T., Rathleff, M. et al. The Strengthening Exercises in Shoulder Impingement trial (The SExSI-trial) investigating the effectiveness of a simple add-on shoulder strengthening exercise programme in patients with long-lasting subacromial impingement syndrome: Study protocol for a pragmatic, assessor blinded, parallel-group, randomised, controlled trial. Trials 19, 154 (2018). https://doi.org/10.1186/s13063-018-2509-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-018-2509-7