
Abstract
Background
Kidney renal clear cell carcinoma (KIRC) represents a significant proportion of renal cell carcinomas and is characterized by high aggressiveness and poor prognosis despite advancements in immunotherapy. Disulfidptosis, a novel cell death pathway, has emerged as a critical mechanism in various cellular processes, including cancer. This study leverages machine learning to identify disulfidptosis-related long noncoding RNAs (DRlncRNAs) as potential prognostic biomarkers in KIRC, offering new insights into tumor pathogenesis and treatment avenues.
Results
Our analysis of data from The Cancer Genome Atlas (TCGA) led to the identification of 431 DRlncRNAs correlated with disulfidptosis-related genes. Five key DRlncRNAs (SPINT1-AS1, AL161782.1, OVCH1-AS1, AC131009.3, and AC108673.3) were used to develop a prognostic model that effectively distinguished between low- and high-risk patients with significant differences in overall survival and progression-free survival. The low-risk group had a favorable prognosis associated with a protective immune microenvironment and a better response to targeted drugs. Conversely, the high-risk group displayed aggressive tumor features and poor immunotherapy outcomes. Validation through qRT‒PCR confirmed the differential expression of these DRlncRNAs in KIRC cells compared to normal kidney cells, underscoring their potential functional significance in tumor biology.
Conclusions
This study established a robust link between disulfidptosis-related lncRNAs and patient prognosis in KIRC, underscoring their potential as prognostic biomarkers and therapeutic targets. The differential expression of these lncRNAs in tumor versus normal tissue further highlights their relevance in KIRC pathogenesis. The predictive model not only enhances our understanding of KIRC biology but also provides a novel stratification tool for precision medicine approaches, improving treatment personalization and outcomes in KIRC patients.
Similar content being viewed by others

Introduction
Approximately 52,360 men and 29,440 women are projected to be diagnosed with kidney cancer in the United States of America in 2023. Kidney cancer is the 10th leading cause of death among men with cancer and the 11th leading cause of death among women with cancer. Survival rates are similar for black and white patients with kidney cancer [1]. Renal cell carcinoma (RCC) is a common tumor of the genitourinary system. It is highly aggressive and has a high mortality rate [2]. Kidney renal clear cell carcinoma (KIRC) is the predominant pathological type of RCC, accounting for approximately 75–80% of RCC cases [3]. KIRC is heterogeneous and has a high metastatic rate and a poor prognosis [4]. Immunotherapy based on immune checkpoint inhibitors (ICIs) has benefited patients with kidney cancer and has significantly better efficacy than conventional cytokine therapy. However, long-term use of the same drugs inevitably generates drug resistance [5]. Therefore, investigating new mechanisms underlying the pathogenesis of kidney cancer and identifying more effective therapeutic targets are necessary for successful treatment.
Disulfidptosis is a novel mechanism of programmed cell death (PCD). It differs from other mechanisms of PCD, such as necroptosis, apoptosis, autophagy and ferroptosis. Disulfidptosis is mediated by high levels of cysteine uptake coupled with inadequate NADPH supply. NADPH depletion leads to the abnormal formation of disulfide bonds in actin cytoskeletal proteins, followed by disulfide stress caused by the excessive accumulation of disulfide molecules, which further affects actin cytoskeletal proteins and leads to the collapse of the actin network and eventually cell death [6]. Specifically, in SLC7A11high cells, glucose depletion leads to a decrease in NADPH production. However, the concentration of cystine does not decrease, resulting in its accumulation. Eventually, abnormal disulfide bonds bind to the actin cytoskeleton, leading to cell death [7]. Actin is a multifunctional cytoskeletal protein that participates in several cellular events, such as transcription, translation, cell morphogenesis, cell mechanics, intracellular transport and disulfide formation [8, 9]. Alterations in the cytoskeleton of plants and animals play an active role in the initiation and regulation of PCD [10, 11]. Therefore, understanding the mechanism of disulfidptosis in tumors is important for inducing cancer cell death.
With the continuous development of biosequencing technology, numerous noncoding RNAs (ncRNAs) have been identified. Long noncoding RNAs (lncRNAs) are a class of ncRNAs that have received substantial attention from scholars in recent years [12]. The expression of the lncRNA NEAT1 is downregulated in RCC and is associated with a poor prognosis. Methylation of NEAT1 results in the upregulation of its expression and decreases the proliferative and migratory abilities of cancer cells [13]. The lncRNA PLK1S1 leads to RCC progression and sorafenib resistance by inhibiting the miR-653/CXCR5 axis [14]. The lncRNA SNHG12 stabilizes SP1 by binding to SP1 and preventing its ubiquitination-dependent hydrolysis, leading to the upregulation of downstream CDCA3, which leads to RCC progression and sunitinib resistance [15]. Chen et al. established a prognostic nomogram based on five m6A-related lncRNAs to predict the overall survival of patients with KIRC and examined the biological functions of m6A-related lncRNAs [16]. Zhang et al. constructed a prognostic signature comprising seven cuproptosis-related lncRNAs that had greater predictive efficacy than clinicopathological parameters and further examined immune characteristics and drug sensitivity [17]. In addition, necrosis-, autophagy- and hypoxia-related lncRNA models have been constructed to predict the prognosis of KIRC patients [18,19,20], providing important guidance for treatment. Although the role of numerous lncRNAs in KIRC has been reported, in-depth studies on lncRNAs in KIRC are warranted owing to the scarcity of clinical samples and the limitations of cellular and animal models.
In a study, mass spectrometry revealed that the tumor microenvironment (TME) of KIRC is characterized by high infiltration of lymphocytes, particularly CD8+PD-1+ T cells [21]. Owing to the low sensitivity of RCC to conventional radiotherapy and chemotherapy, surgery is the primary treatment for RCC [22]. Immunotherapies based on ICIs, such as PD-1/PD-L1 and CTLA-4 inhibitors, mainly target the TME and have revolutionized the treatment of multiple cancers [23]. Changes in the TME of RCC are regulated by the interaction between EHBP1L1 and JAK1 [24]. Single-cell RNA sequencing of KIRC tumors revealed that T-cell depletion was significantly associated with poor prognosis and might serve as a critical factor in the immunosuppression of KIRC tumors [25]. In this study, we investigated the differences in different immune cells and immune functions in the KIRC TME by disulfidptosis-related lncRNAs (DRlncRNAs).
The relevant clinical data of patients with KIRC were obtained from The Cancer Genome Atlas (TCGA) database. R software was used to construct a prognostic model based on DRlncRNAs to improve the prediction of patient prognosis. In addition, the biological functions and signaling pathways of DRlncRNAs, immune cell infiltration, somatic mutations and immunotherapeutic efficacy were examined, and the effects of antitumor drugs on different risk groups were predicted. The findings of this study provide insights into the biological mechanisms of DRlncRNAs in KIRC and may guide the clinical prognosis of patients with KIRC.
Materials and methods
Data collection
The RNA-seq, clinical and somatic mutation data of patients with KIRC, including transcriptomic data from 541 tumor samples, 72 paracancerous samples and 532 clinical data, were extracted from the TCGA database. All patients with KIRC were randomly assigned to the training (n = 373) or test (n = 159) group. The chi-square test was used to estimate differences in the clinical characteristics of patients between the two groups. Disulfidptosis-related genes were obtained from one of the most recently published articles [6].
Construction of a risk model based on disulfidptosis-related lncRNAs
We identified 10 disulfidptosis-related genes by searching the relevant literature [6]. Then, the correlation of the expression pattern between lncRNAs and genes linked to disulfidptosis was evaluated by Pearson correlation coefficients with P < 0.001 and |R|> 0.4. Subsequently, prognostically significant DRlncRNAs were identified via continuous dimensionality reduction. First, 86 DRlncRNAs associated with the prognosis of KIRC patients were identified via univariate Cox analysis. Second, the lasso algorithm was used to reduce dimensionality, and 19 DRlncRNAs associated with overall survival (OS) in patients with KIRC were identified [26]. Third, five DRlncRNAs were identified as significant prognostic factors via multivariate Cox analysis, and a prognostic model was constructed. The risk score of each patient with KIRC was determined using a formula derived from the model, and the patients were subsequently divided into two risk groups based on the median risk score.
Assessment of the accuracy and independence of the risk model and construction of a nomogram
Kaplan–Meier (K–M) curves were plotted to assess the predictive ability of the risk model. Receiver operating characteristic (ROC) curves were plotted to examine the ability of the model to predict 1-, 3- and 5-year survival probabilities. Principal component analysis (PCA) was used for dimensionality reduction and visualization of the results of different subgroups. The risk model was examined using univariate and multivariate Cox regression analyses to assess whether it was a reliable predictor of outcomes. In addition, a nomogram was constructed by integrating various clinical characteristics and risk scores, and calibration curves were plotted to assess the accuracy of the nomogram.
Functional enrichment analysis
Differentially expressed genes (DEGs) were identified between the risk groups based on the following criteria: |log2FC| > 1.0 and p < 0.05. Gene Ontology (GO) analysis was performed to examine the biological functions of the DEGs. The analysis included three aspects, namely, biological processes (BPs), cellular components (CCs) and molecular functions (MFs). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was performed to investigate signaling pathways associated with DEGs. Gene set enrichment analysis (GSEA) was performed to identify signaling pathways in the low- and high-risk groups.
Tumor microenvironment, somatic mutation analysis and drug prediction
The ESTIMATE algorithm was used to evaluate differences in TMEs between the two risk groups [27]. The CIBERSORT algorithm was used to evaluate the infiltration levels of 22 types of immune cells in the two risk groups [28]. The ssGSEA algorithm was used to analyze immune cell infiltration and immune function in different risk groups [29]. The tumor immune dysfunction and exclusion (TIDE) scores were calculated to examine the immune escape of tumor cells and assess their response to ICIs in the two risk groups [30]. Somatic mutation data extracted from the TCGA database were used to analyze the tumor mutational burden (TMB) of patients in the two risk groups and to classify patients into low- and high-TMB groups based on the median TMB score. The ‘oncoPredict’ R package was used to calculate the IC50 values of common antitumour drugs to predict the drug response of patients with KIRC in different risk groups.
Cell culture and quantitative reverse transcription polymerase chain reaction
A human KIRC cell line (786-O, Caki-1) and a human normal control cell line (HK-2) were purchased from Pricella (Wuhan, China). The cells were cultured in RPMI-1640 medium (BI, Israel) supplemented with 10% fetal bovine serum (BI, Israel) and 1% streptomycin and penicillin. The cells were cultured at 37 °C in a humidified environment with 5% CO2. TRIzol (Invitrogen, USA) was used to extract total RNA, and qRT‒PCR was performed to detect the expression of the five DRlncRNAs. PCR was performed using UltraSYBR (CWBIO, China) on an ABI 7500 system under conventional conditions. Glycerol-3-phosphate dehydrogenase (GAPDH) was used as an endogenous reference. The sequences of primers used for PCR are listed in Table 1.
Statistical analysis
All the statistical analyses were performed using R software (version 4.2.1). A t test was used to analyze differences between groups. K‒M analysis and the log-rank test were used to examine differences in survival between the risk groups. Univariate and multivariate Cox regression analyses were performed to identify prognostic factors associated with KIRC. A p value of < 0.05 indicated a statistically significant difference.
Results
Data of patients with KIRC
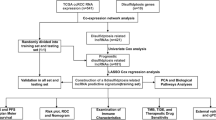
A flow chart demonstrating the study protocol is shown in Fig. 1. A total of 532 patients with KIRC were divided into training (373 patients) and test (159 patients) sets at a ratio of 7:3. The data of patients in the training set were used to identify prognostic DRlncRNAs and establish a prognostic model, whereas the data of patients in the test set were used to verify the accuracy of the model. The two groups did not significantly differ in terms of clinical characteristics, such as age, sex, grade, or TNM stage (p > 0.05) (Table 2).

Flow chart of the study protocol
Risk model based on DRlncRNAs for predicting the prognosis of KIRC
We obtained 10 disulfidptosis-related genes from the literature. Then, the Pearson correlation coefficient method was used to analyze the correlation between lncRNAs in the TCGA database and disulfidptosis-related mRNAs, and 431 DRlncRNAs were identified (Table S1). As shown in Fig. 2A, the Sankey diagram shows that 10 disulfidptosis-related genes and 431 DRlncRNAs were involved in the coexpression network. Univariate Cox regression analysis of OS data from KIRC patients in the training set identified 86 DRlncRNAs associated with OS in KIRC (Table S2). Subsequently, 19 DRlncRNAs that were strongly associated with the prognosis of KIRC patients were identified via LASSO regression analysis (Fig. 2B and C). Finally, five prognostic DRlncRNAs, namely, SPINT1-AS1, AL161782.1, OVCH1-AS1, AC131009.3 and AC108673.3, were used to construct a multivariate Cox regression model (Fig. 2D). The risk score was calculated based on the expression and Cox regression coefficients of these lncRNAs as follows: risk score = SPINT1-AS1 × -0.284849287424872 + AL161782.1 × -0.558120637222694 + OVCH1-AS1 × -0.763337398302014 + AC131009.3 × 0.469858141530083 + AC108673.3 × 0.431142218409321.

Construction of the risk model based on DRlncRNAs. (A) Sankey diagram demonstrating disulfidptosis-related genes and DRlncRNAs. (B, C) Nine DRlncRNAs were identified via LASSO regression analysis. (D) Five DRlncRNAs were used to construct the multivariate Cox regression model. (E) Correlation heatmap of 5 DRlncRNAs and disulfidptosis-related genes included in the multivariate Cox regression model. (F) Distribution of risk scores, survival status and survival time patterns of patients in different risk groups in the training set and the expression heatmap of the 5 DRlncRNAs. OS (G) and PFS (H) of patients in different risk groups in the training set
Furthermore, the correlations between 10 disulfidptosis-related genes and the 5 DRlncRNAs were examined (Fig. 2E). The expression of the five DRlncRNAs in the low- and high-risk groups is shown in Fig. 2F, and the risk score distribution, survival time, and survival status of the two risk groups in the training set were analyzed. The expression of SPINT1-AS1, AL161782.1 and OVCH1-AS1 was greater in the low-risk group than in the high-risk group, indicating that these three DRlncRNAs may serve as protective factors for the prognosis of KIRC patients. However, the expression of AC131009.3 and AC108673.3 was greater in the high-risk group than in the low-risk group, indicating that these two DRlncRNAs may serve as risk factors for KIRC. K‒M curves were subsequently plotted to assess differences in OS and progression-free survival (PFS) between the two risk groups. The results indicated that OS and PFS were better in the low-risk group than in the high-risk group (p < 0.001 for all) (Fig. 2G and H).
Validation of the DRlncRNA-based risk model and PCA
The reliability of the risk model was verified in the test set. Risk curves and scatter plots demonstrated that patients in the low-risk group had lower risk scores, longer survival times and better prognoses than those in the high-risk group (Fig. 3A). In addition, the heatmap validated that SPINT1-AS1, AL161782.1 and OVCH1-AS1 were protective factors for KIRC, whereas AC131009.3 and AC108673.3 were risk factors for KIRC. K‒M analysis revealed that OS was better in the low-risk group than in the high-risk group (p < 0.001) (Fig. 3B). The sensitivity and specificity of the risk score in the test set were assessed using ROC curves. The risk score had a larger area under the curve (AUC) than the other clinical risk indicators in the test set (Fig. 3C).

Validation of the DRlncRNA-based risk model and PCA. (A) Risk score distribution, survival status and survival time patterns of patients in different risk groups in the test set and the expression heatmap of five DRlncRNAs. (B) OS of patients in different risk groups in the test set. (C) ROC curves of clinical risk indicators and risk scores in the test set. (D) ROC curves for predicting 1-, 3- and 5-year OS in the test set. (E) PCA between different risk groups based on DRlncRNAs. (F) PCA between different risk groups based on the risk model
The AUC of the risk model for predicting 1-, 3- and 5-year OS in the test set was > 0.7 (Fig. 3D), suggesting that the risk model based on the five DRlncRNAs has good discrimination efficiency. Subsequently, PCA was performed to assess the predictive performance of 431 DRlncRNAs (Fig. 3E) and the risk model (Fig. 3F). The risk model was effective in distinguishing patients in the low-risk and high-risk groups, indicating a notable difference in sensitivity to disulfidptosis between the two groups.
Independent prognostic analysis and nomogram
Univariate and multivariate Cox regression analyses were performed to determine whether the five DRlncRNAs independently predicted the OS of patients with KIRC. The results of univariate Cox regression analysis showed that age, grade, stage and risk score were significantly associated with OS (p < 0.05 for all) (Fig. 4A). Multivariate Cox regression analysis revealed that the risk score (p < 0.001) independently predicted OS in patients with KIRC (Fig. 4B). These results suggest that the risk model constructed based on the five DRlncRNAs is an independent prognostic factor for KIRC.

Independent prognostic analysis and construction of a nomogram. (A) Forest plot of univariate Cox regression analysis. (B) Forest plot of multivariate Cox regression analysis. (C) ROC curves of clinical risk indicators and risk scores for the entire cohort. (D) ROC curves of the risk model for predicting 1-, 3- and 5-year OS for the entire cohort. (E) Nomogram. (F) Calibration curves of the nomogram
Furthermore, for the entire cohort, the risk model had greater AUC values (AUC = 0.760) than the other clinical risk indicators (Fig. 4C). The AUC of the risk model for predicting 1-, 3- and 5-year survival probabilities in the entire cohort was > 0.7, demonstrating the predictive accuracy and reliability of the model (Fig. 4D). A nomogram was constructed by integrating multiple clinical factors and risk scores to predict the survival of patients with KIRC at 1, 3 and 5 years (Fig. 4E). Subsequently, a calibration curve was plotted to verify the accuracy of the nomogram (Fig. 4F). K‒M curves were plotted to examine the efficiency of the risk model in predicting several clinical traits, such as age, sex and TNM stage. The results indicated that the low-risk group had a better prognosis (Fig. 5).

K‒M analysis of OS in different subgroups based on the clinical characteristics of patients with KIRC in the TCGA cohort. (A) Age ≤ 65 years. (B) Age > 65 years. (C) Female. (D) Male. (E) Stages I–II. (F) Stage III–IV. (G) T1–T2. (H) T3–T4.
Functional enrichment analysis
GO and KEGG enrichment analyses were performed to examine the functions of the 735 DEGs (Table S3). The DEGs were enriched in immune-related BPs, including humoral immune responses, defense responses against bacteria and organic anion transport. In terms of CCs, the DEGs were significantly enriched in apical cells, collagen-containing extracellular matrix and immunoglobulin complexes. In terms of MFs, the DEGs were involved in receptor–ligand activity, antigen binding and immunoglobulin–receptor binding (Fig. 6A).

Functional enrichment analyses. (A) GO. (B) KEGG. (C, D) GSEA.
KEGG enrichment analysis indicated that the DEGs were mainly enriched in pathways associated with cytokine–cytokine receptor interaction, protein digestion and absorption, viral protein interaction with cytokines and cytokine receptors and complement and coagulation cascades (Fig. 6B).
In addition, GSEA revealed that pathways associated with primary immunodeficiency, taste transduction, hematopoietic cell lineage, nod-like receptor signaling and cytokine–cytokine receptor interaction were significantly enriched in the high-risk group (Fig. 6C), whereas pathways associated with apoptosis, drug metabolism, cytochrome p450, fatty acid metabolism, PPAR signaling, valine, leucine and isoleucine degradation and other metabolism-related pathways were significantly enriched in the low-risk group (Fig. 6D). We speculate that disulfidptosis is closely related to immune-related pathways, metabolism and apoptosis.
Immune infiltration landscape analysis and immunotherapeutic efficacy
The TME plays a crucial role in the progression and treatment of KIRC. Therefore, we examined the TME in different risk groups using multiple algorithms. The results of the ESTIMATE algorithm revealed that the immune and ESTIMATE scores of patients were lower in the low-risk group than in the high-risk group (p < 0.05 for all) (Fig. 7A), whereas the stromal scores did not differ between the two groups. Furthermore, the CIBERSORT algorithm was used to evaluate the proportions of 22 types of tumor-infiltrating immune cells. The distribution of immune cells differed between the two risk groups (Fig. 7B). As shown in Fig. 7C, the box plot demonstrated that the abundances of CD8+ T cells, T follicular helper cells, regulatory T cells (Tregs), M0 macrophages, activated mast cells and neutrophils were lower in the low-risk group, whereas those of CD4 resting memory T cells, monocytes, M1 macrophages, M2 macrophages, resting dendritic cells, resting mast cells and eosinophils were greater in the low-risk group (p < 0.05 for all). In addition, the ssGSEA algorithm was used to analyze immune cell infiltration and immune function in different risk groups. The abundance of iDCs, mast cells and neutrophils was greater in the low-risk group, and MHC class I and type II IFN responses were greater in the low-risk group (p < 0.05 for all) (Fig. 7D). Finally, the TIDE algorithm was used to examine the relationship between the risk score and immunotherapy response. The results showed that the high-risk group had a significantly greater potential for immune escape and worse treatment outcomes (p < 0.001) (Fig. 7E).

Immune infiltration analysis and assessment of immunotherapy outcomes. (A) The ESTIMATE algorithm was used to assess differences in immune, stromal and ESTIMATE scores between the two groups. (B, C) The CIBERSORT algorithm was used to evaluate differences in the abundances of 22 types of immune cells between the two groups. (D) The ssGSEA algorithm was used to analyze differences in the proportions of immune cells and immune functions between the two groups. (E) The TIDE algorithm was used to calculate TIDE scores between the two groups
Analysis of the somatic mutation landscape
By comparing the somatic mutation rate between the two risk groups, we found that the mutation rate was lower in the low-risk group (153/199 [76.88%] samples) than in the high-risk group (138/168 [82.14%] samples). The top 15 genes driving the mutations are shown in Fig. 8A. Furthermore, TMB scores were greater in the high-risk group than in the low-risk group (p = 0.012) (Fig. 8B). Patients with KIRC were divided into low- and high-TMB groups based on the median TMB score. K‒M analysis indicated that patients in the low-TMB subgroup had significantly better OS than did those in the high-TMB subgroup (p < 0.001) (Fig. 8C). The TMB and risk score were integrated to compare their efficiency in predicting the prognosis of KIRC patients. K‒M analysis revealed that patients with low TMB and risk scores had the best OS, whereas those with high TMB and risk scores had the worst OS (p < 0.001) (Fig. 8D).

Analysis of the somatic mutation landscape. (A) Mutation distribution in the low-risk group. (B) Mutation distribution in the high-risk group. (C) Differences in TMB scores between different risk groups. (D) K‒M analysis of OS in different TMB groups. (E) K‒M analysis of OS among the four groups based on TMB scores and risk scores
Analysis of drug response
Given the significant differences in prognosis, immune microenvironment and somatic mutation frequency between the two risk groups, we screened for drugs sensitive to precision therapy in patients in the two risk groups. The R package oncoPredict can be used to associate antitumor drugs with biomarkers and to easily predict the response of patients to many anticancer drugs [31]. We used OncoPredict to screen for antitumor drugs for patients with KIRC in different risk groups. The IC50 values of four drugs (axitinib, ibrutinib, osimertinib and ruxolitinib) were lower in the low-risk group, indicating that these drugs are more beneficial for patients with KIRC with low risk scores (Fig. 9A and D). The IC50 values of the other four drugs (crizotinib, lapatinib, linsitinib and nilotinib) were greater in the low-risk group, indicating that these drugs are more effective in patients with KIRC with high risk scores (Fig. 9E and H).

Analysis of therapeutic sensitivity. (A) Axitinib. (B) Ibrutinib. (C) Osimertinib. (D) Ruxolitinib. (E) Crizotinib. (F) Lapatinib. (G) Linsitinib. (H) Nilotinib
The expression pattern of DRlncRNAs
The expression of five DRlncRNAs in KIRC samples and normal samples was first explored in the TCGA database. As shown in Fig. 10A and E, SPINT1-AS1 and AL161782.1 were downregulated, while OVCH1-AS1, AC131009.3 and AC108673.3 were upregulated in KIRC samples compared with normal samples. To validate the reliability of the model, qRT‒PCR was performed to verify the expression of the five DRlncRNAs in normal human renal tubular epithelial cells (HK-2) and KIRC cells (786-O, Caki-1). The expression of SPINT1-AS1 and AL161782.1 was lower in KIRC cells than in normal kidney cells, whereas that of OVCH1-AS1, AC131009.3 and AC108673.3 was greater in KIRC cells than in normal kidney cells (Fig. 10F and J). These results demonstrate the reliability of the risk model constructed based on the five DRlncRNAs.

Exploration of the expression of DRlncRNAs in KIRC. Expression pattern of (A) SPINT1-AS1. (B) AL161782.1. (C) OVCH1-AS1. (D) AC131009.3. (E) AC108673.3 in normal or KIRC samples in the TCGA database. qRT‒PCR was used to detect the expression of (F) SPINT1-AS1. (G) AL161782.1. (H) OVCH1-AS1. (I) AC131009.3. (J) AC108673.3 expression in normal human renal tubular epithelial cells (HK-2) and KIRC cells (786-O, Caki-1). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Discussion
KIRC is a relatively common type of cancer in adults. Because KIRC is not easily detected in the early stages, most patients with KIRC are diagnosed in the middle to late stages and have progressed to metastasis [32]. Few biomarkers are available for the early diagnosis or treatment of KIRC, resulting in a relatively poor prognosis [33]. Immunotherapy can be used to treat patients with kidney cancer. However, two-thirds of patients with advanced disease respond poorly to ICI therapy, possibly because of factors in the TME that help tumor cells evade immunotherapy [34]. To improve the diagnosis of patients with early-stage KIRC and reduce the likelihood of metastasis, we screened for prognosis-related biomarkers for KIRC using bioinformatics tools. These markers may help in the development of more effective immunotherapeutic strategies.
PCD is an active form of cell death mediated by specific genes and proteins. It plays an essential role in maintaining the growth and development of the body and the normal physiological functions of tissues and organs [35]. The PCD mechanisms reported in KIRC include ferroptosis, autophagy and apoptosis. Researchers have attempted to induce PCD through several targets and subsequently kill cancer cells to achieve the goal of treating tumors. Targeted inhibition of ISCA2 reduces HIF-1/2α levels, increases lipid peroxidation, induces iron death and inhibits tumor growth in KIRC [36]. Silencing of the obesity-associated protein FTO enhances N6-methyladenosine (m6A)-mediated autophagic flux and inhibits tumor growth and metastasis in KIRC [37]. In addition, knockdown of KEAP1 inhibits the resistance of KIRC cells to axitinib and promotes apoptosis [38]. Wang et al. constructed a 6-gene signature related to cuprotosis to accurately predict the prognosis of KIRC patients [39]. Recent studies have proposed a novel disulfide stress-mediated PCD mode called disulfidptosis [6].
Excessive accumulation of intracellular disulfide molecules disrupts the actin network because actin cytoskeletal proteins are especially vulnerable to disulfide stress. Studies have demonstrated a close relationship between actin and apoptosis at the cellular and tissue levels. Actin binds to the α-actin protein, predisposing osteoblasts at focal adhesion sites to apoptosis [40]. Apoptotic cells can be expelled by the assembly of F-actin and myosin [41]. The precise alignment of disulfide bonds is essential for maintaining the structural integrity of intracellular cysteine-rich cytokines [42]. Oxidized cysteine residues in actin in sickle cell anemia can form disulfide bonds that reduce the dynamics of actin filaments and lead to disintegration of the erythrocyte cytoskeleton [43]. The thiol–disulfide oxidoreductase PDI1;1 reduces and degrades disulfide bonds in the high-molecular-weight structure of rice actin [44]. In addition, the surface virulence factor IcsA plays an important role in actin-based motility in Shigella through three cysteine residues [45].
In-depth studies have revealed that lncRNAs can either promote or suppress tumors by participating in gene signaling regulation and can serve as promising potential biomarkers [46,47,48]. In one study, eight ferroptosis-associated lncRNAs were used to construct a predictive model that accurately predicted the prognosis of KIRC patients and suggested treatment options [49]. However, the role of DRlncRNAs in KIRC remains elusive. In this study, we identified DRlncRNAs based on disulfidptosis-related genes and investigated how lncRNAs regulating disulfidptosis affect tumor progression and prognosis in KIRC.
Stepwise dimensionality reduction was performed sequentially through lasso and Cox regression analyses to reduce the number of feature parameters and construct a prognostic model. This method has been reported in several studies [50, 51]. Five DRlncRNAs significantly associated with the prognosis of KIRC, namely, SPINT1-AS1, AL161782.1, OVCH1-AS1, AC131009.3 and AC108673.3, were identified and used to construct a prognostic model. Subsequently, a nomogram was developed to improve the prediction of OS in patients with KIRC at 1, 3 and 5 years. Among the five DRlncRNAs, three lncRNAs, OVCH1-AS1, AC131009.3 and AC108673.3, have not been investigated previously, whereas the other two lncRNAs are closely associated with cancer. The lncRNA SPINT1-AS1 has been extensively investigated. It is inconsistently expressed in different cancers and is essential for inhibiting or promoting cancer progression. Initially, the expression of the lncRNA SPINT1-AS1 was found to be elevated in colorectal cancer (CRC) and was negatively associated with the mRNA expression of SPINT1 and the prognosis of CRC patients [52]. Subsequent studies have demonstrated that SPINT1-AS1 promotes CRC progression by suppressing miR-214 and increasing HDGF expression [53]. In addition, SPINT1-AS1 is an important breast cancer (BC) regulator that may interact with the miRNA let-7 a/b/i-5p, and its knockdown inhibits the proliferation and migration of BC cells and promotes the resistance of BC cells to lapatinib [54, 55]. SPINT1-AS1 is upregulated in cervical cancer, inhibits miR-214 via DNM3OS, activates the Wnt/β-catenin signaling pathway and is associated with a poor prognosis [56]. In addition, SPINT1-AS1 expression is downregulated in esophageal squamous cell carcinoma (ESCC), and patients with low expression of SPINT1-AS1 have shorter OS [57]. Furthermore, the expression of SPINT1-AS1 is significantly increased in growth hormone-secreting pituitary adenomas (GH-PAs) and is strongly associated with cancer aggressiveness [58]. In one study, 14 immune-related lncRNAs, including SPINT1-AS1, were used to construct a risk model for predicting the prognosis of patients with melanoma [59]. The lncRNA AL161782.1 is considered a m6A- and cuproptosis-related lncRNA in KIRC [60, 61]. In this study, AL161782.1 was identified as a DRlncRNA. We speculate that it is involved in multiple modes of PCD.
The biological functions and signaling pathways of the five DRlncRNAs were examined via enrichment analysis. The results of GO and KEGG analyses indicated that the DRlncRNAs were associated with several immune and metabolic pathways. Several proteins involved in complement and coagulation cascade reactions are upregulated in the serum of patients with thymomas [62]. DRlncRNAs were found to be associated with complement and coagulation cascades in this study. GSEA revealed that DRlncRNAs can influence immune- and apoptosis-related pathways in KIRC. However, further studies are warranted to verify these findings.
Changes in the TME of kidney cancer may lead to disease progression [63]. We examined the relationship between the risk score and changes in the TME of KIRC patients. Both immune and ESTIMATE scores were significantly greater in the high-risk group. In addition, patients in the high-risk group had greater immune cell infiltration and enhanced immune function, which explains, to some extent, differences in the OS of patients with KIRC between the two risk groups. The NF-κB signaling pathway is significantly activated in infiltrating CD8+ T cells, inducing apoptosis and leading to a worse overall prognosis [64], which is consistent with the results of this study. Tregs have been strongly associated with poor prognosis and poor immunotherapy outcomes in KIRC patients [65]. Th2 cells are a subpopulation of CD4+ T cells that secrete IL-4 and IL-10 to promote tumor growth by suppressing the host immune system [66]. This phenomenon was observed in the high-risk group in this study. We speculate that disulfidptosis mediates alterations in immune function and hence influences the progression of KIRC. However, relevant basic studies demonstrating the relationship between disulfidptosis and immunity in KIRC are lacking. In this study, the abundances of most immune cells and immune function scores were greater in the high-risk group than in the low-risk group, suggesting that immune activity was greater in patients with high risk scores. Furthermore, the TIDE algorithm was used to estimate the relationship between risk score and response to ICIs. The results revealed that the high-risk group had a poorer response to ICI treatment, suggesting that DRlncRNAs are promising biomarkers for predicting the response to ICIs in KIRC patients.
A high TMB is associated with poor survival outcomes and may inhibit immune infiltration in KIRC patients [67]. In this study, the risk score was found to be significantly positively correlated with the TMB. The high-risk-score and high-TMB-score groups had the shortest survival and the worst prognosis, whereas the low-risk-score and low-TMB-score groups had the highest survival rate and the best prognosis. This finding demonstrates the accuracy of the risk score in predicting immunotherapy outcomes in patients with KIRC. Furthermore, we evaluated the sensitivity of patients with KIRC to anticancer drugs in the two risk groups. Axitinib, ibrutinib, osimertinib and ruxolitinib were more effective in patients in the low-risk group, whereas crizotinib, lapatinib, linsitinib and nilotinib were more effective in patients in the high-risk group. These findings may help guide the treatment of patients with KIRC in clinical settings.
This study has several limitations that should be acknowledged. First, while our prognostic model was developed and validated using the TCGA dataset, we were unable to find a suitable external dataset that included all the necessary lncRNAs for validation. This lack of external validation limits the generalizability of our findings, and future studies should prioritize the collection of data on these specific lncRNAs in independent cohorts to further test the reliability and robustness of our model. Second, the biological roles and underlying mechanisms of the candidate DRlncRNAs in KIRC progression were not explored in this study. Future research should focus on validating the prognostic value of these lncRNAs using both in vitro and in vivo experiments to provide more comprehensive biological evidence to support the statistical findings of our study.
Conclusion
In summary, this study is the first to report the role of DRlncRNAs in KIRC. A prognostic model was constructed based on DRlncRNAs using data from the TCGA-KIRC cohort. The findings of this study offer prospects for the diagnosis and treatment of KIRC.
Data availability
No datasets were generated or analysed during the current study.
References
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. Cancer J Clin. 2023;73(1):17–48.
Li G, Xie ZK, Zhu DS, Guo T, Cai QL, Wang Y. KIF20B promotes the progression of clear cell renal cell carcinoma by stimulating cell proliferation. J Cell Physiol. 2019;234(9):16517–25.
Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, et al. Renal cell carcinoma. Nat Rev Dis Primers. 2017;3:17009.
Zhai W, Zhu R, Ma J, Gong D, Zhang H, Zhang J, et al. A positive feed-forward loop between LncRNA-URRCC and EGFL7/P-AKT/FOXO3 signaling promotes proliferation and metastasis of clear cell renal cell carcinoma. Mol Cancer. 2019;18(1):81.
Liu W, Ren D, Xiong W, Jin X, Zhu L. A novel FBW7/NFAT1 axis regulates cancer immunity in sunitinib-resistant renal cancer by inducing PD-L1 expression. J Exp Clin Cancer Res. 2022;41(1):38.
Liu X, Nie L, Zhang Y, Yan Y, Wang C, Colic M, et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol. 2023;25(3):404–14.
Machesky LM. Deadly actin collapse by disulfidptosis. Nat Cell Biol. 2023;25(3):375–6.
Ren W, Zhao W, Cao L, Huang J. Involvement of the Actin Machinery in programmed cell death. Front Cell Dev Biol. 2020;8:634849.
Zhang Y, Chang SKC. Color and texture of surimi-like gels made of protein isolate extracted from catfish byproducts are improved by washing and adding soy whey. J Food Sci. 2022;87(7):3057–70.
Franklin-Tong VE, Gourlay CW. A role for actin in regulating apoptosis/programmed cell death: evidence spanning yeast, plants and animals. Biochem J. 2008;413(3):389–404.
Smertenko A, Franklin-Tong VE. Organisation and regulation of the cytoskeleton in plant programmed cell death. Cell Death Differ. 2011;18(8):1263–70.
Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18(1):5–18.
Chen J, Liao X, Cheng J, Su G, Yuan F, Zhang Z, et al. Targeted methylation of the LncRNA NEAT1 suppresses malignancy of renal cell carcinoma. Front Cell Dev Biol. 2021;9:777349.
Li W, Yang D, Zhang Y, Zhao S, Li D, Liu M. Long non–coding RNA PLK1S1 was associated with renal cell carcinoma progression by interacting with microRNA–653 and altering C–X–C chemokine receptor 5 expression. Oncol Rep. 2020;44(5):1985–96.
Liu Y, Cheng G, Huang Z, Bao L, Liu J, Wang C, et al. Long noncoding RNA SNHG12 promotes tumour progression and sunitinib resistance by upregulating CDCA3 in renal cell carcinoma. Cell Death Dis. 2020;11(7):515.
Yu J, Mao W, Sun S, Hu Q, Wang C, Xu Z, et al. Identification of an m6A-Related lncRNA signature for Predicting the prognosis in patients with kidney renal clear cell carcinoma. Front Oncol. 2021;11:663263.
Li D, Wu X, Song W, Cheng C, Hao L, Zhang W. Clinical significance and immune landscape of cuproptosis-related lncRNAs in kidney renal clear cell carcinoma: a bioinformatical analysis. Ann Transl Med. 2022;10(22):1235.
Luo Y, Zhang G. Identification of a Necroptosis-Related Prognostic Index and Associated Regulatory Axis in kidney renal clear cell carcinoma. Int J Gen Med. 2022;15:5407–23.
Yu J, Mao W, Xu B, Chen M. Construction and validation of an autophagy-related long noncoding RNA signature for prognosis prediction in kidney renal clear cell carcinoma patients. Cancer Med. 2021;10(7):2359–69.
Chen H, Pan Y, Jin X, Chen G. Identification of a four Hypoxia-Associated Long non-coding RNA signature and establishment of a Nomogram Predicting Prognosis of Clear Cell Renal Cell Carcinoma. Front Oncol. 2021;11:713346.
Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, et al. Immune Atlas Clear Cell Ren Cell Carcinoma Cell. 2017;169(4):736–49. e18.
Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. 2019;380(12):1116–27.
Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discov. 2022;21(7):509–28.
Pan Y, Shu G, Fu L, Huang K, Zhou X, Gui C, et al. EHBP1L1 drives Immune Evasion in Renal Cell Carcinoma through binding and stabilizing JAK1. Adv Sci (Weinh). 2023;10(11):e2206792.
Hu J, Chen Z, Bao L, Zhou L, Hou Y, Liu L, et al. Single-cell transcriptome analysis reveals Intratumoral Heterogeneity in ccRCC, which results in different clinical outcomes. Mol Ther. 2020;28(7):1658–72.
Zhang Z, Wang S, Zhu Z, Nie B. Identification of potential feature genes in non-alcoholic fatty liver disease using bioinformatics analysis and machine learning strategies. Comput Biol Med. 2023;157:106724.
Yu S, Wang Y, Peng K, Lyu M, Liu F, Liu T. Establishment of a Prognostic signature of Stromal/Immune-Related genes for gastric adenocarcinoma based on ESTIMATE Algorithm. Front Cell Dev Biol. 2021;9:752023.
Guan M, Jiao Y, Zhou L. Immune Infiltration Analysis with the CIBERSORT Method in Lung Cancer. Dis Markers. 2022;2022:3186427.
Wang J, Xia W, Huang Y, Li H, Tang Y, Li Y, et al. A vasculogenic mimicry prognostic signature associated with immune signature in human gastric cancer. Front Immunol. 2022;13:1016612.
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550–8.
Maeser D, Gruener RF, Huang RS. oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform. 2021;22(6):34260682.
Barata PC, Rini BI. Treatment of renal cell carcinoma: current status and future directions. CA Cancer J Clin. 2017;67(6):507–24.
De Meerleer G, Khoo V, Escudier B, Joniau S, Bossi A, Ost P, et al. Radiotherapy for renal-cell carcinoma. Lancet Oncol. 2014;15(4):e170–7.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168(4):707–23.
Patra S, Patil S, Klionsky DJ, Bhutia SK. Lysosome signaling in cell survival and programmed cell death for cellular homeostasis. J Cell Physiol. 2023;238(2):287–305.
Green YS, Ferreira Dos Santos MC, Fuja DG, Reichert EC, Campos AR, Cowman SJ, et al. ISCA2 inhibition decreases HIF and induces ferroptosis in clear cell renal carcinoma. Oncogene. 2022;41(42):4709–23.
Xu Y, Zhou J, Li L, Yang W, Zhang Z, Zhang K, et al. FTO-mediated autophagy promotes progression of clear cell renal cell carcinoma via regulating SIK2 mRNA stability. Int J Biol Sci. 2022;18(15):5943–62.
Huang H, Zhang J, Jiang P, Xu X, Huang F, Zhao B, et al. FXR1 facilitates axitinib resistance in clear cell renal cell carcinoma via regulating KEAP1/Nrf2 signaling pathway. Anticancer Drugs. 2023;34(2):248–56.
Zhang F, Lin J, Feng D, Liang J, Lu Y, Liu Z, et al. Cuprotosis-related signature predicts overall survival in clear cell renal cell carcinoma. Front Cell Dev Biol. 2022;10:922995.
Triplett JW, Pavalko FM. Disruption of alpha-actinin-integrin interactions at focal adhesions renders osteoblasts susceptible to apoptosis. Am J Physiol Cell Physiol. 2006;291(5):C909–21.
Rosenblatt J, Raff MC, Cramer LP. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr Biol. 2001;11(23):1847–57.
Hasipek M, Grabowski D, Guan Y, Alugubelli RR, Tiwari AD, Gu X, et al. Therapeutic targeting of protein disulfide isomerase PDIA1 in multiple myeloma. Cancers (Basel). 2021;13(11):34071205.
Farah ME, Sirotkin V, Haarer B, Kakhniashvili D, Amberg DC. Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskeleton (Hoboken). 2011;68(6):340–54.
Onda Y, Okino T. Thiol-disulfide oxidoreductase PDI1;1 regulates actin structures in Oryza sativa root cells. FEBS Lett. 2022;596(23):3015–23.
Qin J, Hong Y, Morona R, Totsika M. Cysteine-dependent conformational heterogeneity of Shigella flexneri Autotransporter IcsA and implications of its function. Microbiol Spectr. 2022;10(6):e0341022.
Meng K, Lu S, Li YY, Hu LL, Zhang J, Cao Y, et al. LINC00493-encoded microprotein SMIM26 exerts anti-metastatic activity in renal cell carcinoma. EMBO Rep. 2023;24(6):e56282.
Zhang MX, Zhang LZ, Fu LM, Yao HH, Tan L, Feng ZH, et al. Positive feedback regulation of lncRNA PVT1 and HIF2alpha contributes to clear cell renal cell carcinoma tumorigenesis and metastasis. Oncogene. 2021;40(37):5639–50.
Tan C, Du H, Wang Y, Zhao J, Cheng X, Lan H. LncRNA GABPB1-IT1 inhibits the tumorigenesis of renal cancer via the miR-21/PTEN axis. J Biochem Mol Toxicol. 2023;37(4):e23288.
Lai J, Miao S, Ran L. Ferroptosis-associated lncRNA prognostic signature predicts prognosis and immune response in clear cell renal cell carcinoma. Sci Rep. 2023;13(1):2114.
Zhang Z, Shi Y, Zhu Z, Fu J, Liu D, Liu X, et al. Characterization of myeloid signature genes for predicting prognosis and immune landscape in ewing sarcoma. Cancer Sci. 2023;114(4):1240–55.
Zhang X, Wu H, Niu J, Hu Y, Zhang W, Chang J, et al. A novel mitochondria-related gene signature in esophageal carcinoma: prognostic, immune, and therapeutic features. Funct Integr Genomics. 2023;23(2):109.
Li C, Li W, Zhang Y, Zhang X, Liu T, Zhang Y, et al. Increased expression of antisense lncRNA SPINT1-AS1 predicts a poor prognosis in colorectal cancer and is negatively correlated with its sense transcript. Onco Targets Ther. 2018;11:3969–78.
Fang Y, Yang Q. Specificity protein 1-induced serine peptidase inhibitor, Kunitz Type 1 antisense RNA1 regulates colorectal cancer cell proliferation, migration, invasion and apoptosis through targeting heparin binding growth factor via sponging microRNA-214. Bioengineered. 2022;13(2):3309–22.
Zhou T, Lin K, Nie J, Pan B, He B, Pan Y, et al. LncRNA SPINT1-AS1 promotes breast cancer proliferation and metastasis by sponging let-7 a/b/i-5p. Pathol Res Pract. 2021;217:153268.
Xiang Z, Song S, Zhu Z, Sun W, Gifts JE, Sun S, et al. LncRNAs GIHCG and SPINT1-AS1 are crucial factors for Pan-cancer cells sensitivity to Lapatinib. Front Genet. 2019;10:25.
Song H, Liu Y, Liang H, Jin X, Liu L. SPINT1-AS1 drives cervical Cancer Progression via repressing miR-214 Biogenesis. Front Cell Dev Biol. 2021;9:691140.
Shen FF, Pan Y, Yang HJ, Li JK, Zhao F, Su JF, et al. Decreased expression of SPINT1-AS1 and SPINT1 mRNA might be independent unfavorable prognostic indicators in esophageal squamous cell carcinoma. Onco Targets Ther. 2019;12:4755–63.
Yin H, Zheng X, Tang X, Zang Z, Li B, He S, et al. Potential biomarkers and lncRNA-mRNA regulatory networks in invasive growth hormone-secreting pituitary adenomas. J Endocrinol Invest. 2021;44(9):1947–59.
Li FW, Luo SK. Identification and construction of a Predictive Immune-related lncRNA signature model for Melanoma. Int J Gen Med. 2021;14:9227–35.
Liu L, Zhuang M, Tu XH, Li CC, Liu HH, Wang J. Bioinformatics analysis of markers based on m(6) a related to prognosis combined with immune invasion of renal clear cell carcinoma. Cell Biol Int. 2023;47(1):260–72.
Hong P, Huang W, Du H, Hu D, Cao Q, Wang Y, et al. Prognostic value and immunological characteristics of a novel cuproptosis-related long noncoding RNAs risk signature in kidney renal clear cell carcinoma. Front Genet. 2022;13:1009555.
Li J, Qi G, Liu Y. Proteomics analysis of serum from thymoma patients. Sci Rep. 2023;13(1):5117.
Zhang X, Qin X, Yu T, Wang K, Chen Y, Xing Q. Chromatin regulators-related lncRNA signature predicting the prognosis of kidney renal clear cell carcinoma and its relationship with immune microenvironment: a study based on bioinformatics and experimental validation. Front Genet. 2022;13:974726.
Kourtis N, Wang Q, Wang B, Oswald E, Adler C, Cherravuru S, et al. A single-cell map of dynamic chromatin landscapes of immune cells in renal cell carcinoma. Nat Cancer. 2022;3(7):885–98.
Bai D, Feng H, Yang J, Yin A, Qian A, Sugiyama H. Landscape of immune cell infiltration in clear cell renal cell carcinoma to aid immunotherapy. Cancer Sci. 2021;112(6):2126–39.
Romagnani S. Th1/Th2 cells. Inflamm Bowel Dis. 1999;5(4):285–94.
Zhang C, Li Z, Qi F, Hu X, Luo J. Exploration of the relationships between tumor mutation burden with immune infiltrates in clear cell renal cell carcinoma. Ann Transl Med. 2019;7(22):648.
Acknowledgements
The authors sincerely acknowledge the publicly available TCGA (https://portal.gdc.cancer.gov/) database.
Funding
This study was supported by the Major Project of Natural Science Research in Colleges and Universities in Jiangsu Province (20KJA320005), the Open Project Program of Jiangsu Key Laboratory of Zoonosis (No. R2015), and the Nanjing Medical University Kanda College Research and Development Fund Program (KD2023KYJJ163).
Author information
Authors and Affiliations
Contributions
Kangjie Xu: Conceptualization, Methodology, Investigation, Software, Writing – original draft. Dongling Li: Conceptualization, Methodology, Investigation, Software, Writing – original draft. Hua Dai: Conceptualization, funding acquisition, project administration, resources, supervision. Hang Sun: Conceptualization, funding acquisition, project administration, resources, supervision. Kangkang Ji: Data curation, Investigation, Software, Writing – original draft. Yanhua Zhang: Data curation, Formal analysis, Investigation, Writing – original draft. Minglei Zhang: Data curation, Formal analysis, Investigation, Writing – review & editing. Hai Zhou: Formal analysis, Software, Validation, Visualization, Writing – review & editing. Xuefeng Hou: Formal analysis, Software, Validation, Visualization, Writing – review & editing. Jian Jiang: Software, Validation, Visualization, Writing – review & editing. Zihang Zhang: Validation, Visualization, Writing – review & editing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, K., Li, D., Ji, K. et al. Disulfidptosis-associated LncRNA signature predicts prognosis and immune response in kidney renal clear cell carcinoma. Biol Direct 19, 71 (2024). https://doi.org/10.1186/s13062-024-00517-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13062-024-00517-7